

Abstract
In neurons, local protein synthesis in synaptodendritic microdomains has been implicated in the growth and plasticity of synapses. Prerequisites for local translation are the targeted transport of RNAs to distal sites of synthesis in dendrites and translational control mechanisms to limit synthesis to times of demand. Here we identify dendritic BC1 RNA as a specific repressor of translation. Experimental use of internal ribosome entry mechanisms and sucrose density gradient centrifugation showed that BC1-mediated repression targets translation at the level of initiation. Specifically, BC1 RNA inhibited formation of the 48S preinitiation complex, i.e., recruitment of the small ribosomal subunit to the messenger RNA (mRNA). However, 48S complex formation that is independent of the eukaryotic initiation factor 4 (eIF4) family of initiation factors was found to be refractory to inhibition by BC1 RNA, a result that implicates at least one of these factors in the BC1 repression pathway. Biochemical experiments indicated a specific interaction of BC1 RNA with eIF4A, an RNA unwinding factor, and with poly(A)-binding protein. Both proteins were found enriched in synaptodendritic microdomains. Significantly, BC1-mediated repression was shown to be effective not only in cap-dependent translation initiation but also in eIF4-dependent internal initiation. The results suggest a functional role of BC1 RNA as a mediator of translational control in local protein synthesis in nerve cells.
- neuronal nontranslatable RNAs
- localized RNAs
- local protein synthesis
- dendritic translation
- postsynaptic microdomains
- synaptic plasticity
Diverse types of neuronal mRNAs are transported to distal target sites such as postsynaptic dendritic microdomains where they are presumed to be translated into cognate proteins on site (for review, see Kindler et al., 1997; Tiedge et al., 1999; Kiebler and DesGroseillers, 2000; Wells et al., 2000; Greenough et al., 2001; Job and Eberwine, 2001b; Richter, 2001; Steward and Schuman, 2001). Characterized by highly elongated dendritic and axonal processes that form large numbers of synaptic connections, nerve cells have been suggested to rely on local protein synthesis for an effective management of their mosaic postsynaptic protein repertoires in dendrites. Experience-dependent, site-specific modulations of synaptic protein complements through local synthesis are thus thought to provide a basis for long-lasting plastic changes of synaptic form and function (Tiedge et al., 1999; Job and Eberwine, 2001b).
The notion of postsynaptic translation has been strengthened in recent years by the discovery of various neuronal RNAs that are selectively localized to dendrites. Dendritic mRNAs encode proteins that belong to different classes, including cytosolic proteins and cytoskeletal components, as well as membrane-associated and membrane-integrated proteins (for review, see Kiebler and DesGroseillers, 2000; Job and Eberwine, 2001b; Richter, 2001). According to a recent estimate (Eberwine et al., 2001), the family of dendritic mRNAs is composed of several hundred members.
Components of the translational machinery have been identified in dendritic domains (Tiedge and Brosius, 1996; Torre and Steward, 1996;Gardiol et al., 1999). Dendritic translation has been documented in physically isolated dendrites (Torre and Steward, 1992) and in cultured neurons (Crino and Eberwine, 1996). Local translation has also been shown to be a requirement for synapse formation (Schacher and Wu, 2002). Recent data further suggest that protein synthesis in dendrites can be subject to modulation by neuronal activity, receptor activation, and neurotrophic action (Steward and Halpain, 1999; Kacharmina et al., 2000; Scheetz et al., 2000; Aakalu et al., 2001; Greenough et al., 2001; Job and Eberwine, 2001a). The available evidence, in summary, is in support of a model in which a select group of mRNAs is transported to dendrites, subsequent to which they can be translated, on demand, in specific postsynaptic microdomains where the cognate proteins are required (Tiedge et al., 1999; Job and Eberwine, 2001b).
This model, although attractive, relies on a number of premises that have not been addressed. Paramount among them is the issue of translational control. To prevent inappropriate protein synthesis at the wrong place or at the wrong time, the translational activity of any dendritic mRNA will have to be tightly controlled during the sequential steps of targeted transport, postsynaptic localization, and regulated local translation (Job and Eberwine, 2001b). A key question in this regard is raised by the assumption that many dendritic mRNAs may remain translationally silent after they have reached their postsynaptic target sites, until such time that an appropriate signal is received. What is the molecular mechanism of such translational repression, and how is it modulated?
In the present report, we identify dendritic BC1 RNA as a translational repressor. It has been shown previously that this RNA is specifically and rapidly transported to dendrites (Muslimov et al., 1997) and that somatodendritic BC1 expression levels are subject to activity-dependent modulation (Muslimov et al., 1998). We now report that BC1 RNA is a specific repressor of translation initiation in both cap-dependent and internal entry modes. The combined data indicate that nontranslatable BC1 RNA plays a functional role in translational control of gene expression in neurons.
MATERIALS AND METHODS
RNAs. Plasmid pBCX607 was used to generate full-length BC1 RNA as described previously (Cheng et al., 1996;Muslimov et al., 1997). Plasmids pSP6-U4 and pSP6-U6 (Hausner et al., 1990) were used for the in vitro transcription of U4 and U6 RNAs, respectively, as described (Muslimov et al., 1997). Yeast tRNA was purchased from Sigma (St. Louis, MO). Plasmid pTub-A98/TA2 was kindly provided by Dr. J. Brosius (University of Münster, Münster, Germany). In this vector, the full-length α-tubulin cDNA insert is immediately followed by an uninterrupted stretch of 98 A residues. It was linearized withXbaI or XhoI, and in vitro transcribed with T7 RNA polymerase, to yield programming mRNA encoding α-tubulin either with or without a 3′ 98-residue poly(A) tail, respectively.
Plasmid pBDCG (kindly provided by Dr. J. Carson, University of Connecticut Health Center, Farmington, CT) was used to produce polyadenylated blue fluorescent protein/encephalomyocarditis virus-internal ribosome entry site/green fluorescent protein (BFP/EMCV–IRES/GFP) dicistronic mRNA as described (Kwon et al., 1999). To generate a monocistronic version, plasmid monocistronic green was derived from pBDCG by partial digestion with XbaI andXmaI to remove segment nucleotide 28–753. It was linearized with SapI and transcribed with SP6 RNA polymerase to produce polyadenylated EMCV-IRES/GFP mRNA. Plasmid pCSFV(1–442).NS′(A) was used to generate polyadenylated classical swine fever virus-IRES/truncated influenza virus nonstructural protein (CSFV-IRES/NS′) programming mRNA. Derived from plasmid pCSFV(1–442).NS′ (Pestova et al., 1998) by insertion of an A98 segment at position 1305, it was linearized with EcoRI for in vitro transcription with T7 RNA polymerase. All programming mRNAs were used polyadenylated, unless noted otherwise. Whenever desired, mRNAs were capped by in vitro transcription in the presence of 0.3 mm m7G(5′)ppp(5′)G (Stratagene, La Jolla, CA).
Expression and purification of recombinant proteins. Recombinant eukaryotic initiation factor 4A (eIF4A) was expressed from plasmid pET(His6-eIF4A) in EscherichiaBL21(DE3) and purified as described (Pestova et al., 1996a). Recombinant eIF4G (central domain, aa 697–1076) was analogously generated from pET28(His6-eIF4G697–1076) (Lomakin et al., 2000).
Recombinant poly(A)-binding protein (PABP) was generated from vector pET3B.PABP-His as described previously (Khaleghpour et al., 2001). A C-terminal domain (aa 462–633) of PABP was generated from vector pGex2T.PABPaa462–633 (Imataka et al., 1998). Analogously, an N-terminal domain (aa 1–182) of PABP, containing RNA recognition motif (RRM) domains 1 and 2, was generated from vector pGex2T.PABPaa1–182. Expressed as glutathione S-transferase (GST) fusion proteins, PABP domains were purified on glutathione-Sepharose beads (Amersham Biosciences, Piscataway, NJ) as described (Smith and Johnson, 1988).
Translation assays. Rabbit reticulocyte lysates (RRLs) were purchased from Ambion (Austin, TX) or Roche (Indianapolis, IN), andin vitro translation reactions were performed according to the instructions of the manufacturer. Lysate, reaction buffer,35S-methionine (∼1200 Ci/mmol; NEN, Boston, MA), and respective programming mRNA were incubated for 1 hr at 30°C in the presence of BC1 RNA or other small RNAs, as indicated. Reaction mixtures were treated with 0.1 mg/ml RNaseA for 10 min, and translation products were separated by SDS-PAGE, using 10% acrylamide gels. Gels were dried and subjected to autoradiography to visualize protein bands. Signal intensities of bands were quantified using a Storm 860 phosphorimaging system with ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
The integrity of programming mRNAs that were used in this work was verified in time course experiments with32P-labeled transcripts under otherwise identical reaction conditions. No RNA degradation was observed in any of these control experiments.
Analysis of ribosomal complexes. To analyze 48S and 80S complexes, we used sucrose density gradient centrifugation according to previously established protocols (Gray and Hentze, 1994; Pestova et al., 1996a). In vitro translation reactions were performed as described above, except that the reaction mixture did not initially contain mRNA and methionine was not radiolabeled. The reaction mixture was preincubated at 30°C for 15 min with translational inhibitor guanylyl imidodiphosphate (GMP-PNP; 1.2 mm) or cycloheximide (0.8 mm). Small RNAs (e.g., BC1 RNA, U4 RNA) were used at 600 nm. Subsequently,32P-labeled programming mRNA (50 ng) was added, and incubation continued for another 5 min at 30°C. Complexes were resolved by centrifugation through a 5–25% sucrose gradient in SG buffer (100 mm KCl, 2 mmDTT, 2 mm magnesium acetate, 20 mm Tris-HCl, pH 7.5) for 3 hr at 4°C at 30,000 rpm with a Beckman SW41 rotor. Twenty-five fractions were collected per tube, starting from the bottom. The radioactivity of fractions was determined by Cerenkov counting.
Electrophoresis mobility shift assay.32P-labeled RNA probes (50,000 cpm per reaction, ∼10 ng) were heated for 10 min at 70°C, cooled for 5 min at room temperature, and then incubated together with proteins in binding buffer (300 mm KCl, 5 mm MgCl2, 2 mm DTT, 5% glycerol, 20 mmHEPES, pH 7.6) for 20 min at room temperature. If unlabeled competitor RNAs were used, they were treated analogously but preincubated with proteins for 10 min before labeled RNAs were added to the reaction. Reaction time was increased to 40 min if simultaneous binding to more than one protein was analyzed. RNA–protein complexes were subsequently resolved on 5% polyacrylamide gels (60:1 polyacrylamide/bisacrylamide) and analyzed by autoradiography as described (Gu and Hecht, 1996;Thomson et al., 1999).
Brain extracts. Brains were dissected from adult Sprague Dawley rats and immediately frozen in liquid nitrogen. Brains were resuspended in 2 ml per brain of buffer A (100 mmNaCl, 0.5 mm dithiothreitol, 3 mm MgCl2, 0.5 mm phenylmethylsulfonyl fluoride, 0.5 μg/ml leupeptin, 1 μg/ml aprotinin, 50 mm Tris-HCl, pH 8.0) and homogenized slowly on ice with a motor-driven homogenizer (Kontes, Vineland, NJ). The homogenate was centrifuged at 5000 ×g for 15 min. The supernatant was mixed with 0.1 volume of buffer B (2.5 M NaCl, 500 mm Tris-HCl, pH 8.0). After further centrifugation at 14,000 × g for 1 hr at 4°C, the supernatant was snap-frozen in liquid nitrogen and stored at −70°C.
Immunodepletion of brain extracts. Brain extracts (60 μl) were incubated with 20 μl of anti-GST-PABP (aa 462–633) (Imataka et al., 1998) for 3 hr at 4°C with gentle rotation. Subsequently, 15 μl of protein-A agarose (Roche) suspension was added to the mixture and incubated, with rotation, at 4°C overnight. Complexes were collected by centrifugation at 12,000 × g for 20 sec (Zhang et al., 2001). The immunodepleted brain extracts were then used for electrophoretic mobility shift assays (EMSAs) as described above.
Supershift assay. 32P-labeledin vitro transcribed BC1 RNA (50,000 cpm per reaction, ∼1 ng) was heated for 10 min at 70°C and cooled for 5 min at room temperature. The RNA was then incubated with brain extract (30–40 μg) or immunodepleted brain extract in binding buffer for 20 min at room temperature. In competition experiments, unlabeled BC1 RNA (2000-fold excess) was added 10 min before the binding reaction. Mixtures containing brain extract were then incubated with an anti-GST-PABP antibody (raised against a fusion protein containing PABP aa 462–633) (Imataka et al., 1998) or an anti-GST control antibody for 3 hr at room temperature. To minimize unspecific binding, samples were incubated with heparin (5 mg/ml) for 10 min at room temperature. As in EMSA, complexes were resolved on 4% native polyacrylamide gels and analyzed by autoradiography.
Immunocytochemistry with hippocampal neurons in primary culture. Immunocytochemistry was performed as described (Tiedge and Brosius, 1996). Primary antibodies were used at the following dilutions: anti-eIF4A, 1:50; anti-eIF4G, 1:50; anti-PABP, 1:50; anti-synaptophysin, 1:500. Polyclonal anti-eIF4A, anti-PABP, and anti-eIF4G antibodies have been described previously, and their respective specificities have been established (Wakiyama et al., 2000). A monoclonal anti-synaptophysin antibody was purchased from Synaptic Systems (Göttingen, Germany). Secondary antibodies were used as follows: biotinylated anti-rabbit (Amersham), 1:200; anti-mouse labeled with fluorescein isothiocyanate (Jackson ImmunoResearch, West Grove, PA), 1:25. Biotinylated secondary antibodies were decorated with streptavidin-conjugated rhodamine (5 μg/ml; Jackson ImmunoResearch). Control experiments to ascertain unspecific background labeling were performed as follows. (1) In the case of polyclonal antibodies, preimmune or nonimmune serum was substituted for the primary antibody. (2) In the case of antibodies directed against GST fusion proteins, an anti-GST antibody was used as a primary antibody. (3) Background labeling was further ascertained by incubation in the absence of a primary antibody. Confocal images were acquired with a Radiance 2000 Plus confocal laser scanning microscope (Bio-Rad, San Francisco, CA) attached to an Axioskop 2 microscope (Zeiss, Thornwood, NY).
RESULTS
BC1 RNA is a specific repressor of translation
In our initial experimental approach directed at the functional role of BC1 RNA in translational regulation, we used the RRL cell-free system to probe the competence of BC1 RNA as a modulator of translation. In untreated RRLs (i.e., reticulocyte mRNA transcripts not removed by nuclease), translation of endogenous mRNAs was inhibited by BC1 RNA in a concentration-dependent manner (Fig.1A,B). Results from these experiments were quantified by phosphorimaging. Analysis of several experiments showed that the presence of BC1 RNA at a concentration of 320 nm resulted in a decrease of translation efficiency by 70–80%. Such a reduction was observed with all protein bands that were resolved by SDS-PAGE, a result indicating that BC1-mediated translational repression was not restricted to particular mRNAs. However, in clear contrast to BC1 RNA, other small nontranslatable RNAs (e.g., U4 and U6 RNAs, tRNAs), used at similar or higher concentrations, had no effect on translation efficiency (Fig. 1C). The results demonstrate that BC1 RNA is a specific repressor of translation that is effective in the submicromolar concentration range.

BC1 RNA is a repressor of translation in the submicromolar concentration range. Protein products were labeled by35S-methionine incorporation, using the RRL system, and were visualized by SDS-PAGE and autoradiography. A, Translation of endogenous RRL mRNAs was inhibited by increasing concentrations of BC1 RNA. Relative signal intensities of the major band were quantified by phosphorimaging and are listed for each lane. The signal intensity generated in the absence of BC1 RNA was assigned a relative value of 1. B, Results from three experiments, quantified by phosphorimaging, showed that the signal of the major protein band was reduced by 72% at 320 nm BC1 RNA [one-way ANOVA, p < 0.001; Scheffe's multiple comparison post hoc analysis (comparison with 0 nm BC1 RNA control): **p < 0.01 for 40 nm BC1 RNA, ***p < 0.001 for other groups]. Signal intensities of other protein bands were similarly reduced by 70–80%. Note that the x-axis is exponential. C, No inhibition of translation was observed in the presence of control RNAs, including U4 and U6 RNAs, and tRNAs. D, When capped and polyadenylated α-tubulin mRNA was used as a programming mRNA, translation was similarly inhibited in the same BC1 concentration range. Each experiment shown inC and D was performed at least twice.
These results were confirmed with lysates in which endogenous RRL transcripts had been removed by nuclease treatment before translation experiments. Using capped and polyadenylated α-tubulin mRNA as a programming mRNA in these experiments, we established that BC1 RNA (but not nuclear U4 RNA or other control RNAs) inhibited cap-dependent translation to the same degree and in the same submicromolar concentration range as shown above (Fig. 1D). Uncapped or nonadenylated programming mRNAs were not efficiently translated (data not shown); translation of capped but nonadenylated α-tubulin mRNA appeared to be less susceptible to BC1-mediated inhibition than capped and polyadenylated programming mRNA, although this could not be reliably established because of lower overall translational efficiencies. Therefore, all subsequent experiments were performed with polyadenylated programming mRNA, unless noted otherwise. Furthermore, it should be noted that BC200 RNA, the primate counterpart of rodent BC1 RNA (Tiedge et al., 1993), used in the same nanomolar concentration range, was found to inhibit translation as effectively as BC1 RNA (data not shown).
In summary, the above data indicate that BC1 RNA and BC200 RNA act as specific repressors of translation. They raise the question as to which step in the translation pathway is targeted in BC1-mediated repression and which factor(s) BC1 RNA is interacting with in the course of such repression.
BC1 RNA inhibits formation of the 48S preinitiation complex
Eukaryotic translation can be subdivided into the three sequential phases of initiation, elongation, and termination. Frequently, it is the initiation phase that is targeted in translation regulation mechanisms (Gingras et al., 1999). We therefore hypothesized that in repressing translation, BC1 RNA interacts with the translational machinery at the level of initiation. We tested this hypothesis as follows.
Cap-dependent translation initiation typically begins with the assembly of the 40S small ribosomal subunit, eIF1A, eIF3, and an eIF2/GTP/Met-tRNAi complex, to form a 43S preinitiation complex. In the next step, the 43S complex is recruited to the mRNA and translocates (“scans”) to the AUG start codon where it forms a stable 48S pre-initiation complex. This recruitment step, often the rate-limiting one in initiation and frequently also the target of regulation, is mediated by the eIF4 group of factors. The m7GpppN cap at the 5′ end of the mRNA is recognized by the eIF4E subunit of eIF4F. eIF4E is bound to eIF4G, a central coordinator of initiation that also associates with eIF3 and eIF4A, an RNA helicase that unwinds secondary structure. (The heterotrimeric complex of eIF4A, eIF4E, and eIF4G constitutes eIF4F.) Finally, after release of initiation factors from the 48S preinitiation complex, the 60S ribosomal subunit joins to form the 80S complex (for review, see Gingras et al., 1999; Hershey and Merrick, 2000; Pestova et al., 2001; Dever, 2002).
To dissect functional interactions of BC1 RNA with the translation initiation mechanism, we experimentally visualized different stages in translation initiation by arresting the mechanism at that stage and by subsequently resolving stable complexes by sucrose density gradient centrifugation. As described previously (Gray and Hentze, 1994), recruited 43S preinitiation complexes will stall at the initiator AUG, and 48S complexes will therefore accumulate, if the subsequent step of initiation factor dissociation (which depends on the hydrolysis of GTP bound to eIF2) is blocked by the nonhydrolyzable GTP analog GMP-PNP. Analogously, 80S ribosomal initiation complexes can be detected by using cycloheximide to inhibit elongation: ribosomes will be arrested at the start site, resulting in the accumulation of 80S complexes (Fig.2A).

BC1 RNA inhibits 48S and 80S complex assembly in cap-dependent initiation. A, A schematic diagram summarizes the steps in translation initiation that lead to the successive formation of 48S and 80S complexes. Steps that are targeted by inhibitors GMP-PNP and cycloheximide are indicated byarrows. The heterotrimeric complex eIF4F consists of eIF4A, eIF4E, and eIF4G. The helicase activity of eIF4A is stimulated by eIF4B. In addition, eIF4A is also present in free, monomeric form. [For more detailed diagrams of the translation initiation pathway, seeGingras et al. (1999), Hershey and Merrick (2000), and Dever (2002.)]B, 32P-labeled capped and polyadenylated α-tubulin mRNA was used as a programming mRNA in the presence of cycloheximide to visualize 80S complexes. At 600 nm BC1 RNA, 80S complex formation was found to be reduced by 61 ± 5% (measured from the slope of the ribonucleoprotein complex peak; 3 experiments). C, Analogously, assembly of 48S preinitiation complexes was visualized by using GMP-PNP. At 600 nm BC1 RNA, 48S complex formation was found to be reduced by 81 ± 5% (measured from the slope of the ribonucleoprotein complex peak; 3 experiments). D, In contrast to BC1 RNA, U4 RNA at the same concen- tration had no effect on 48S complex assembly.E, Formation of 48S complexes on nonadenylated α-tubulin programming mRNA was inhibited in the presence of BC1 RNA to an extent similar to polyadenylated α-tubulin mRNA (compare withC). Assembled complexes were resolved by sucrose density gradient centrifugation. Sedimentation was from right toleft. Fractions from top parts of the gradient have been omitted for clarity. Tub(A) mRNA, Polyadenylated (A98) α-tubulin mRNA; Tub mRNA, nonadenylated α-tubulin mRNA.
We first used cycloheximide to visualize assembly of 80S complexes with a capped programming mRNA encoding α-tubulin (Fig.2B). Full-length BC1 RNA, used at 600 nm, significantly reduced 80S complex formation, indicating that translation initiation was inhibited at or before this step. Next we used GMP-PNP to visualize formation of 48S preinitiation complexes. As with 80S complex formation, the presence of 600 nm BC1 RNA resulted in a significant reduction of 48S complex assembly (by 81% on average) (Fig. 2C). In contrast to BC1 RNA, U4 RNA at the same concentration had no effect on the formation of 48S complexes (Fig. 2D). These data confirm that the BC1-mediated inhibition of initiation complex formation was specific. Finally, we asked whether the inhibition of translation initiation by BC1 RNA was dependent on the adenylation status of the programming mRNA. This did not seem to be the case because we observed no difference in the extent of BC1-mediated inhibition of 48S complex formation depending on whether the programming α-tubulin mRNA was polyadenylated or nonadenylated (Fig.2E).
Taken together, these results indicate that BC1 RNA specifically represses formation of the 48S preinitiation complex (and, consequently, of the 80S complex). They are consistent with the notion that BC1 RNA inhibits recruitment of the 43S complex to the mRNA and/or its translocation to the AUG start site.
BC1 RNA represses translation through interaction with initiation factors of the eIF4 group
Having shown that BC1 RNA inhibits assembly of the 48S preinitiation complex, we next sought to pinpoint the target site(s) of BC1 RNA in that part of the translation initiation pathway that leads to 48S complex formation. For this purpose, we used a functional test in which we took advantage of different types of viral IRES translation initiation mechanisms.
Internal ribosome entry provides an alternative to the cap-dependent initiation mechanism: the small ribosomal subunit binds to an IRES, either at or upstream of the AUG start codon, in an end-independent manner (for review, see Jackson, 2000; Hellen and Sarnow, 2001; Pestova et al., 2001). Viral internal ribosome entry initiation mechanisms differ from each other in their need for canonical initiation factors. Two major subtypes of viral internal entry mechanisms can be distinguished. The first one is exemplified by the EMCV and other picornavirus IRESs. Formation of the 48S complex at the EMCV IRES requires the same set of canonical initiation factors as the cap-dependent mechanism except for eIF4E, the cap-binding protein (Pestova et al., 1996a,b). Translation commences at the AUG at the 3′ border of the IRES: thus, no scanning is necessary, but eIF4A is required to melt mRNA secondary structure for effective ribosomal recruitment. A second subtype of internal entry, exemplified by the hepatitis C virus IRES and the CSFV and related pestivirus IRESs, uses a much simpler mechanism (Pestova et al., 1998). This type of IRES binds directly to the 40S ribosomal subunit in a mechanism that does not require any of the factors of the eIF4 group.
The two described internal entry mechanisms were used for a functional dissection of translation initiation repression by BC1 RNA. We first asked whether such repression was cap dependent. To address this question, we used an uncapped programming mRNA (encoding GFP) in which internal entry was mediated by the EMCV IRES. BC1 RNA effectively repressed translation of this mRNA (Fig.3A). Phosphorimaging quantification of six experiments showed that on average, BC1 RNA decreased translation efficiency by ∼79% at 320 nm (Fig. 3B). This reduction is very similar in extent to the one observed above for capped programming mRNAs. As in cap-dependent translation, U4 RNA had no effect on translation efficiency (Fig. 3C). Similar results were obtained with other programming mRNAs and with dicistronic constructs. In the example shown in Figure 3D, the first cistron was preceded by a 5′ cap, whereas the second cistron was preceded by an EMCV IRES. BC1 RNA inhibited both cap- and IRES-mediated translation in this system. Translation from the IRES-dependent cistron, being more efficient in the absence of BC1 RNA, was also more susceptible to BC1-mediated repression. This result suggests that the EMCV IRES has a higher dependence on a factor/activity that is inhibited by BC1 RNA. It is interesting to note in this context that translation mediated by this IRES is also more strongly inhibited by trans-dominant eIF4A mutants than cap-dependent translation (Pause et al., 1994). Finally, analogous experiments with human BC200 RNA revealed that this RNA repressed translation in very much the same manner. Translation initiated by internal entry at the EMCV IRES was inhibited by BC200 RNA by 73% at 270 nm (data not shown).

BC1 RNA inhibits translation initiated by the EMCV IRES. A, The programming mRNA encoded GFP, contained an EMCV IRES in the 5′ untranslated region, and was used uncapped.B, Results from six experiments, quantified by phosphorimaging, showed that translation was repressed by 79% at 320 nm BC1 RNA [one-way ANOVA, p < 0.001; Scheffe's multiple comparison post hoc analysis (comparison with 0 nm BC1 RNA control): ***p < 0.001 for all groups]. C, As a control, the same mRNA was translated in the presence of U4 RNA.D, Both cap-initiated and IRES-initiated translation from a dicistronic programming mRNA were repressed by BC1 RNA. The first, cap-dependent cistron encoded blue fluorescent protein (BFP). An EMCV IRES preceded the second, GFP-encoding cistron.
The results indicate that BC1-mediated translational repression is not cap/eIF4E-dependent because translation initiated through internal entry via the EMCV IRES mechanism is equally inhibited. Are other members of the eIF4 family of translation initiation factors required for BC1-mediated translational repression? We addressed this question by taking advantage of the CSFV IRES system. Figure4A shows that BC1 RNA was not effective in repressing translation if internal entry was mediated by the CSFV IRES. Quantification by phosphorimaging revealed no significant change in translational efficiency with increasing concentrations of BC1 RNA (Fig. 4B). Control RNAs such as U4 RNA (Fig. 4C) were equally ineffectual. It is concluded that translation initiation by internal entry using the CSFV IRES mechanism effectively bypasses BC1-mediated translational repression.
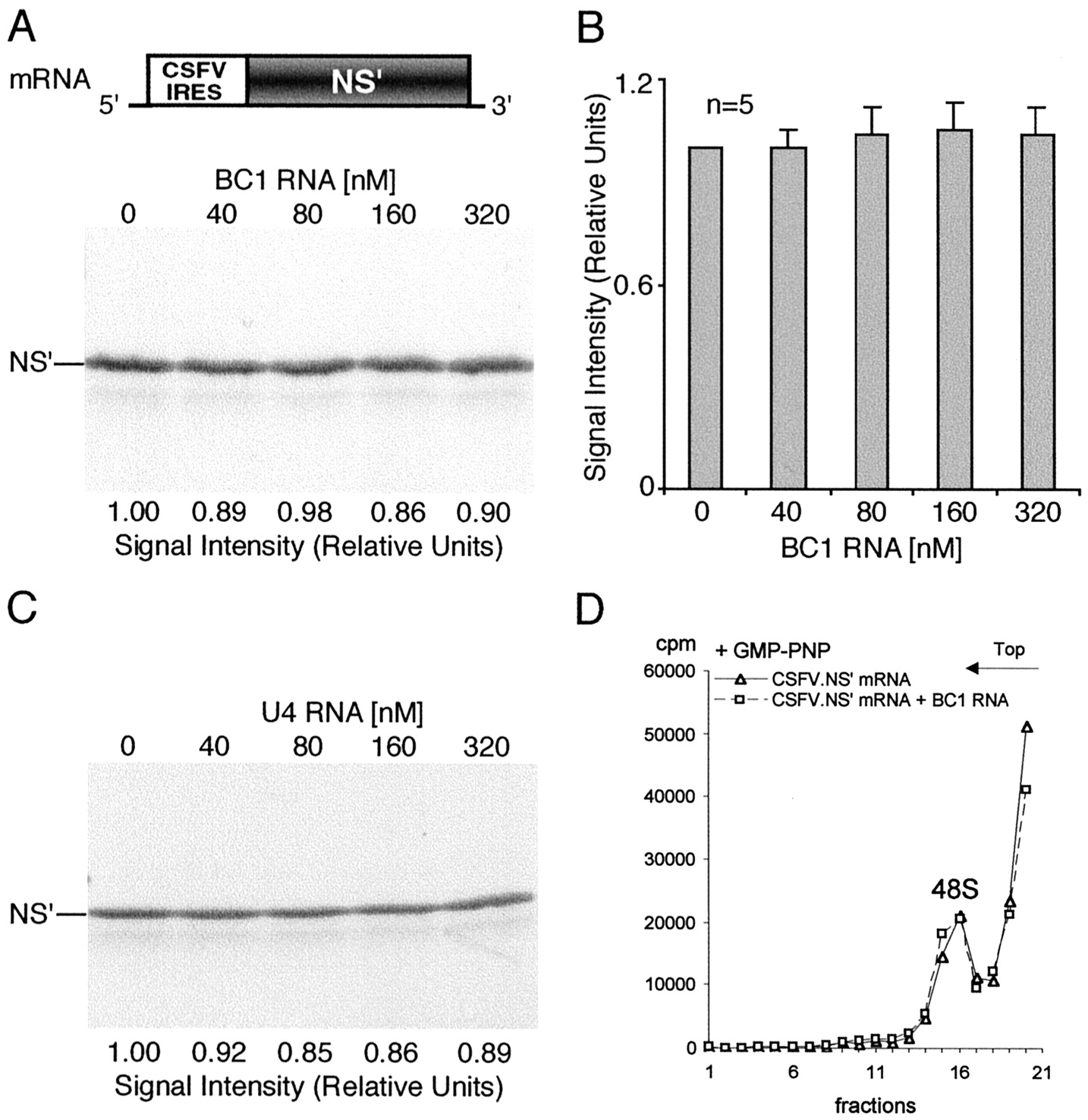
Translation and 48S complex formation mediated by the CSFV IRES are refractory to repression by BC1 RNA. The uncapped but polyadenylated programming mRNA encoded a truncated version of the influenza virus nonstructural protein (NS′).A, B, Translation efficiency was not significantly altered by increasing concentrations of BC1 RNA (one-way ANOVA, p = 0.9694; n = 5).C, Nuclear U4 RNA also failed to affect translation initiated from the CSFV IRES. D, Assembly of 48S complexes mediated by the CSFV IRES was refractory to inhibition by BC1 RNA (3 experiments). 48S complexes were assembled in the presence of GMP-PNP and resolved by sucrose density gradient centrifugation as described above (see also Fig. 2).
These results were confirmed and extended by sucrose density gradient centrifugation analysis. BC1 RNA was found not to repress formation of either 48S complexes (Fig. 4D) or 80S complexes (data not shown) if internal entry occurred at the CSFV IRES. This result confirms the notion that translation initiated via the CSFV IRES mode is refractory to BC1-mediated repression. Mechanisms that are common to both the CSFV IRES and the EMCV IRES mode can therefore be ruled out as candidate targets for BC1-mediated translational repression. These include all elongation and termination steps as well as most steps in the initiation pathway, such as, for example, formation of the ternary eIF2/GTP/Met-tRNAi complex, prerequisite for 48S complex assembly (for review, see Hellen and Sarnow, 2001; Pestova et al., 2001).
Initiation on the CSFV IRES differs from both EMCV IRES-mediated and cap-dependent initiation in that there is no requirement for any of the members of the eIF4 group of factors (Pestova et al., 1998). Of these factors, eIF4G and eIF4A are required for 48S complex assembly in the EMCV-type internal entry mode but not in the CSFV-type internal entry mode (Pestova et al., 1996a; Pestova et al., 1998). In addition, PABP also qualifies as a potential BC1 target because it enhances initiation mediated by the EMCV IRES (Michel et al., 2001; Svitkin et al., 2001). The combined results therefore effectively limit the pool of likely BC1 target candidates to these aforementioned factors.
eIF4A and PABP interact directly with BC1 RNA
Functional analysis was thus used to narrow down potential target sites for BC1-mediated inhibition in the translation initiation pathway and, consequently, potential BC1 interacting factors in the translation initiation machinery. In the next step, we applied biochemical methods for a direct analysis of BC1–protein interactions with those candidates.
Using EMSAs with recombinant proteins, we probed binding of BC1 RNA to eIF4A, eIF4G, and PABP. Because the central domain of eIF4G has been shown previously to bind to the EMCV IRES (Pestova et al., 1996b), we first examined potential interactions of BC1 RNA with this domain. We were unable to detect any specific binding of BC1 RNA to the central eIF4G domain (aa 697–1076; data not shown). In contrast, EMSA analysis revealed specific binding of BC1 RNA to eIF4A (Fig.5A). Specificity was demonstrated by the fact that preincubation with unlabeled BC1 RNA effectively abolished the mobility shift. Conversely, unlabeled irrelevant RNAs such as random-sequence vector RNA or tRNAs were not effective in competing with BC1 RNA for binding to eIF4A in these assays (Fig. 5A). In the presence of such noncompeting RNAs, the eIF4A-induced mobility shift was resolved as a duplex band. This observation is interpreted to indicate that under these conditions, two BC1/eIF4A complexes were migrating at slightly different mobilities.

BC1 RNA binds to translational factors eIF4A and PABP. EMSA experiments were performed with 32P-labeled BC1 RNA. A, BC1 RNA was incubated with eIF4A in the absence or presence of unlabeled competitor RNAs. Unlabeled BC1 RNA, but not unlabeled random sequence (RS) RNA or tRNAs, competed for binding to eIF4A and effectively abolished the mobility shift.B, BC1 RNA produced a band shift with full-length PABP. Effective competition was seen with unlabeled BC1 RNA but not with unlabeled U4 RNA or U6 RNA. C, Simultaneous incubation of BC1 RNA with eIF4A and PABP (N-terminal segment) produced a more substantial mobility shift than incubation with either protein alone.D, In rat brain extracts, BC1 RNA was observed to be shifted to two bands of lower mobility (lane 1). An antibody specific for PABP (lane 2), but not a control antibody against GST (lane 3), produced a supershift with BC1 RNA. Conversely, the regular mobility shift of BC1 RNA was reduced in brain extracts that had been immunodepleted of PABP; note the reduction in intensity of the major BC1 RNA complex bands and the appearance of a band at higher mobility (lane 5).BE, Brain extract; ID BE, PABP-immunodepleted brain extract.
In addition, we found that BC1 RNA bound specifically to PABP (Fig.5B). Again, specificity was ascertained in EMSA competition experiments in which unlabeled BC1 RNA effectively competed for binding, whereas irrelevant RNAs did not. Simultaneous exposure of BC1 RNA to both eIF4A and PABP in EMSA experiments produced a larger shift than exposure to either eIF4A or PABP alone (Fig. 5C), indicating that binding of these two proteins to BC1 RNA was not mutually exclusive. In addition, using an antibody specific for PABP, we found that the mobility shift that is observed with BC1 RNA in rat brain extracts was specifically “supershifted” to further reduced mobility (Fig. 5D). Conversely, if the same antibody was used to immunodeplete brain extracts of PABP, the mobility shift of BC1 RNA was now predominantly observed at increased mobility (Fig. 5D). Taken together, the results suggest that BC1 RNA interacts specifically with eIF4A and PABP.
eIF4A, eIF4G, and PABP are localized in dendrites
Because BC1 RNA is targeted to dendrites, any interaction with eIF4A and PABP would obviously require the presence in dendrites of these proteins as well. In addition, eIF4G would also be needed in its role of a scaffolding protein that interacts with both eIF4A and PABP (for review, see Gingras et al., 1999; Jackson, 2000; Dever, 2002). It was therefore necessary to probe for the presence of these three proteins in dendrites. To this end, we applied immunocytochemistry, in conjunction with confocal laser scanning microscopy (CLSM), to hippocampal neurons in culture (Tiedge and Brosius, 1996). The results presented in Figure 6 illustrate that eIF4A, eIF4G, and PABP were detectable in dendrites at substantial levels. (No significant labeling was detected along axonal shafts for any of these factors.) Throughout dendrites, labeling patterns for all three proteins were of heterogeneous, particulate nature, often giving a punctate appearance. On average, such labeling clusters were observed less frequently in distal dendritic segments than in proximal segments. The results indicate that eIF4A, eIF4G, and PABP are distributed along dendrites in a heterogeneous, clustered manner.

Factors eIF4A, eIF4G, and PABP are enriched in synaptodendritic microdomains of hippocampal neurons in culture. Neurons were labeled (red fluorescence) for eIF4G (A), for PABP (B), or for eIF4A (C). Cells were double labeled with an antibody against synaptophysin (green fluorescence). Boxed dendritic segments are shown at three times higher magnification in insets. Note the clustered appearance of dendritic labeling signals for all three factors. Such clusters were often but not always observed in apposition to synaptophysin puncta. D, Control experiments were performed in an identical manner except that incubation with primary antibodies was omitted. Scale bar, 10 μm.
Are such dendritic clusters associated with synaptic structures? To address this question, immunocytochemical experiments were performed in dual-labeling mode, using in parallel an antibody against synaptophysin, a marker protein for synaptic vesicles and thus for presynaptic specializations (Jahn et al., 1985). This antibody has been shown previously to identify presynaptic specializations as discrete puncta in mature hippocampal neurons in culture (Fletcher et al., 1991,1994). Using CLSM, we detected such puncta prominently displayed along dendritic extents, typically at decreasing frequency in more distal segments (Fig. 6). Subpopulations of eIF4A, eIF4G, and PABP labeling clusters were seen in spatial association with synaptophysin puncta. Such association was best observed in distal dendrites, where cluster densities were not so high as to obscure resolution by excessive overlap (Fig. 6). Red (eIF4A, eIF4G, or PABP) and green (synaptophysin) labeling clusters were often seen in direct apposition to each other, the latter typically of more superficial appearance. Some, but not all, apposing red/green puncta pairs apparently overlapped to some degree, evidenced by narrow yellow interface areas. Because green puncta identify axonal presynaptic specializations, it is concluded that such apposing red clusters correlate with postsynaptic dendritic compartments.
In summary, the results indicate a differential intradendritic localization of eIF4A, eIF4G, and PABP clusters, with some of those clusters positioned in postsynaptic microdomains underneath, or in the direct vicinity of, presynaptic axonal specializations. We suggest that in dendrites, such synapse-associated clusters serve in the local synthesis of dendritic proteins (such as CaMKIIα) (Burgin et al., 1990) that are enriched in postsynaptic compartments, whereas extrasynaptic eIF4A, eIF4G, and PABP clusters preferentially participate in the synthesis of dendritic proteins (such as MAP2) (Garner et al., 1988) that are not synapse associated.
DISCUSSION
Modulation of synaptic activity may result in long-term structural and functional changes at the synapse. Some of such changes are likely to be orchestrated through mechanisms of local protein synthesis in postsynaptic dendritic microdomains (for review, see Tiedge et al., 1999; Kiebler and DesGroseillers, 2000; Wells et al., 2000; Greenough et al., 2001; Job and Eberwine, 2001b; Richter, 2001; Steward and Schuman, 2001). Translational control of gene expression at the synapse would require that local regulatory mechanisms are in place to ensure that requisite proteins are being synthesized not only at the correct synapse but also at the right time (Job and Eberwine, 2001b). Thus, to prevent postsynaptic protein synthesis at inappropriate times, translation of local mRNAs will have to be repressed, until such time that synthesis of cognate proteins is required. For postsynaptic mRNAs, translational repression may therefore often be the default status because some of the local repression mechanisms may operate on a synapse-wide basis. Alternatively or in addition, depending on local requirements, translational modulation may be selective for certain classes of mRNAs, or even for individual mRNAs.
We now identify dendritic BC1 RNA as a specific repressor of translation. BC1 RNA is a nontranslatable small neuronal RNA that does not contain a protein coding sequence (for review, see Brosius and Tiedge, 1995, 2001). It has been localized previously to dendrites (for review, see Brosius and Tiedge, 2001) where it was found enriched in postsynaptic compartments, colocalized with a subset of neuronal mRNAs that are selectively delivered to dendrites (Chicurel et al., 1993). It was on the basis of such and other evidence that BC1 RNA has been hypothesized to function as a translational modulator (Brosius and Tiedge, 2001). In this paper, we have functionally dissected the translational repression competence of BC1 RNA. Taking advantage of IRES systems with differential factor requirements, and of sucrose density gradient centrifugation techniques to resolve initiation complexes, we show that BC1 RNA represses translation by inhibiting initiation at the level of 48S complex assembly.
Formation of the 48S preinitiation complex is the rate-limiting step in translation initiation under most circumstances (for review, seeGingras et al., 1999; Hershey and Merrick, 2000). BC1-mediated translational repression therefore targets a strategic position in the initiation pathway. Our combined functional data further indicate that BC1-mediated translational repression operates through the eIF4 family of initiation factors because internal initiation by the CSFV IRES mechanism, which does not require any of these factors, effectively bypasses this repression. A key factor in the recruitment of the 43S preinitiation complex to the mRNA is eIF4F, a heterotrimeric complex composed of eIF4E, a cap-binding protein, eIF4A, an ATP-dependent RNA helicase, and eIF4G, a large scaffolding protein (for review, seeGingras et al., 1999; Jackson, 2000; Pestova et al., 2001). The data reported here show that BC1-mediated repression is cap-independent (and therefore eIF4E-independent). On the other hand, EMSA assays indicate a specific interaction of BC1 RNA with eIF4A, the unwinding factor that is presumed to melt down secondary structure in the 5′ region of the mRNA. It is possible that secondary structure elements within BC1 RNA (Rozhdestvensky et al., 2001) are responsible for the eIF4A interaction. In contrast to other initiation factors that have been estimated to be present at intracellular concentrations in the submicromolar range, eIF4A has been shown to be more abundant (Pause et al., 1994). The subpopulation of eIF4A that is part of the holo-eIF4F complex appears to function as the physiological RNA helicase (Gingras et al., 1999). Inhibition of eIF4A-containing eIF4F by BC1 RNA can be expected to prevent the 43S preinitiation complex from being recruited to the mRNA and thus from forming a stable 48S complex. Translation would thus be repressed just before the point at which the system commits itself to initiation.
Another layer of control may be provided by the interaction of BC1 RNA with PABP. [Recently, PABP has also been observed to be associated with BC1 ribonucleoprotein particles (Muddashetty et al., 2002; West et al., 2002).] PABP binds poly(A) tails of mRNAs and, at the same time, interacts with the eIF4G component of eIF4F, thereby effectively circularizing the mRNA (Tarun and Sachs, 1996; Imataka et al., 1998). It is assumed that such dual interaction forms the basis for PABP-dependent stimulation of translation; however, details of this mechanism remain poorly understood (Gingras et al., 1999; Hershey and Merrick, 2000; Sachs, 2000). Although we observed that BC1-mediated repression is effective in poly(A)-enhanced translation, but less so in poly(A)-independent translation, 48S complex formation was clearly repressed by BC1 RNA regardless of whether the programming mRNA was polyadenylated. In the latter assay, complex formation is analyzed on previously uninitiated mRNAs such that by definition, terminating ribosomes cannot be present to recycle. However, PABP has also been reported to activate translation in a poly(A)-independent manner, presumably via functional interactions with eIF4F (Le et al., 1997;Otero et al., 1999). It is tempting to speculate that BC1 RNA, by interacting with eIF4A and PABP, targets a functional link between these two proteins that is required for efficient initiation.
It should be noted that sequence similarity between rodent BC1 RNA and primate BC200 RNA (Tiedge et al., 1993) is restricted to the 3′ domain and the central A-rich domain, the latter a potential target for PABP binding. Interactions of relevant factors with these domains therefore may be assumed to be underlying BC1-mediated translational repression. This hypothesis, to be substantiated in future research, would propose that in the tripartite domain structure of BC1 RNA (Brosius and Tiedge, 1995; Rozhdestvensky et al., 2001), the 5′ domain is competent to specify dendritic transport (Muslimov et al., 1997), whereas the central and 3′ domains play functional roles in translational repression.
A hallmark of the BC1 repression mechanism is the fact that it is effective with cap-dependent initiation as well as with internal initiation of the EMCV type. It has been reported recently that a number of dendritic mRNAs may be translated in a cap-independent manner, and it has been suggested that IRES-mediated postsynaptic translation of such mRNAs may allow for differential modulation in response to synaptic activation (Pinkstaff et al., 2001). Prerequisite for such a scenario would be a mechanism to control IRES-mediated postsynaptic translation, a function that BC1 RNA is well positioned to fulfill. However, the nature and mechanism of action of dendritic IRESs remain to be established.
Clearly, differential modulation of postsynaptic protein synthesis would require the functional interplay of more than one translational control pathway. How, for instance, would BC1-mediated repression be reversed at times of demand? We have localized eIF4A, eIF4G, and PABP to synaptodendritic compartments using confocal microscopy, and it is certainly possible that the functionality of one or several of these factors in dendrites is subject to activity-dependent modulation. For example, the phosphorylation status of the eIF4G component of eIF4F (Raught et al., 2000) may affect the binding affinity of the complex toward BC1 RNA, and thus the ability of the RNA to repress translation. How could translational derepression be selective for certain classes or types of mRNAs? In several cases, for example during cell growth, in response to cell stress, and during apoptosis, cellular IRES-mediated translation has been shown to remain operational at times when cap-dependent translation has shut down (for review, see Hellen and Sarnow, 2001). Differential repression/derepression of cap-mediated versus IRES-mediated modes of translation has not yet been described in neurons. However, mechanisms have been reported for the selective modulation of cap-dependent local translation in dendrites (for review, see Wells et al., 2000; Job and Eberwine, 2001b). In one example, the rapamycin-sensitive kinase mTOR has been implicated in long-term hippocampal plasticity in a pathway that may involve dendritic eIF4E and eIF4E binding proteins 1 and 2 (Raught et al., 2001; Tang et al., 2002). In a second example, the cytoplasmic polyadenylation pathway has been suggested to provide a further means of selective translational modulation. Activity-dependent cytoplasmic polyadenylation may increase translatability of mRNAs, such as dendritic CaMKIIα mRNA, that contain cytoplasmic polyadenylation elements (CPEs). Conversely, the default state of CPE-containing mRNAs appears to be translational repression, mediated through an eIF4E-dependent mechanism (Wu et al., 1998; Wells et al., 2001; Cao and Richter, 2002) (for review, see Wells et al., 2000).
The combined evidence indicates that local translational control in dendrites is likely to be a multitiered network of intersecting pathways. At one level, the BC1-dependent mechanism is proposed to repress cap-dependent translation as well as translation mediated by internal ribosome entry of the EMCV type. This mechanism may involve most or all mRNAs at the synapse. Derepression at this level would be prerequisite to initiate translation of both capped and IRES-containing mRNAs. However, such derepression would not necessarily be sufficient to stimulate all types of cap-dependent translation because pathways such as those discussed above may remain repressed. In this model, additional derepression at the level of such individual pathways would result in the selective translational activation of specific synaptic mRNAs or classes of synaptic mRNAs. We thus suggest that simultaneous activation or derepression of several interdependent translational control pathways is required to orchestrate activity-modulated synthesis of postsynaptic proteins in local microdomains. A further level of complexity is added by the fact that the translational repressor BC1 RNA is itself subject to activity-dependent regulation (Muslimov et al., 1998). Such long-term, reversible modulation of overall BC1 levels may contribute to a cell-wide upregulation or downregulation of dendritic protein synthesis as a function of the physiological state of the cell.
With the significance of functional, nontranslatable RNAs in cellular structure and function being increasingly appreciated, the traditional view of RNAs as mere passive carriers of information is in obvious need of amendment. Nontranslatable RNAs have been implicated in various cellular functions (for review, see Storz, 2002); some micro-RNAs, for example, may participate in translational control, albeit in mechanisms that are clearly distinct from the BC1 pathway. Functional RNAs may exist in much larger numbers than hitherto assumed, and it is likely that genes encoding such RNAs, far from being mere remnants of an early RNA world, are continually being generated in eukaryotic species (Brosius and Tiedge, 1996; Kuryshev et al., 2001; Eddy, 2002; Wang et al., 2002). We therefore submit that nontranslatable RNAs in nerve cells not only function as determinants of neuronal functionality and plasticity, but at the same time serve as a driving force in neural species diversification.
Footnotes
This work was supported in part by fellowships from Consiglio Nazionale delle Ricerche and Istituto Pasteur–Fondazione Cenci Bolognetti (A.I.), by National Science Foundation Grant 0110834 (C. U. Hellen), by a New York City Council Speaker's Fund for Biomedical Research Grant (I.A.M.), and by National Institutes of Health Grant NS34158 (H.T.). We thank C. U. Hellen and V. G. Kolupaeva for advice and discussion, J. Brosius, J. Carson, and T. V. Pestova for plasmids, H. Asmussen and G. Banker for hippocampal neurons in culture, and J. Weedon for help with statistical analyses.
Correspondence should be addressed to Henri Tiedge, Department of Physiology and Pharmacology, State University of New York, Health Science Center at Brooklyn, 450 Clarkson Avenue, Brooklyn, NY 11203. E-mail: tiedge{at}hscbklyn.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}