

Abstract
The molecular mechanism of long-term memory has been extensively studied in the context of the hippocampus-dependent recent memory examined within several days. However, months-old remote memory maintained in the cortex for long-term has not been investigated much at the molecular level yet. Various epigenetic mechanisms are known to be important for long-term memory, but how the 3D chromatin architecture and its regulator molecules contribute to neuronal plasticity and systems consolidation is still largely unknown. CCCTC-binding factor (CTCF) is an 11-zinc finger protein well known for its role as a genome architecture molecule. Male conditional knock-out mice in which CTCF is lost in excitatory neurons during adulthood showed normal recent memory in the contextual fear conditioning and spatial water maze tasks. However, they showed remarkable impairments in remote memory in both tasks. Underlying the remote memory-specific phenotypes, we observed that female CTCF conditional knock-out mice exhibit disrupted cortical LTP, but not hippocampal LTP. Similarly, we observed that CTCF deletion in inhibitory neurons caused partial impairment of remote memory. Through RNA sequencing, we observed that CTCF knockdown in cortical neuron culture caused altered expression of genes that are highly involved in cell adhesion, synaptic plasticity, and memory. These results suggest that remote memory storage in the cortex requires CTCF-mediated gene regulation in neurons, whereas recent memory formation in the hippocampus does not.
SIGNIFICANCE STATEMENT CCCTC-binding factor (CTCF) is a well-known 3D genome architectural protein that regulates gene expression. Here, we use two different CTCF conditional knock-out mouse lines and reveal, for the first time, that CTCF is critically involved in the regulation of remote memory. We also show that CTCF is necessary for appropriate expression of genes, many of which we found to be involved in the learning- and memory-related processes. Our study provides behavioral and physiological evidence for the involvement of CTCF-mediated gene regulation in the remote long-term memory and elucidates our understanding of systems consolidation mechanisms.
Introduction
The formation of long-term memory in the brain involves dynamic gene regulation through multiple layers of mechanisms. This has been established based on studies focusing on the recent long-term memory, which is typically examined one to several days after learning (Lee et al., 2007; Huang et al., 2013; Nagayoshi et al., 2017). However, memory is believed to be further processed into remote long-term memory over several weeks after the initial consolidation, during which the major brain region preserving the memory is shifted from the hippocampus to the cortex; this process is called systems consolidation (Frankland et al., 2004; Wang et al., 2006). During systems consolidation, cortical neural circuits bearing memory traces are reorganized and strengthened, leading to stabilization of the memory for long-term storage.
For the expression of remote memory, anterior cingulate cortex (ACC) has been noted to be particularly important among several brain regions (Einarsson et al., 2015). Several studies have shown that ACC is critical for remote fear and spatial memories (Frankland et al., 2004; Teixeira et al., 2006). More recent studies have shown that optogenetic inhibition of ACC (Goshen et al., 2011) and a disruption of spine growth in ACC (Restivo et al., 2009) block the contextual fear memory, suggesting that structural modifications in ACC are important for remodeling of long-range cortical connections during systems consolidation. Some previous studies have shown that epigenetic mechanisms are involved in systems consolidation. For example, cortical DNA methylation and histone acetylation participate in the formation and/or maintenance of remote memory (Miller et al., 2010; Yu et al., 2011; Peixoto and Abel, 2013). However, much of systems consolidation process still remains elusive, and the exact molecular underpinning of how remote memory is regulated is yet to be discovered.
Recently, 3D genome architecture has been receiving increasing attention in the research on epigenetic mechanisms (Bonev and Cavalli, 2016). 3D genome organization allows chromatin interaction within the topologically associating domains, which are genomic regions that frequently make contacts within themselves (Pombo and Dillon, 2015). This provides important structural bases for gene regulation, thereby contributing to the cell type-specific gene expression (Bouwman and de Laat, 2015). In both mouse models and human patients, the disruption of 3D chromatin architecture has been shown to be associated with neuropsychiatric diseases, such as Alzheimer's disease, post-traumatic stress disorder, and schizophrenia (Medrano-Fernández and Barco, 2016). However, the role of 3D chromatin architecture in remote memory has not been addressed to date.
Among several chromatin architecture regulators, CTCF is one of the most well-characterized proteins. CTCF exhibits an insulator activity, which guards genes from inappropriate chromatin interactions (Gaszner and Felsenfeld, 2006). CTCF recognizes the consensus sequence CCGCGNGGNGGCAG using 11 zinc finger motifs (Kim et al., 2007) and can bind to various sequences through combinations of its zinc fingers (Filippova et al., 1996). Chromatin immunoprecipitation experiments combined with high-throughput sequencing (ChIP-seq) have mapped CTCF binding activities in diverse tissues, revealing that CTCF can bind to 55,000–65,000 sites on the mammalian genome (Ong and Corces, 2014). CTCF binds to enhancers, gene promoters, and gene bodies to regulate genome architecture (Holwerda and de Laat, 2013; Nichols and Corces, 2015), and its binding site occupancy is modulated by epigenetic processes, such as DNA methylation (Maurano et al., 2015). As many of the CTCF binding sites are located near genes (Hirayama et al., 2012), CTCF forms chromatin loops with other transcription factors, such as cohesion, and regulate transcription (Guo et al., 2012). Also, it has been shown that the orientation of CTCF-binding sites is highly correlated to chromatin loops and 3D organization of the genome (Guo et al., 2015). As CTCF-mediated genome architecture mediates nuclear processes, such as enhancer-promoter interactions and alternative splicing (Kornblihtt, 2012; Ren et al., 2017), CTCF acts as a linker protein that connects the genome architecture to its function.
In this study, we examined two CTCF-deficient mouse models to explore a novel role of CTCF in systems consolidation.
Materials and Methods
Animals
CTCF conditional knock-out (cKO) mice were generated by crossing CTCFfl/+;CaMKIIαCre/+ with CTCFfl/+;CaMKIIα+/+. Heterozygous CTCF (HT) mice were generated by crossing CTCFfl/+ with VgatCre/+. Littermates that did not carry the Cre transgene or the floxed CTCF were used as controls. The 12- to 15-week-old adult male and female mice were used for the molecular, behavioral, and electrophysiological experiments. All animals were housed under a 12 h light/dark cycle with food and water provided ad libitum. The Animal Care and Use Committee of Seoul National University approved the animal protocols.
Behavioral tests
Male mice at 12–15 weeks of age were used for the behavioral analyses in this study. The behavioral experiments were performed essentially following our previous study (Kim et al., 2016). For the Morris water maze (MWM), mice were handled daily for 3 min over a week before training. The water maze was a gray cylinder-shaped tank (140 cm diameter, 100 cm height) placed in a room with multiple spatial cues and dim light. Water mixed with white paint (19°C–21°C) was filled up to 1 cm above the escape platform (10 cm diameter). Mice were trained for four trials per day with 1 min intertrial intervals. Mice were placed into the different edge points of the maze in each four trials, facing the inner wall of the tank, and tracked using the Ethovision software (Noldus). The order of releasing point was changed daily. When mice reached the platform within 60 s, they were removed from the maze and returned to the transport cage. When they failed, they were guided to or placed on the platform and were subsequently removed from the maze. After 5 d of training, a probe test was performed after removing the platform. Mice were placed at the center of the maze and tracked for 1 min. One more session of training was performed after the probe test. To check the remote memory, the probe test was performed 4 weeks after the last training. For the contextual fear conditioning (CFC), mice were placed into the fear conditioning chamber (Coulbourne). After 148 s, they received foot shocks (2 s, 0.75 mA) twice with 30 s interval. After 30 s, they were returned to the home cage. Contextual fear memory was tested by placing the mice again in the conditioning chamber and measuring the freezing levels (immobility) for 4 min using Freeze Frame software (Coulbourne).
Electrophysiology
Hippocampal fEPSP recording.
fEPSP recordings were performed as described previously (Park et al., 2014). After anesthetization with isoflurane, mice were decapitated, and their brains were removed. Transverse hippocampal slices were sectioned 400 μm thick using a vibratome (Leica). The slices were retained at 32°C for 30 min during the recovery period and then incubated at 28°C until the experiment. All incubation chambers were submerge-fashioned and the ACSF (124 mm NaCl, 2.5 mm KCl, 1 mm NaH2PO4, 25 mm NaHCO3, 10 mm glucose, 2.6 mm CaCl2, 1.3 mm MgSO4) was oxygenated with 95% O2 and 5% CO2 and perfused at 1 ml/min throughout the experiment. fEPSPs were recorded from the Schaffer collaterals of CA1. Stimuli were given every 30 s using concentric bipolar electrodes (MCE-100; Kopf Instruments), and the responses were recorded using a glass pipette electrode filled with ACSF (1 mΩ). Field potentials were amplified, low-pass filtered (GeneClamp 500; Molecular Devices), and then digitized (NI PCI-6221; National Instruments) for measurement. Data were monitored, analyzed online, and reanalyzed offline using the WinLTP program (WinLTP; winltp.com, University of Bristol, Bristol, UK). For the LTP and LTD experiments, stimulation was provided at the intensity that produces ∼40% of the slice's maximum slope. Two responses elicited per minute were averaged and expressed relative to an average of the 20 min baseline responses. Theta burst stimulation protocols were used to induce E-LTP and L-LTP (five pulses of 100 Hz repeated five times at 5 Hz; 10 s intertrain interval used for E-LTP; 10 min intertrain interval for L-LTP). The fEPSP response average of the last 5 and 10 min of the E-LTP and L-LTP experiments were used to compare the level of synaptic plasticity between the groups.
ACC field potential recording using multielectrode array.
For ACC multielectrode array experiments, three or four 300-μm-thick coronal brain slices after the corpus callosum connection were sectioned using a vibratome. The slices were incubated in a submerged chamber at room temperature until the experiment. ACSF (124 mm NaCl, 2.5 mm KCl, 1 mm NaH2PO4, 25 mm NaHCO3, 10 mm glucose, 2.5 mm CaCl2, 1 mm MgSO4) was oxygenated with 95% O2 and 5% CO2 and perfused at 2–3 ml/min throughout the experiment. The MED64 system (Panasonic) was used as previously described (Kang et al., 2012). A slice was placed on the MED64 probe (MED-P515A, 8 × 8 array, interpolar distance 150 μm, Panasonic) and perfused with ACSF at 28°C-30°C. The electrical stimulation (1–20 μm, 0.2 ms) was given to a channel in the deep layer region. MED64 Mobius was used for data acquisition and analysis. One pulse was given per minute, and the data were averaged every 4 min. The percentages of the last 4 min (E-LTP) and 8 min (L-LTP) fEPSP slopes were normalized to the averaged value of the 20 min baseline.
Western blot
Mouse brain tissues were homogenized in RIPA buffer with the protease inhibitor mixture (Roche). The protein concentrations were measured using the BCA reagents (Thermo Fisher Scientific), and the equal amounts of proteins across animals were subjected to SDS-PAGE and transferred to nitrocellulose membrane. The CTCF protein was detected using the antibody (Abcam), and their levels were normalized to the GAPDH (Ambion) measured in the same lanes. The chemiluminescence of ECL substrate catalyzed by HRP-conjugated to the secondary antibody was detected and measured by ChemiDoc system (BMS).
qRT-PCR
Total RNA from tissues or cell culture was extracted using TRIZOL or RNAiso and reverse-transcribed using Superscript III following the manufacturers' instructions. Using the cDNA as templates, quantitative PCR was performed using SyBR premix ExTaqII (Takara) on ABI7300. The 2−dCt method was used to measure the relative mRNA level of each gene of interest.
RNA extraction and sequencing
Total RNA from cultured cortical neurons was extracted, and the integrity and quality of the extracted RNA were assessed by BioAnalyzer. The standard protocol (Illumina) was used to make sequencing libraries for RNA-Seq. Using gel electrophoresis, ∼300 bp fragments were isolated and amplified by PCR and sequenced using the HiSeq 2000 (Illumina) in the paired-end sequencing mode (2 × 101 bp).
RNA-Seq read processing and differential gene expression test
RSEM (RNA-Seq by Expectation Maximization, version 1.2.28) (Li and Dewey, 2011) was used to align the raw sequencing reads to the mm10 mouse genome. Only uniquely and properly mapped read pairs were used for further analysis. To assess gene expression levels, the transcript per million measure was calculated (Wagner et al., 2012) using the read counts of each gene annotated in Ensembl release 82 (Yates et al., 2016). The EdgeR package (Robinson et al., 2010) in R was used to identify the differentially expressed genes between the CTCF knockdown (KD; shCTCF) and wild-type (WT; shLacZ) samples. Differentially expressed genes were defined as those with changes of at least 1.2-fold between samples at a false discovery rate of 5%. Gene ontology term enrichment analysis on the differentially expressed genes was executed through ToppGene Suite (https://toppgene.cchmc.org/) (Chen et al., 2009), and the network analysis was performed using QIAGEN's Ingenuity Pathway Analysis software (IPA, QIAGEN, www.qiagen.com/ingenuity).
Experimental design and statistical analysis
All experiments were performed in a blind fashion. We used the minimum number of mice that can produce statistical validity. For CTCF cKO mice experiments, we used 8–16 male mice for behavioral tests, 6–10 female mice for electrophysiology, and 3 male and 4 female mice for qRT-PCR experiments. For CTCF HT mice experiments, we used 15–31 male mice for behavioral tests and 10 female mice for electrophysiology.
Data were represented by the mean ± SEM. For each variable, comparison of two groups was made using Student's t test. Two-way ANOVA and post hoc Bonferroni test were used for further comparisons.
Results
Generation of CTCF cKO mice with CTCF deletion in forebrain excitatory neurons
Previously, Hirayama et al. (2012) demonstrated that the deletion of CTCF in cortical and hippocampal neurons during postnatal development causes abnormal neuronal development, obvious growth retardation, and early lethality within 4 weeks after birth. Because the early lethality prevents examining mice in behavioral and electrophysiological tests, we generated CTCF cKO mice by crossing the floxed CTCF line with CaMKIIa-Cre line, which expresses Cre recombinase in the forebrain excitatory neurons starting from 4 to 5 weeks of age. This enabled us to circumvent the lethal effect of postnatal CTCF deletion, and our CTCF cKO mice were viable at least until ∼8 months of age with no apparent health abnormalities. We confirmed that CTCF expression is sufficiently reduced in the cortical and hippocampal excitatory neurons of the 12-week-old mice using immunohistochemistry (Fig. 1A,B) and qRT-PCR (Fig. 1C,D). The residual mRNA in the CTCF cKO mice shown in Figure 1C, D is probably due to the intact mRNA expression in inhibitory neurons and glial cells. The CTCF cKO mice did not exhibit any abnormal level of anxiety or locomotion (data not shown).

CTCF cKO mice have reduced expression of CTCF. A, B, Immunohistochemistry analysis showed that CTCF cKO mice have reduced CTCF protein expression in the ACC and hippocampus. Blue represents DAPI. Red represents NeuN. Green represents CTCF. C, D, qRT-PCR analysis confirmed that CTCF mRNA level is reduced in CTCF cKO mice (WT, n = 4; cKO, n = 3; C, WT, 1 ± 0.0471; cKO, 0.5094 ± 0.025; unpaired t test; p = 0.0004; D, WT, 0.9674 ± 0.076; cKO, 0.6512 ± 0.102; unpaired t test; p = 0.0257). *p < 0.05, ***p < 0.001.
Impaired remote memory in CTCF cKO mice
To examine whether the neuronal CTCF is needed for the formation of long-term memory, we trained CTCF cKO mice on hippocampus-dependent memory tasks. Subsequently, we tested the mice at two different time points: 1 d or 4 weeks after the behavioral training. When the CTCF cKO mice were first tested 1 d after the training in the CFC, they displayed comparable freezing levels to the WT mice (Fig. 2A). Similarly, in the MWM, the CTCF cKO mice exhibited normal spatial reference memory during the probe test (Fig. 2C,D). The CTCF cKO mice showed a delay in locating the hidden platform during the training (Fig. 2B), but it is unlikely that they had not learned the location of the platform because their performance level on the day 6 probe test was comparable with that of WT. Instead, the delay in the escape latency is likely due to the impaired swimming speed of the CTCF cKO mice (Fig. 2E,H). It is plausible to assume that CTCF mutant mice may have a mild motor deficit that has contributed to slower swimming speed but not to the performance on memory tests. After 4 weeks, we retested the same mice and found that the CTCF cKO mice have a significantly impaired remote memory. In the CFC, the CTCF cKO mice exhibited a lower level of freezing than the WT controls (Fig. 2A). Similarly, the CTCF cKO mice displayed reduced target quadrant duration and a lower count of platform crossing during the probe test (Fig. 2F,G). These results suggest that the CTCF in the adult forebrain excitatory neurons is not necessary for the formation of recent memory but is indispensable for the remote memory.

CTCF cKO mice display impaired cortex-dependent memory. A, In the CFC test, CTCF cKO mice showed intact recent fear memory but impaired remote fear memory (WT, n = 8; cKO, n = 9; two-way repeated-measures ANOVA; effect of interaction, F(1,15) = 9.831, p = 0.0068; Bonferroni post hoc test for day 29, p < 0.01). B, During the training phase of the MWM, CTCF cKO mice showed longer escape latency on days 4–6 (WT, n = 14; cKO, n = 11; two-way repeated-measures ANOVA, effect of interaction, F(5,115) = 4.072, p = 0.0019; Bonferroni post hoc tests, day 4, p < 0.01; day 5, p < 0.01; day 6, p < 0.05). C–E, In the recent memory probe test of the MWM test, CTCF cKO mice performed comparably with WT but showed slower swimming speed (C, two-way ANOVA, effect of interaction of genotype and quadrant, F(3,92) = 1.246, p = 0.2976; D, WT, 3.071 ± 0.5593; cKO, 1.818 ± 0.5191; unpaired t test; p = 0.1225; E, WT, 20.89 ± 0.7795; cKO, 18.04 ± 1.063; unpaired t test; p = 0.0370). TQ, Target quadrant; OQ, opposite quadrant; AQ, adjacent quadrant. F–H, When the same mice were tested 3 weeks after training, CTCF cKO mice displayed loss of spatial memory with less number of platform crossing and still showed slower swimming speed (F, two-way ANOVA, effect of interaction of genotype and quadrant, F(3,92) = 3.551, p = 0.0175; G, WT, 5.429 ± 0.7317; cKO, 2.364 ± 0.4724; unpaired t test; p = 0.0031; H, WT, 21.17 ± 0.6098; cKO, 17.16 ± 0.6548; unpaired t test; p = 0.0002). *p < 0.05, **p < 0.01, ***p < 0.001.
Impaired cortical synaptic plasticity in the CTCF cKO mice
Next, we performed electrophysiological experiments to verify the underlying mechanisms of the remote memory deficit at the synaptic level. We chose hippocampal and ACC field recordings to compare the contribution of hippocampal and cortical LTP in the remote memory process. In the hippocampal slice recording, SC-CA1 E-LTP and L-LTP were induced normally and the potential levels stably lasted for 1 and 3 h, respectively (Fig. 3A,B). We performed input–output curve and confirmed that hippocampal basal transmission is normal in CTCF cKO mice (Fig. 3C). Together, these results suggest that the hippocampal deletion of CTCF does not affect the electrophysiological properties. Because these results are in line with the normal recent memory shown in CTCF cKO mice, we tested the cortical plasticity, which is a physiological trace of remote memory (Frankland and Bontempi, 2005). In the CTCF cKO ACC slices, E-LTP was normal (Fig. 3D), whereas L-LTP appeared significantly impaired with a considerably lower potentiation level (Fig. 3E). Moreover, the basal transmission was also downregulated in CTCF cKO mice (Fig. 3F), indicating that CTCF has a region-specific role in regulating the basal transmission level. The slice recording results indicate that CTCF deletion specifically disrupts the cortical synaptic plasticity, which leads to impaired performance in remote memory behaviors. Our results provide physiological evidence of the shift from the hippocampus to the cortex as a core region in processing the remote memory.

CTCF cKO mice exhibit impaired cortical synaptic plasticity. A, Hippocampal E-LTP was normal in CTCF cKO mice (WT, n = 13; cKO, n = 8; average of fEPSP slopes for the last 5 min; WT, 140.8 ± 5.8%; cKO, 136.8 ± 8.6%; unpaired t test; p = 0.6947). B, CTCF cKO mice did not exhibit any impairment in the theta burst stimulation-induced hippocampal L-LTP, and the potentiation level was maintained for 3 h at a comparable level with WT (WT, n = 5; cKO, n = 4; average fEPSP slopes for the last 10 min; WT, 144.0 ± 10.4%; cKO, 138.1 ± 16.7%; unpaired t test; p = 0.7635). C, Input–output curve was normal in CTCF cKO mice (WT, n = 11; cKO, n = 8; repeated-measures two-way ANOVA, effect of genotype, F(1,17) = 0.0929; p = 0.7641). D, CTCF cKO mice displayed normal cortical E-LTP (WT, n = 16; cKO, n = 12; average of fEPSP slopes for the last 4 min; WT, 122.6 ± 2.5%; cKO, 120.6 ± 3.2%; unpaired t test; p = 0.6190). E, CTCF cKO mice showed a significant deficit in cortical L-LTP with a substantially decreased potentiation level after induction (WT, n = 10; cKO, n = 6; average fEPSP slopes for the last 8 min; WT, 141.7 ± 8.1%; cKO, 116.4 ± 6.9%; unpaired t test; p = 0.0498). F, Cortical basal transmission was attenuated in CTCF cKO mice (WT, n = 8; cKO, n = 8; repeated-measures two-way ANOVA, effect of genotype, F(1,14) = 7.364; p = 0.0168). *p < 0.05.
CTCF in inhibitory neurons also participate in remote memory maintenance
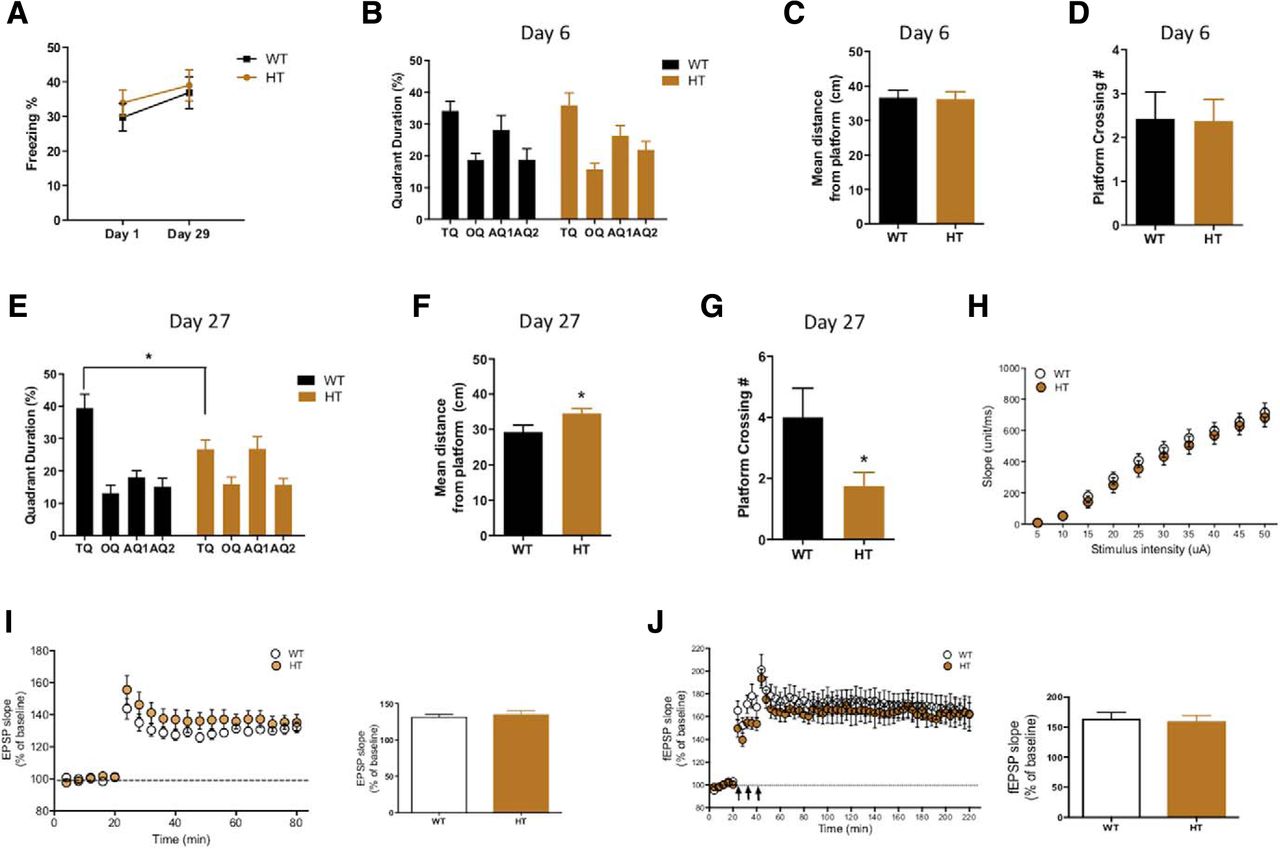
Having found that the CTCF deletion in excitatory neurons induces remote memory deficits, we further examined the effect of CTCF deletion in the inhibitory neurons. We crossed the floxed CTCF line with the Vgat;Cre line to induce inhibitory neuron-specific deletion of CTCF during development. However, we found that homozygous CTCF KO mice were embryonic lethal and only HT mice survived. We concluded that, as in the previous report (Hirayama et al., 2012), the homozygous deletion of CTCF in inhibitory neurons during development has a lethal effect on the animals and confirmed that CTCF is crucial for development. We used the CTCF HT mice for the experiments. As with the CTCF cKO mice, we trained the CTCF HT mice in the MWM and CFC and examined recent and remote memory. In the CFC, the CTCF HT mice exhibited normal freezing level in both recent and remote memory tests (Fig. 4A). However, in the MWM, the CTCF HT mice displayed a significant impairment in finding the platform with a higher number of platform crossings in the probe test (Fig. 4E–G). The recent spatial memory was completely intact in the CTCF HT mice (Fig. 4B–D). These results indicated that the CTCF HT mice have a deficit in spatial remote memory and that CTCF in the inhibitory neurons is also involved in systems consolidation. The partial impairment may be due to an assumedly mild effect of the HT CTCF deletion compared with the homozygous deletion. The cortical basal transmission and LTP appeared normal in the CTCF HT mice (Fig. 4H–J), which indicate that CTCF dysfunction in inhibitory neurons did not affect the synaptic plasticity or postsynaptic potential of excitatory neurons.

CTCF HT mice show a partial deficit in remote memory. A, CFC recent memory and remote memory were normal in CTCF HT mice (WT, n = 15; HT, n = 16; two-way ANOVA; effect of interaction, F(1,58) = 0.0631; p = 0.8026). B–D, CTCF HT mice performed comparably with WT in the recent memory test of the MWM (WT, n = 7; HT, n = 8; B, two-way ANOVA; effect of interaction of genotype and quadrant, F(3,52) = 0.4176; p = 0.7411; C, WT, 36.7 ± 2.126; cKO, 36.24 ± 2.155; unpaired t test; p = 0.8835; D, WT, 2.429 ± 0.6117; HT, 2.375 ± 0.4978; unpaired t test; p = 0.9463). E–G, Three weeks later, the CTCF HT mice showed a significant memory deficit in the MWM probe test. Quadrant duration, distance from platform, and number of platform crossings were all impaired (WT, n = 7; HT, n = 8; E, two-way ANOVA; effect of interaction of genotype and quadrant, F(3,52) = 4.882; p = 0.046; Bonferroni post hoc test for TQ (target quadrant). p = 0.0124; F, WT, 29.23 ± 1.985; HT, 34.55 ± 1.372; unpaired t test; p = 0.0423; G, WT, 4 ± 1.750; HT, 1.750 ± 0.4532; unpaired t test; p = 0.0442). H, Basal transmission was normal in ACC of CTCF HT mice (WT, n = 10; HT, n = 11; two-way ANOVA; effect of interaction, F(9,171) = 0.2952; p = 0.9753). I, E-LTP was intact in ACC of CTCF HT mice (WT, n = 7; HT, n = 6; average fEPSP slopes for the last 4 min; WT, 132.1 ± 3.252%; HT, 135.1 ± 5.005%; unpaired t test; p = 0.6123). J, L-LTP was also normal in ACC of CTCF HT mice (WT, n = 6; HT, n = 4; average fEPSP slopes for the last 8 min; WT, 164.2 ± 10.12%; HT, 159.8 ± 9.665%; unpaired t test; p = 0.7700). *p < 0.05.
Gene expression profile changes upon CTCF deletion in cortical neuron culture
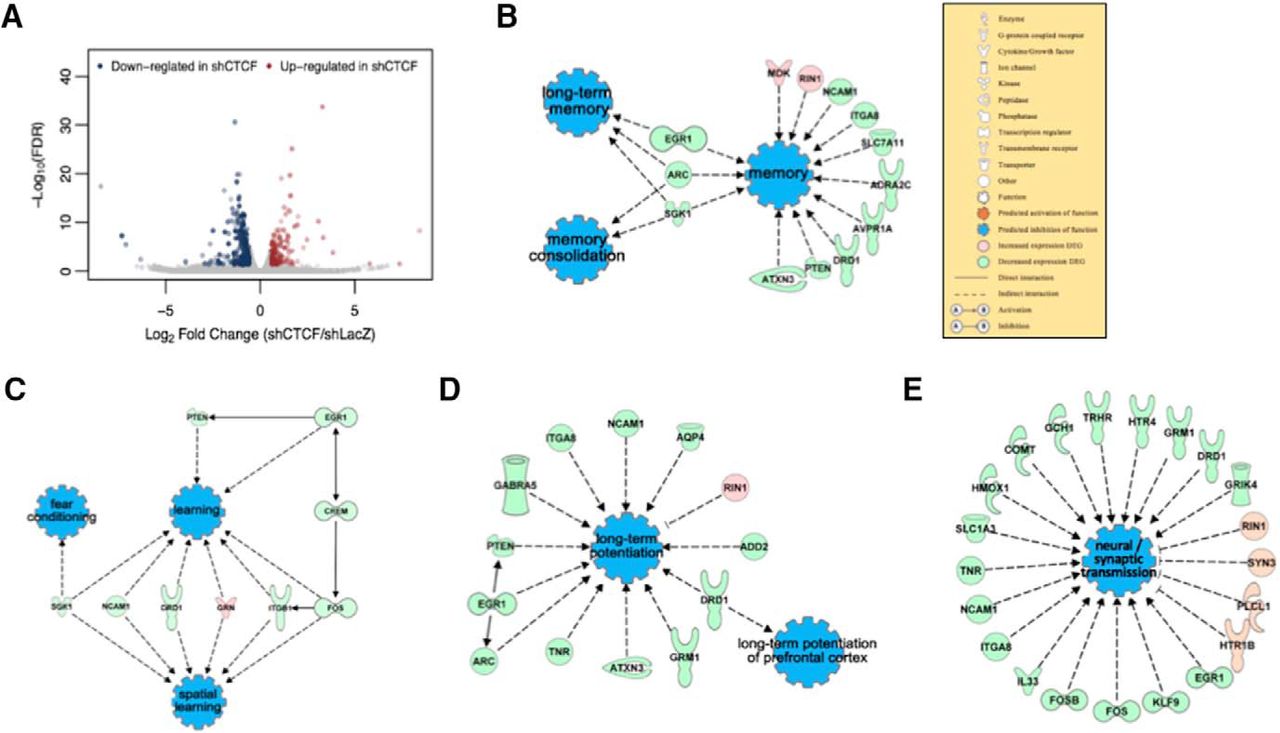
To investigate the molecular changes underlying the phenotypes of the CTCF cKO and HT mice, we explored the variations in the gene expression profile. It is well established that appropriate gene expression is necessary for memory (Igaz et al., 2004; Peixoto and Abel, 2013). We induced CTCF KD in primary mouse cortical cultures using adeno-associated virus (∼67% mRNA reduction) and performed RNA sequencing (RNA-seq). We found a total of 394 differentially expressed genes (DEGs) in CTCF KD neurons with the fold change cutoff set to >1.5. Of the 394 DEGs, 146 (37%) genes were upregulated, whereas 248 (63%) genes were downregulated. This indicated a shift in the overall gene expression to a downregulating direction (Fig. 5A), which was similar to previous results of CTCF deletion studies (Wan et al., 2008; Hirayama et al., 2012). To analyze the DEGs from RNA-seq with unifying terminologies, we performed gene ontology analysis, which is a major bioinformatics technique that uses gene annotations to hierarchically classify the genes and their functions (Ashburner et al., 2000). We found that many of our DEGs are involved in cell adhesion, neuroactive ligand-receptor interaction, and calcium binding. More importantly, we found that genes that are involved in learning and memory, synapse assembly, and cognition were enriched in our DEG list (Table 1), suggesting that they were improperly expressed in the CTCF-deficient cells. For more detailed investigation, we performed IPA to connect the DEGs in pathways under certain terminologies, and we found that the IPA results exactly reflected the behavioral and electrophysiological phenotypes of our CTCF cKO and HT mice. Memory and learning were predicted to be functionally inhibited, and, specifically, long-term memory and memory consolidation appeared to be inhibited (Fig. 5B,C). The expression of most of the DEGs functionally involved in LTP and synaptic transmission was decreased, leading to the inhibition of the two functions (Fig. 5D,E). Furthermore, in the DEG list, we found a highly frequent appearance of the clustered Pcdh family isoforms. There was a total of 27 Pcdh genes, which made up 10% of the DEG list (Table 2). This pattern was similar to the previous microarray analysis data from Hirayama et al. (2012), indicating that CTCF critically regulates the neuronal Pcdh expression in adult as well as developmental brain. We performed qRT-PCR experiments using the ACC tissues from CTCF cKO mice and ratified that the effects of CTCF deletion in the cortical culture are similar to those of CTCF deletion in vivo (Fig. 6A–E). Moreover, we performed qRT-PCR for the same genes using the hippocampal tissues from CTCF cKO mice to compare the results with that of ACC. We found that expressions of Drd1, Pcdhα4, Pcdhβ13, and PcdhγA12 are significantly decreased or show a tendency of decrease (Fig. 6G–J), as they were in ACC. However, the expression of RhoU was comparable with that of WT in hippocampus (Fig. 6F). This finding represents a gene expressional difference between hippocampus and ACC caused by CTCF dysfunction, supporting our conclusion that CTCF plays region-specific roles for regulation of remote memory. Together, the RNA-seq data and bioinformatics analysis suggest that CTCF deletion alters the expression of a set of genes, which leads to defects in the cortical plasticity and remote memory in CTCF-deficient mice.

RNA-seq data from CTCF KD cortical culture reveal differentially expressed genes. A, Volcano plot showed that more genes were downregulated than upregulated in the DEG list. B–E, IPA showed connections of DEGs for four functions: memory, learning, LTP, and synaptic transmission. They were all predicted to be inhibited.
List of the GO terms and categories in order of the smallest to the largest q value
List of the downregulated Pcdh isoformsa

qRT-PCR of 5 DEGs shows differential results in ACC and hippocampus. A–E, qRT-PCR of ACC tissues from CTCF cKO mice confirmed the RNA-seq data of gene downregulation (WT, n = 4; cKO, n = 3; A, normalized expression of RhoU mRNA; WT, 1 ± 0.04013%; cKO, 0.7669 ± 0.05816%; unpaired t test; p = 0.0187; B, normalized expression of Drd1 mRNA; WT, 1 ± 0.07316%; cKO, 0.6552 ± 0.1332%; unpaired t test; p = 0.0584; C, normalized expression of Pcdhα4 mRNA; WT, 1 ± 0.06878%; cKO, 0.4386 ± 0.07655%; unpaired t test; p = 0.0029; D, normalized expression of Pcdhβ13 mRNA; WT, 1 ± 0.02351%; cKO, 0.3466 ± 0.02203%; unpaired t test; p < 0.0001; E, normalized expression of PcdhγA12 mRNA; WT, 1 ± 0.04239%; cKO, 0.4336 ± 0.02648%; unpaired t test; p = 0.0001). F–J, qRT-PCR of hippocampal tissues from CTCF cKO mice showed a different pattern of gene expression (WT, n = 4; cKO, n = 3; F, normalized expression of RhoU mRNA; WT, 1 ± 0.05991%; cKO, 0.95 ± 0.07146%; unpaired t test; p = 0.6132; G, normalized expression of Drd1 mRNA; WT, 1 ± 0.05873%; cKO, 0.7701 ± 0.114%; unpaired t test; p = 0.1090; H, normalized expression of Pcdhα4 mRNA; WT, 1 ± 0.3823%; cKO, 0.593 ± 0.3689%; unpaired t test; p = 0.4907; I, normalized expression of Pcdhβ13 mRNA; WT, 1 ± 0.07615%; cKO, 0.6268 ± 0.06089%; unpaired t test; p = 0.0155; J, normalized expression of PcdhγA12 mRNA; WT, 1 ± 0.0923%; cKO, 0.6785 ± 0.0057%; unpaired t test; p = 0.0322). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
How long-lasting memory is formed and maintained in the brain through systems consolidation is a critical question extensively explored in neurobiology. However, the mechanisms are still largely in veil. To investigate how memory traces are retained for a long time, it is important to examine long-lasting gene-expression changes. Our CTCF cKO mice bearing the deletion of CTCF in mature excitatory neurons developed normally with impaired remote memory and cortical late-LTP. Similarly, the CTCF deletion in inhibitory neurons caused a remote memory-specific deficit in the spatial memory behavioral task. In accordance with these results, the mRNA level of genes related to learning and memory were downregulated in CTCF-deficient neurons. Together, these results suggest that CTCF has a critical function in regulation of long-term memory storage and maintenance in both excitatory or inhibitory neurons.
In this study, we also provide evidence that CTCF may have a differential role in hippocampus and ACC for remote memory reregulation. Our qRT-PCR results showed that the expression of RhoU is regulated by CTCF in ACC but not in hippocampus. This is in line with our behavioral and physiological results from CTCF-deficient mice because remote memory formation is known to more heavily rely on cortical networks (Takehara-Nishiuchi et al., 2006) and functional plasticity in cortical regions (Headley and Paré, 2017). Further analysis on CTCF's gene expressional regulation in hippocampus and ACC will provide an insight into region-specific mechanisms in systems consolidation. Frankland et al. (2001) provided one of the early studies on molecular mechanisms of permanent cortical memory. Using α-CaMKII+/− mutant mice, they revealed that fear and spatial remote memories depend on persistent activation of cortical traces and that α-CaMKII-dependent LTP is required for cortical memory consolidation. More recently, Rossetti et al. (2017) discovered that CaMKII is not only required for memory formation but also for memory storage and LTP maintenance through a memory erasure experiment. Similarly, we showed in this study that CTCF-dependent gene expression in ACC is important for cortical synaptic plasticity and remote memory consolidation. As with CaMKII, further analysis on time-dependent CTCF-induced gene expression will reveal how CTCF is involved in different phases of consolidation process and stabilization of remote memory.
Recently, Sams et al. (2016) reported on the role of CTCF in hippocampus-dependent memory. They created CTCF cKO mice using a similar strategy of crossing the floxed CTCF mice with CaMKIIa-Cre line to inhibit the CTCF expression in postmitotic excitatory neurons. However, their results were partially and yet significantly different from our present results. These discrepancies may be due to several factors, and we think that it is noteworthy to go over the differences. While Sams et al. (2016) similarly reported memory-related deficits in the CTCF cKO mice, they showed that the Cre-dependent CTCF deletion started at 1 week of age and a significant decrease of the protein level was reached by 8 weeks of age. However, our CaMKIIa-Cre line started its Cre expression at 4 weeks of age as previously reported (Tsien et al., 1996; Liu et al., 2010), and a sufficient level of protein reduction was only reached at 12 weeks of age (Fig. 1C). Therefore, for most of the experiments, Sams et al. (2016) used 10- to 12-week-old CTCF cKO mice, whereas we used 12- to 15-week-old mice, which had avoided developmental effects of the gene deletion. Furthermore, Sams et al. (2016) reported that their CTCF cKO mice died after 17 weeks of age with weight loss, whereas our CTCF cKO mice were viable for >40 weeks of age and exhibited no apparent health abnormalities. In addition to physical phenotypes, the memory-related deficits also appeared to be different between the two studies. The CTCF cKO mice in the Sams et al. (2016) study exhibited impaired hippocampal LTP with disrupted recent memory in the cued fear conditioning and MWM tests. However, our CTCF cKO mice exhibited normal hippocampus-dependent phenotypes with dramatically impaired cortex-dependent remote memory. Moreover, while the RNA-seq data showed a similar result in Pcdh expression change, Sams et al. (2016) showed a higher number of upregulated genes, and our present study had a higher number of downregulated genes in the DEG list. In summary, Sams et al. (2016) focused on investigating the effect of CTCF deletion early in life and its roles in the hippocampus, whereas we focused on the effect of CTCF deletion in adulthood and its roles in the cortex-dependent remote memory. The differences in the results also may arise from the difference in the mouse lineage background, as it has been previously reported that recombination patterns and according phenotypes of the same Cre mouse line may differ due to the genetic background and breeding strategies (Fex et al., 2007; Gil-Sanz et al., 2015). Also, for the RNA-seq experiment, Sams et al. (2016) used hippocampal tissue from 10-week-old mice, whereas we used CTCF KD cortical cultures, which can also account for the difference in the data. The early lethality observed in the previous study (Sams et al., 2016) may be due to the apoptosis of pyramidal cells, which was not detected in our mice at the age used in this study. Therefore, further investigations are needed to find the exact mechanisms underlying the differences between the two studies. Also, it may be interesting to explore the age-dependent role of CTCF in neurons because the timing of the protein deletion accounts for the main difference between the two CTCF cKO mouse lines. It may be plausible to think that CTCF changes its main regulatory function in the brain with the age.
To our knowledge, this is the first study to reveal the CTCF function in excitatory and inhibitory neurons of mammalian adult brain in relation to remote memory. We have found a novel association between CTCF, a central molecule for 3D genome architecture, and remote memory. Our study provides a valuable set of data for future studies. For example, our CTCF-deficient mice can be used as a remote memory-impairment mouse model to study the difference in the mechanisms of recent and remote memory. Also, human genome is known to have many CTCF binding sites (Kim et al., 2007), and it has been reported that several de novo mutations in CTCF have been found in individuals with intellectual disability (Gregor et al., 2013). Moreover, RNA-seq of inhibitory neurons will be able to more accurately define CTCF's role in inhibitory neurons. In this study, we performed LTP field recording to examine how CTCF dysfunction in inhibitory cells affects excitatory cells and found that LTP (fEPSP) was normal. A future experiment, such as patch-clamp recording of inhibitory neurons, may reveal that CTCF dysfunction alters electrical properties or functional connectivity of inhibitory neurons. Further investigations on DEGs from our RNA-seq data may provide more information on how CTCF-mediated genome architecture supports permanent memories to last and help develop therapeutics for patients suffering from disorders with long-term memory deficits, such as dementia.
Footnotes
This work was supported by the National Research Foundation of Korea, Korean Government Grant NRF-2012R1A3A1050385 to B.-K.K. J.-i.K. was supported by the POSCO T.J. Park Foundation. N.-K.Y. was supported by L'Oreal Korea-UNESCO Women in Science Fellowship and L'Oreal-UNESCO Rising Talent Award.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Bong-Kiun Kaang, Department of Biological Sciences, College of Natural Sciences, Seoul National University, 1 Gwanangno, Gwanak-gu, Seoul 08826, South Korea. kaang{at}snu.ac.kr





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}