

Abstract
Neurons are highly differentiated cells that normally never enter a cell cycle; if they do, the result is usually death, not division. For example, cerebellar granule neurons in staggerer and lurcher mutant mice initiate a cell cycle-like process just before they die. E2F1 is a transcription factor that promotes cell cycle progression. Because E2F1 is also involved in apoptosis, we bred double mutants (E2f1−/−; staggerer and E2f1−/−; lurcher) to assess its role in the cell cycle-related death of cerebellar granule cells in vivo. We found neither granule cell cycle initiation nor cell death was significantly altered in either double mutant. However, after postnatal day 10, neurons throughout the CNS of E2f1−/− and E2f1+/− animals were found to express cell cycle proteins and replicate their DNA. Whereas Map2 and synapsin1 staining are little altered, there is a reduction of calbindin in Purkinje cell dendrites at 1 year of age, suggesting that the mutant cells also undergo a slow, subtle atrophy. These events are cell autonomous, because cultured E2f1−/− cortical neurons “cycle” in vitro, whereas wild-type neurons do not. Our results suggest that, in mature CNS neurons, E2F1 functions as a cell cycle suppressor.
Introduction
In the developing CNS, once young neurons leave the neurogenic regions of the ventricular and subventricular zone, they will never again complete a full cell cycle. Yet evidence is mounting that adult nerve cells are not permanently postmitotic (for review, see Herrup and Yang, 2007). During target-related cell death, for example, death is preceded by DNA synthesis and cell cycle protein expression (Herrup and Busser, 1995). Similarly, in adult onset neurodegenerative diseases, neuronal death is often preceded by the neurons reengaging a cell cycle-like process. Human Alzheimer's disease (AD) and its mouse models are perhaps the best studied examples of this intersection between the cell cycle and cell death processes (Vincent et al., 1996; McShea et al., 1997; Nagy et al., 1997; Arendt et al., 1998; Busser et al., 1998; Herrup et al., 2004). Although they begin a cell cycle-like process, there is no report of a neuron proceeding to M-phase.
The E2F family of transcription factors plays a pivotal role in cell proliferation, differentiation, and apoptosis through transcription regulation (Ishida et al., 2001; Muller et al., 2001; Ren et al., 2002; Stanelle et al., 2002). Among the eight structurally related E2F members, E2F1, E2F2, and E2F3a are transcriptional activators, an activity that is blocked by their association with the retinoblastoma (RB) tumor suppressor. This association is lost after RB is phosphorylated by cyclin-dependent kinases. Cell cycle promotion is the most frequently cited function of E2F, but deregulation of E2F1 can also induce apoptosis in different cellular contexts (Wu and Levine, 1994; Denchi and Helin, 2005; DeGregori and Johnson, 2006). For example, E2F1 is elevated in the brains of AD and Down's syndrome patients (Liu and Greene, 2001; Motonaga et al., 2001), and E2f1-deficient neurons in culture are resistant to stimuli leading to cell cycle-related neuronal death (Giovanni et al., 2000; O'Hare et al., 2000). Similarly, in RB-deficient mice, there is profound neuronal loss in the CNS, and E2f1−/−; Rb1−/− double mutants show that this loss is E2F dependent (Tsai et al., 1998; Ziebold et al., 2001). Indeed, given the key role played by these proteins in cell division and apoptosis, it is surprising that E2F1, E2F2, or E2F3 knock-out mice are born as viable fertile animals. In older E2f1−/− mice, paradoxically, cellular dysplasia and tumor formation has been described in several tissues.
Given the role of cell cycle events in target-related cell death and the proapoptotic role of E2F1 in several situations, we predicted that E2F1 would be involved in cerebellar granule cell death in both staggerer and lurcher mutant mice. This prediction proved incorrect, because neither cell cycle activity nor cell death in these mutants is rescued by removing E2F1. Instead, we found ectopic cell cycle events in many other neurons throughout E2f1−/− mutant brain. These events include constitutive expression of proteins normally only found in cycling cells as well as DNA replication. We suggest that E2F1 works as cell cycle suppressor in differentiated neurons.
Materials and Methods
Animals
A breeding colony of mice with a targeted disruption of the E2f1 gene (Field et al., 1996) was established from founders obtained from The Jackson Laboratory (Bar Harbor, ME). Generation of mutants was achieved through the mating of heterozygous E2f1 males and females. The mice were maintained on a mixed (C57BL/6J × 129/S4) background. Genotyping was performed on DNA extracted from tail biopsies using PCR techniques as described previously (Field et al., 1996).
For E2f1−/−; staggerer (Rorasg) double-mutant mice, we first crossed E2f1−/− mice with Rorasg/+ mice to get E2f1+/−; Rorasg/+ mice, and then intercrossed the double heterozygotes to get double mutants. We used a similar method to create E2f1−/−; lurcher (Grid2Lc) mice. We first crossed E2f1−/− mice with Grid2s+/Lc mice to get E2f1+/−; Grid2+/Lc mice. Then, we crossed E2f1+/−; Grid2+/Lc with E2f1−/−; Grid2+/+ mice to get E2f1−/−; Grid2+/Lc mice. Genotyping of the staggerer locus was performed on DNA extracted from tail biopsies using PCR techniques described previously. Grid2+/Lc mice were identified based on their ataxia.
Animals were housed in either the Case Western Reserve University Medical School Animal Resource Center or the Laboratory Animals Services Center of Rutgers University. Both facilities are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All procedures for animals were approved by the Institutional Animal Care and Use Committee of the two universities.
Histology
After animals were deeply anesthetized with Avertin (0.02 cc/mg body weight), they were transcardially perfused with PBS, followed by 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB). The brain was dissected and transferred to fresh 4% paraformaldehyde at 4°C overnight. The brains were then cryoprotected by sinking in 30% sucrose/PB at 4°C overnight. After bisecting along the midline, the brains were embedded in OCT, and 10 μm cryostat sections were cut and allowed to air dry on SuperPlus glass slides overnight. All slides were stored at −80°C.
Single and double immunocytochemistry
The proliferating cell nuclear antigen (PCNA) mouse monoclonal antibody and rabbit polyclonal antibody (DakoCytomation, High Wycombe, UK) were diluted 1:300 in 10% goat serum/PBS blocking buffer before use. Rat anti-bromodeoxyuridine (BrdU) (1:100; Abcam, Cambridge, UK) was used to detect DNA replication. A rabbit polyclonal cyclin A antibody raised against the C-terminal domain of cyclin A2 was used at a dilution of 1:500 (ab 7956; Abcam). The mouse monoclonal antibodies anti-Tuj1 (1:1000; Covance, Princeton, NJ) and anti-NeuN (1:300; Chemicon, Temecula, CA) were used as neuronal markers. The mouse monoclonal calbindin antibody at a dilution of 1:500 (Sigma, St. Louis, MO) was used as a Purkinje cell marker.
Double fluorescence immunocytochemistry was used on the mouse cryosections. Sections were first rinsed in PBS, followed by pretreatment in 0.1 m citrate buffer for 6–8 min at 95°C. After the slides had cooled in buffer for 45 min at room temperature, slides were rinsed in PBS. Sections were incubated in 10% goat serum in PBS to block nonspecific binding for 1 h at room temperature. All primary antibodies were diluted in PBS containing 0.5% Triton X-100 and 10% goat serum and then were applied to sections and incubated overnight at 4°C. After rinsing in PBS, they were incubated for 2 h with secondary antibody, which was conjugated with various fluorescent Alexa dyes (dilution, 1:1000; Invitrogen, San Diego, CA). The sections were then rinsed in PBS and reincubated in 10% goat serum blocking solution for 1 h, followed by addition of the second primary antibody (raised in a different species from the first primary antibody) for a second overnight incubation at 4°C. Sections were then rinsed in PBS, and both secondary antibodies conjugated with different fluorescence dyes were applied to the sections for 2 h at room temperature. After rinsing, all sections were mounted in PBS/glycerol under a glass coverslip.
Fluorescent in situ hybridization
Three mouse-specific DNA probes were generated from bacterial artificial chromosomes (BACs), which carried unique mouse genomic DNA sequences. One of the DNA probes (480C6) was made from the region that encodes the β-amyloid precursor protein located on mouse chromosome 16. The other two probes were generated from overlapping BACs (170L21 and 566) containing the structural gene for mouse aldolase C on mouse chromosome 11. Each of the three probes covers 100–300 kb. They were labeled by standard nick translation protocols using digoxygenin-labeled dUTP.
After labeling, probes were concentrated with mouse Cot-1 DNA (Invitrogen) to block hybridization to repetitive sequences. Before hybridization, all tissue sections were rinsed in PBS and then pretreated with 30% pretreatment powder (Oncor, Gaithersburg, MD) for 15 min at 45°C followed by treatment with protease (0.25 mg/ml; Oncor) for 25 min at 45°C. After rinsing in 2× SSC, the slides were dehydrated through graded alcohols and allowed to air dry. Labeled probe was applied to the individual sections, which were then covered with a glass coverslip and sealed with rubber cement. To denature DNA, slides were heated at 90–92°C on a heated block for 12 min and hybridized with probe overnight at 37°C. After rinsing in 50% formamide/2× SSC at 37°C for 15 min, the slides were transferred to 0.1× SSC buffer for 30 min at 37°C and then rinsed in 0.5 m phosphate buffer with 0.5% NP-40 (PN buffer) at room temperature. To block nonspecific antibody binding, 10% goat serum in PBS was applied to the sections, followed by incubation in mouse anti-digoxygenin primary antibody (dilution, 1:200; Boehringer Mannheim, Indianapolis, IN) for 30 min at 37°C. After rinsing, secondary anti-mouse antibody conjugated with Alexa 488 (dilution, 1:250) was applied to the sections for 30 min at 37°C. Slides were rinsed three times in PN buffer, and the sections were counterstained with either 4′,6′-diamidino-2-phenylindole (DAPI) or propidium iodide and covered with a coverslip.
The number of spots of hybridization in each nucleus was determined at 1000× magnification under fluorescent illumination. Images were captured on a Leitz (Wetzlar, Germany) research microscope equipped with a digital camera (Prog C14). Purkinje cells were identified on the basis of the size of their nucleus and their position in the Purkinje cell layer. Cortical neurons were identified on the basis of the size and the position of their nucleus. For each of genotypes [postnatal day 30 (P30) E2f1+/+ and E2f1−/−], 60 cortical neurons from layer II and layer III were randomly picked and scored from six different areas for each section. A total of 12 sections was scored for each genotype.
Cell counts
For each of the three genotypes (P30 E2f1+/+, E2f1+/−, and E2f1−/−), we began at one end of the Purkinje cell layer within a given section and continued counting until the other end of layer was reached. All calbindin-positive cells were scored for the presence or absence of the cell cycle marker. Only cells with a discernable portion of their nucleus in the section were scored. Two animals were scored for each genotype.
For each cortex of the two genotypes (P20 E2f1+/+ and E2f1−/−), we examined six sections, each ∼400 μm from the previous, for each brain. In each section, six fields were randomly picked and scored for the presence or absence of the cell cycle marker from all the DAPI-positive nuclei. A total of 24 sections was scored for each genotype.
Primary neuronal cultures
Pregnant dams were killed by cervical dislocation and the uteri were dissected and placed on ice during the embryo harvest procedure. For E2F1-deficient cultures, all embryos from an E2f1−/− × E2f1−/− mating were treated separately. Embryos were collected in ice-cold PBS–glucose, and the cortical lobes were dissected out. Meninges were removed and the cortices were placed in trypsin-EDTA for 15 min at 37°C. The tissue was removed from the trypsin solution and placed in DMEM with 10% FBS to inactivate the trypsin, followed by transfer to Neurobasal media supplemented with B-27, penicillin/streptomycin, and l-glutamine (2 mm). Tissue was triturated 10 times through a 5 ml pipette and allowed to settle to the bottom of a 15 ml conical tube for ∼1 min. Cells in solution above the tissue pellet were removed and used in all subsequent procedures. Aliquots of the suspension were stained with trypan blue to allow counts of live and dead cells. The culture substrate used was poly-l-lysine (0.05 mg/ml) coated on plates or glass coverslips. Cells were grown in 24-well plates at a density of 50,000 cells/well. All cultures were grown for a minimum of 5 d in vitro (DIV) before any treatment. To assess cell cycle activity, BrdU was added to the culture to obtain a final concentration of 10 μm. After 5 d, cultures were fixed with 4% paraformaldehyde in phosphate buffer at room temperature for 30 min, and then washed and stored in PBS. All experiments were performed on a minimum of three litters; each condition was examined in triplicate.
Immunoblotting
E2f1−/− littermates were killed by cervical dislocation. Brains were rapidly dissected and placed in lysis buffer (20 mm Tris, 1% Triton X-100, 100 mm NaCl, 40 mm NaF, 1 mm EDTA, 1 mm EGTA, 1 mm Na3VO4, aprotinin, leupeptin, and PMSF) on ice and homogenized. Lysates were then sonicated for 15 s and centrifuged at 13,000 rpm for 15 min at 4°C. Supernatant was collected and protein was measured by the Bradford method. Equal amounts of protein (10 or 15 μg) in Laemmli buffer were boiled for 7 min. The samples were run on SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes as described previously (Combs et al., 2001). Antibodies used for immunoblotting were as follows: mouse anti-PCNA (1:200; DakoCytomation), mouse anti-actin (1:2000; Sigma), donkey anti-mouse HRP (1:2000; Jackson ImmunoResearch, West Grove, PA). Blots were developed with ECL (Amersham Biosciences, Piscataway, NJ) according to the instructions of the manufacturer.
Results
Granule cell death-by-cycle in staggerer and lurcher mice is E2F1 independent
Staggerer and lurcher are two mutations of mice that suffer postnatal granule cell loss in the cerebellum (Sidman et al., 1962; Caddy and Biscoe, 1979). The recessive mutation staggerer is the result of the inactivation of the Rora gene encoding an orphan retinoid receptor (Hamilton et al., 1996). The dominant mouse mutation lurcher is caused by a point mutation in the orphan glutamate receptor gene, Grid2 (Zuo et al., 1997). Both mutants have severe ataxia because of the cell-autonomous loss and/or deformation of Purkinje cells. In both mutants, there is a well defined cell cycle reentry process that occurs before the granule cells die (Herrup and Busser, 1995). Because the E2F1 protein is involved in both cell cycle and cell death, we sought to learn whether it played a role in staggerer or lurcher granule cell death. We produced double mutant animals (E2f1−/−; Rorasg/sg and E2f1−/−; Grid2+/Lc) by the crosses described in Materials and Methods. The cerebella of these animals and their littermate controls were examined at P30. At this age, the internal granule cell layer (IGL) of single mutant mice (staggerer or lurcher) is thin and sparsely populated because of the death of most of the granule cells (Fig. 1A–C). Significantly, the size of cerebellum of the E2f1 double mutant mice was not noticeably different from that seen in staggerer or lurcher alone (Fig. 1D,E). Thus, the deletion of the E2f1 gene provides no protection against granule cell loss in either mutation.

Deletion of the E2f1 gene does not protect granule cells from target-related cell death in either staggerer or lurcher cerebellum. Sagittal sections of P30 cerebellum were stained by cresyl violet. Compared with wild-type cerebellum (A), note the thinning of the granule cell layer in staggerer (B) and lurcher (C) mutants caused by the death of most granule cells. Double mutants with E2f1, either E2f1−/−; Rorasg/sg (D) or E2f1−/−; Grid2+/Lc (E) reveal no change in the overall morphology of the cerebellum. Scale bar, 200 μm.
It is possible that, despite the coincidence of cell cycle and cell death events in the granule cells, the two are not linked mechanistically. Thus, although the absence of E2F1 does not rescue the granule cell death in either staggerer or lurcher, we wanted to know whether the granule cell cycle events (CCEs) preceding the cell death were affected. We immunostained sections from double mutant animals with antibodies against a Purkinje cell marker, calbindin, and PCNA, a commonly used cell cycle marker and a component of the DNA polymerase complex that is elevated in S-phase and remains high through G2 (Bravo et al., 1987). As reported (Herrup and Busser, 1995), in Rorasg/sg or Grid2+/Lc mouse cerebella, there are PCNA-positive cells in IGL (Fig. 2A,G). We also saw PCNA staining in some mutant Purkinje cells, but this was quite weak (Fig. 2C,I). In the cerebellum of E2f1 double mutant mice, the pattern of PCNA staining in the IGL was unchanged (Fig. 2D,J). Thus, E2F1 deficiency alters neither the program of cell death nor the appearance of cell cycle events in the granule cells of staggerer or lurcher mice.

Deletion of the E2f1 gene does not suppress ectopic cell cycle events in either staggerer or lurcher cerebellum. Sagittal sections of P30 cerebellum from different genotypes were double labeled with the antibodies for the cell cycle marker PCNA (green) and the Purkinje cell marker calbindin (red). A–C, E2f1+/+; Rorasg/sg. D–F, E2f1−/−; Rorasg/sg. G–I, E2f1+/+; Grid2+/Lc. J–L, E2f1−/−; Grid2+/Lc. The pattern of PCNA staining in the granule cell layer of the double mutant mice is similar to that seen in single mutant animals. In the Purkinje cells, however, PCNA staining is much brighter in double mutant mice (D, J) than in single mutant animals (A, G). Scale bars, 25 μm.
Cell cycle-related protein expression in E2f1−/− mouse brain
During the course of these studies, we noticed that the levels of PCNA immunostaining of Purkinje cells were higher in double mutant mice compared with those observed in the single mutant mice (Fig. 2F,L). This enhanced cell cycle protein expression in Purkinje cells prompted us to investigate whether loss of E2F1 alone has any effect on cell cycle control in the cerebellar neurons. Mice of three genotypes (E2f1+/+, E2f1+/−, and E2f1−/−) were examined by immunohistochemistry. As expected, we saw no PCNA staining in any wild-type Purkinje cell (Fig. 3A). To our surprise, however, we found PCNA expression in the nuclei of substantial numbers of Purkinje cells in the E2f1−/− mouse brain (Fig. 3E). The same unexpected expression of PCNA was also found in heterozygous, E2f1+/−, animals (Fig. 3C). This indicates that the loss of even one allele of the E2f1 gene can induce ectopic CCEs in mouse Purkinje cells. To determine the extent of this phenomenon, we counted the number of calbindin/PCNA double-positive cells. We found that nearly one-half of the E2f1−/− Purkinje cells had high levels of PCNA protein in their nuclei and, furthermore, that there was no significant difference between E2f1 heterozygous and null mice (data not shown). To expand this finding, we used a second cell cycle maker, cyclin A (a partner of the Cdk2 and Cdk1 kinases and an S–G2 phase marker). As with PCNA, cyclin A appeared in both E2f1 heterozygous and null Purkinje cells. Unlike PCNA, however, the subcellular localization of cyclin A was not nuclear, as expected, but rather cytoplasmic (Fig. 3D,F). This can be seen as cell body staining (asterisks) as well as in a punctate configuration suggestive of a process such as an axon. No cyclin A or PCNA-labeled Purkinje cells were found in control animals (Fig. 3B).

Ectopic cell cycle protein expression in E2f1-deficient Purkinje cells. Mice of three E2f1 genotypes were examined by immunostaining. A, B, E2f1+/+. C, D, E2f1+/−. E, F, E2f1−/−. A, C, E, P30 sagittal cerebellar sections were labeled with the Purkinje cell marker, calbindin (red), and the cell cycle marker, PCNA (green). No PCNA staining is found in wild-type animals (A). However, PCNA is expressed in the nuclei of E2f1+/− (C) and E2f1−/− (E) Purkinje cells. B, D, and F are double immunostained with PCNA (green) and cyclin A (red). Both PCNA and cyclin A staining are found in the Purkinje of E2f1 heterozygotes (D) and mutant (F) mouse brains. No cyclin A or PCNA was expressed in wild-type animal (B). Scale bar, 25 μm.
The unusual pattern of cell cycle protein expression led us to examine other regions of the E2f1−/− mouse brain. No cyclin A- and PCNA-positive cells were found in the wild-type cortex (Fig. 4A–C); however, we found PCNA and cyclin A expressed in the nerve cells of several neuronal populations in E2f1−/− animals and in heterozygotes. Perhaps the most prominent of these sites was the neocortex (Fig. 4D–I). We validated the morphological impression that the “cycling” cells are neurons by double staining sections for both cyclin A and the neuron-specific antigen, NeuN (Fig. 4J–L). The subcellular localization of the cell cycle proteins was similar to that seen in Purkinje cells: PCNA in the nucleus as expected and cyclin A ectopically expressed in the perikaryon and cell process. We also found variation among the six layers of neocortex. There were more cell cycle-positive neurons in layers II and III than in layers V and VI. We found no cell cycle protein reexpression in the neurons of hippocampus (data not shown). Western blot analysis of PCNA protein levels also revealed an increase in PCNA protein levels in both E2f1−/− animals and in heterozygotes (Fig. 4M). Although the cells shown in Figures 3 and 4 are from brains of 1-month-old animals, the unusual protein expression patterns do not appear to be transient. We saw similar quantitative and qualitative PCNA and cyclin A staining patterns in adult mice (see below).

Reappearance of cell cycle proteins in E2F1-deficient neocortex. Sagittal sections of P30 mouse brain from three different E2f1 genotypes (E2f1+/+; E2f1+/−; E2f1−/−) were double labeled with PCNA (green) and cyclin A (red) antibodies. As in cerebellum, PCNA and cyclin A were reexpressed in both the heterozygous E2f1+/− (D–F) and homozygous E2f1−/− (G–I) brain. There was no cell cycle protein detected in wild-type animal (A–C). The subcellular localization of these two cell cycle proteins was the same as seen in cerebellum, PCNA in the nucleus and cyclin A in cytoplasm. The stained cells are neurons as revealed by double labeling with the neuronal maker, NeuN, and the cell cycle marker, cyclin A (J–L). M is a Western blot revealing the increase of PCNA levels in both E2f1+/− and E2f1−/− mouse cortex. Scale bar, 25 μm.
Neuronal DNA replication in the E2f1−/− brain
Cell cycle-related protein reexpression alone is not sufficient to rigorously distinguish DNA replication from DNA repair (Kuan et al, 2004; Herrup and Yang, 2007). Therefore, we used fluorescence in situ hybridization (FISH) to probe the ploidy of the neuronal nuclei. We used three different mouse BAC probes to detect unique genomic sequences on two different mouse chromosomes, 16 and 11. The results with probe 480C6, which recognizes sequences on chromosome 16, are shown in Figure 5. We found many Purkinje cells and neocortical neurons with three or four hybridization signals (three to four spots) in both mutant and heterozygous mice (Fig. 5B,C,E,F). In no case did we find an example of a neuron in which there were more than four spots of hybridization. In wild-type animals, virtually all neurons in comparable regions had two or fewer hybridization signals (Fig. 5A,D). This indicates that there has been true DNA replication in E2f1+/− and E2f1−/− neurons. The distribution of the “hyperploid” neurons matched our immunohistochemistry findings well. For example, there were substantially more neurons with three or four hybridization signals in layers II and III than in layers V and VI of neocortex.

DNA replication occurs in neurons of the E2f1+/− and E2f1−/− brain, as revealed by FISH. A, D, E2f1+/+. B, E, E2f1+/−. C, F, E2f1−/−. Evidence for DNA replication was readily observed in both Purkinje cells (A–C) and cells of neocortex (D–F). In wild-type animals (A, D), there were two or fewer hybridization signals, whereas in either heterozygous (B, E) or homozygous (C, F) mutant mice, there were many cells with three or four hybridization signals (insets in B, C, E, F: high magnification of the nuclei indicated with the arrows). Confocal microscopy reveals that the spots of hybridization are clearly within the nuclei. Wild-type cells are illustrated in the left two images of the pairs (Ga, Gc), whereas E2f1 mutant cells are shown in the right two images (Gb, Gd). ML, Molecular layer; PC, Purkinje cell layer; IGL, internal granule cell layer. Scale bar, 10 μm.
Cell counts (for details, see Materials and Methods) further validate these impressions. In wild-type animals, the number of cells with three or four spots in layers II and III was ∼5% of the total nuclei. In the same location in the E2f1−/− brains, >20% of the nuclei had three or four spots of hybridization. We were unable to achieve reliable immunocytochemistry in conjunction with our FISH protocol, precluding the positive identification of the cell type in which the hyperploid signals were found. Our qualitative impression is that many, if not most, of the three- and four-spot cells in the wild-type animals were smaller and likely to be non-neuronal. Some of the E2f1−/− hyperploid cells were also of this class, but many more were found in large nuclei that are characteristic of neurons. In the cerebellum, the position and large size of the Purkinje cell nuclei made positive identification of the cell type more reliable than in neocortex. On occasion, in both wild-type and E2F1-deficient mice, we found cells with more than two hybridization signals. As in cerebral cortex, the nuclei of these cells tended to be quite small, however, and their location was outside the Purkinje cell layer. We are confident that these are not Purkinje cell nuclei. FISH data obtained with another pair of BAC probes (170L21 and 566) recognizing sequences on mouse chromosome 11 gave similar results (data not shown). In the aggregate, our data demonstrate that nerve cells in the E2f1−/− brain have undergone DNA replication.
Age of onset of the cell cycle events in the E2F1−/− brain
The high prevalence of the CCEs in the 1 month E2f1−/− brain raises the possibility that these nerve cells had never left the cell cycle and thus represent a developmental problem rather than a problem of cell cycle regulation in the postnatal CNS neuron. To address this question, we examined E2f1+/+, E2f1+/−, and E2f1−/− mice at different ages from birth to adulthood. We used double-labeled immunohistochemistry to detect PCNA and cyclin A in those mice brains. As late as P10 we found no evidence of ectopic neuronal CCE in either cerebellum or neocortex. This was the case for all three genotypes (Fig. 6). In these younger animals, the robust mitotic activity in the cells of the external granule cell layer of the cerebellum serves as a positive control. In this mitotically active layer, we found strong PCNA and cyclin A staining. There were also some positive cells in the internal granule cell layer at P10 (Fig. 6A–C), but these likely reflect the normal CCEs of granule cells during target related cell death (Herrup and Busser, 1995). By P20, however, there was a dramatic increase of PCNA- and cyclin A-positive Purkinje cells in both the E2f1−/− mutant and the heterozygote (Fig. 6D–F). The number of positive cells was comparable with that found in P30 brain (Fig. 3D,F). The subcellular localization of PCNA and cyclin A staining was the same as that found in the P30 mice: PCNA in the nucleus and cyclin A in the cytoplasm.
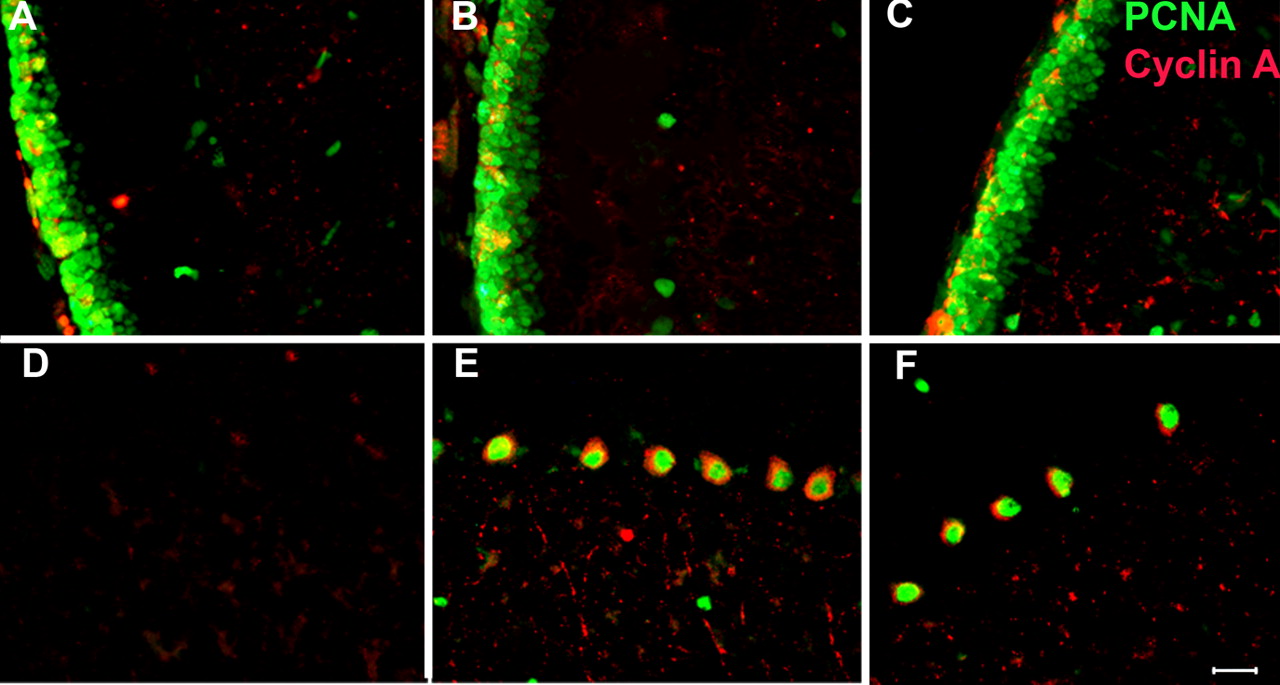
Ectopic cell cycle events in the developing cerebellum of E2F1-deficient mice. Brains of P10 (A–C) and P20 (D–F) animals of three different genotypes were stained with PCNA (green) and cyclin A (red) antibodies. A, D, Wild-type brain. B, E, E2f1+/−. C, F, E2f1−/−. No cell cycle events are apparent in the young animals (A–C), although the mitotic cells in the external granule cell layer serve as a positive control. By P20, PCNA and cyclin A stained many Purkinje cells in both E2f1+/− and E2f1−/− mice but not in wild-type controls. Scale bar, 25 μm.
In neocortex, the picture was similar to that found in the cerebellum. At P10, we could see some PCNA- and/or cyclin A-positive cells in all three genotypes, but these did not appear to be neuronal cells (as assessed by their nuclear morphology). Furthermore, in these cells, cyclin A was nuclear in subcellular localization as it should be in normal cycling cells (Fig. 7A–C). By P20, both mutant and heterozygous mice had abundant numbers of PCNA and cyclin A double-positive neuronal cells. In these cells, the localization of the protein was the familiar pattern of PCNA in the nucleus and cyclin A in the cytoplasm including some processes (Fig. 7E,F). The involvement with cell cycle processes was significant; 35% of the cells in the E2F1 null neocortex were cell cycle positive, which was much higher than the percentage found in wild type. We found no PCNA or cyclin A immunostaining in wild-type neuronal cells (Fig. 7D).

Ectopic cell cycle events in the developing neocortex of E2F1-deficient mice. P10 (A–C) and P20 (D–F) neocortical cells were stained with antibodies to cyclin A (red) and PCNA (green). A, D, Wild-type brain. B, E, E2f1+/−. C, F, E2f1−/−. PCNA and cyclin A were not expressed in any neuronal cells at P10, whereas by P20 PCNA and cyclin A were positive in both E2f1+/− and E2f1−/− brain. Already at P20, the subcellular localization of the cell cycle proteins was same as P30; PCNA was nuclear, and cyclin A was cytoplasmic. Scale bar, 25 μm.
As discussed above, the appearance of these cell cycle proteins is not a time-limited developmental event. PCNA and cyclin A are found in neurons of both the cerebellum and cortex of E2f1−/− animals as old as 1 year (Fig. 8D,F). As at 1 month, no cell cycle-positive neurons were detected in the control brain. The strong suggestion of these images is that the cycling cells of the E2f1−/− brain maintain abnormally high levels cell cycle proteins throughout their life. The normal size of the adult brain and the apparently normal neuronal density suggests that these cells neither progress to finish cell division, thus increasing neuronal cell number, nor die. To determine whether the cycling cells suffered consequences from the ectopic protein expression, we examined the dendrites of the E2f1−/− Purkinje cells closely using calbindin. At low magnification, the calbindin staining of the E2f1−/− Purkinje cells (Fig. 8F) looks less intense than that in wild type (Fig. 8E). At high magnification in the confocal microscope, there is a clear loss of dendritic mass that appears to be accompanied by a decrease in the density of the dendritic spines found on the tertiary branches (Fig. 8G–J). Staining with Map2 or synapsin protein, however, revealed few if any differences between mutant and wild type. This suggests that the changes in calbindin staining may reflect only the deployment of the calcium binding protein itself, rather than a structural atrophy of the Purkinje cell dendrite. Thus, despite the apparent constitutive presence of cell cycle proteins and their hyperploid genomic content, the Purkinje cells of the E2f1−/− cerebellum appear to predominantly retain their morphological integrity (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).

Cell cycle protein reexpression in 1-year-old E2f1−/− brain. Wild-type (A, C, E, G, I) and E2f1−/− mutant (B, D, F, H, J) brains were immunostained to examine cell cycle activity and Purkinje cell dendritic structure. PCNA (green) and cyclin A (red) antibodies revealed that the mutant Purkinje cells remained robustly stained for both cell cycle proteins (B, D), whereas both were absent in comparable regions of wild-type controls. A, B, Cortex. C, D, Cerebellum. Calbindin staining (E–J) reveals the fine details of the Purkinje cells. Even at 1 year of age, wild-type Purkinje cells (E, G) retain richly branched and densely spined dendritic arbors, whereas the arbors of the E2f1−/− dendrites (F, H, J) appeared less dense. G–J, Confocal micrographs of calbindin-stained molecular layer showing the details of the Purkinje cell dendritic arbor. Scale bars: A–F, 25 μm; G–J, 10 μm.
Cell cycle events in cultured E2f1−/− primary neurons
Studies of the ectopic appearance of neuronal cell cycles have shown that they can be triggered by extrinsic events such as the failure of trophic support (Herrup and Busser, 1995; Park et al., 1997) or by intrinsic problems such as a compromised cell cycle checkpoint system (Yang and Herrup, 2005) or both (Lipinski et al., 2001; MacPherson et al., 2003). Because the activation of cell cycle events in an E2F1-deficient brain is unexpected, we sought to determine whether this was an intrinsic, cell-autonomous response of the mutant neurons or, instead, a response to an extrinsic failure elsewhere in the animal. Cultures of dissociated embryonic day 16.5 (E16.5) neurons were plated in minimal medium (Neurobasal) at relatively low density on poly-l-lysine-coated coverslips. Identical cultures were prepared from wild-type and homozygous E2f1−/− embryos. Replicate cultures were analyzed every 5 d. For each time point, BrdU (10 μm) was added to the medium 5 d before fixation to assess DNA synthesis. Cultures were stained by immunocytochemistry for either PCNA expression or BrdU incorporation as well as either TuJ1 or Map2 as neuronal cell type markers.
In vivo (Fig. 7) cell cycle events appeared in cortical neurons between P10 and P20. For neurons harvested at E16.5, these dates are the equivalent of 13 and 23 DIV. In cultured wild-type neurons, at 5 DIV, there was no neuronal PCNA expression apparent in any culture and only a small amount of BrdU incorporation, which we believe is a reflection of residual mitotic activity from the newly harvested ventricular zone cells (Fig. 9A). After this brief period of BrdU incorporation, the neurons in the wild-type cultures remained mitotically inactive for the entire culture period (Fig. 9C,E). In the E2f1−/− cultures, however, significant amounts of PCNA synthesis and BrdU incorporation are detected at 5, 10, and 15 DIV (Fig. 9B,D,F,G). These are the temporal equivalents of days P2, P7, and P12 in vivo. Thus, in dissociated cultures, the appearance of cell cycle events is triggered by a factor intrinsic to the mutant neurons themselves. The timing of cell cycle initiation, however, is set by an extrinsic cue. This cue (or the loss of an inhibitory influence) must appear on an accelerated timescale in vitro.

Ectopic cell cycle events in cultured dissociated primary neurons from E16.5 mouse neocortex. Neurons were cultured for 5 (A, B), 10 (C, D), and 15 (E, F) DIV and fixed. BrdU was added to the culture medium for 5 d before fixation. Fixed cells were double-labeled with BrdU (green) and TuJ1 (red) antibodies. A, C, E, Wild type. B, D, F, E2f1−/−. In wild-type cultures, few neuronal (TuJ1+) cells were positive for BrdU staining at 5 DIV, and none was present at 10 or 15 DIV. In the E2f1−/− cultures, in contrast, there were substantial numbers of such cells at all three time points, with an apparent peak in BrdU incorporation between 5 and 10 DIV. Shown in G is the percentage of BrdU-positive neurons (Tuj1-positive cells) at 5, 10, and 15 DIV. Scale bar, 100 μm.
Discussion
Mature neurons represent a population of differentiated cells that are believed to have exited from the cell cycle forever once they have left the ventricular or subventricular zone and begun differentiation. Yet ectopic cell cycle events in mature neurons do occur and are related to neuronal degeneration in human neurodegeneration diseases such as Alzheimer's (Vincent et al., 1996; McShea et al., 1997; Nagy et al., 1997; Busser et al., 1998), Parkinson's (Jordan-Sciutto et al., 2003; Hoglinger et al., 2007), amyotrophic lateral sclerosis (Ranganathan et al., 2001; Ranganathan and Bowser, 2003), and others (Osuga et al., 2000; Jordan-Sciutto et al., 2002). Abortive neuronal CCEs have also been found in mouse neurodegenerative models. These include the retinoblastoma knock-out mouse (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992), T-antigen transgenic mice (al-Ubaidi et al., 1992; Feddersen et al., 1992), as well as in granule cells of staggerer and lurcher mice (Herrup and Busser, 1995). In other cases, the mouse neurons in question engage in cell cycle reentry, but no neuronal cell loss is reported (Yang and Herrup, 2005; Yang et al., 2006). In the aggregate, the data indicate that neurons need to constantly hold their cell cycle in check to sustain their normal function and possibly their viability.
In a typical cycling cell, E2F1 helps to control the G1/S transition. When it is deregulated, by overexpression or by release from its binding to RB, it can initiate cell cycle reentry both in vivo and in vitro. Elimination of E2f1 can reduce ectopic S-phase entry and apoptosis in Rb-null neurons in the embryonic CNS (Tsai et al., 1998). Our results suggest that E2F1 also plays a role in the regulation of the cell cycle in the postnatal CNS neurons as well, but the direction of its influence, cell cycle suppression, appears reversed from that found in other systems.
Cultured E2f1−/− granule cells have been reported to be slower to die when exposed to low K+ (O'Hare et al., 2000), toxic concentrations of the Aβ peptide (Giovanni et al., 2000), or kainite (Smith et al., 2003). Furthermore, E2F1-null mice are resistant to neurotoxin-induced dopaminergic cell death than their wild-type littermates (Hoglinger et al., 2007). These findings of a protective influence of the E2F1-deficient state are in contrast to our findings in staggerer and lurcher mice. Here, target-related granule cell death is preceded by a well characterized ectopic S-phase entry, yet deletion of E2F1 in either mutant has no effect on either granule cell cycle reentry or cell death. The different conclusions arrived at in these reports merit consideration. One obvious difference is the nature of the assays. All other tests examined granule cells in vitro, whereas we analyzed the same cells in vivo. Also, the triggers for granule cell death in the various reports are different. In staggerer and lurcher mice, granule cells die because of a loss of trophic support from Purkinje cells. The other studies used specific toxins (often at high concentrations) and this might well engage a different cell death pathway. As it has been shown that the function of E2F1 is context dependent, this difference could be crucial. In vitro, for example, E2F1 has been shown to either increase or decrease oncogenic transformation, depending on the conditions of the assay (Melillo et al., 1994; Johnson, 2000; Lee and Farnham, 2000).
Our finding of differentiated neurons with cell cycle-related protein expression and significant DNA replication in vivo is quite consistent with the original reports of the E2f1−/− knock-out mice (Field et al., 1996; Yamasaki et al., 1996). Both groups highlight the different roles played by E2F1 in different tissues. In exocrine tissues such as pancreas and salivary gland, it appears to act as a tumor suppressor, whereas in other tissues it functions more as a cell cycle promoter. And the mice die with significant numbers of tumors in their bodies
The finding of ectopic cell cycle events in the neurons of the early postnatal E2f1−/− brain is reminiscent of the situation in the atm−/− mouse. In this mouse model of ataxia-telangiectasia, only ∼12% of the ATM-deficient Purkinje cells and a few striatal neurons engage in cell cycle activity (Yang and Herrup, 2005), whereas in the E2f1−/− mouse, nearly one-half of the Purkinje cells show cell cycle protein reexpression. Despite these quantitative differences, there are similarities in the cell cycle phenotypes of the E2f1 and atm mutants that deserve notice. There is a striking similarity in the time window during which cell cycle reentry occurs. Both begin between P10 and P20 and quickly rise to a percentage involvement that remains relatively constant throughout the life of the animal. An additional similarity is that, with both the atm and E2f1 mutations, ectopic cell cycle events are found in both heterozygous and homozygous neurons. This implies that the tumor suppressor role of both genes functions as a recessive trait, possibly indicating a threshold of activity that is needed to fully suppress the neuronal cell cycle. The similarities between the neuronal phenotypes of the two mutations may have even deeper roots because studies have shown that E2F1, like ATM, is involved in DNA repair and cell cycle checkpoint functions. The level of E2F1 is increased when cells are treated with DNA damaging agents, and recent studies have shown that the stability of E2F1 is highly sensitive to phosphorylation by either ATM or checkpoint kinase 2 (Chk2) (Lin et al., 2001; Stevens and La Thangue, 2003). This suggests a model in which ATM acts in part to regulate E2F1 stability in normal neurons. When the atm gene is deleted, the levels of E2F1 are reduced and in cells in which the effective levels drop sufficiently (by about one-half, as suggested by the heterozygote data) the nerve cells reenter the cell cycle. Deletion of E2f1 itself simply expands the extent of the ectopic cell cycle events. It would be predicted the levels of E2F1 would be reduced in cell cycle-positive atm−/− neurons.
FISH staining in cerebral cortex of the E2f1−/− mice showed that ∼20% of the cells were hyperploid. This confirms a true cell cycle was at least begun in those postmitotic neurons. We note, however, that ∼5% of cells in the wild-type layers II and III had three or four hybridization signals. This is not consistent with previous work on AD mouse models (Yang et al., 2006) or the results from human pathological material (Yang et al., 2001). In these previous studies, wild-type neurons displayed no multiple hybridization signals. There are several differences of note between the mouse models studied. First, the genetic background is different. Our E2f1 mice are maintained on a mixed (C57BL/6J × 129/S4) background. The previous study used C57BL/6J as control. The age of the animals investigated was different as well [prepuberal (P30) in the current study, adult or aged mice (6–12 months) in the previous work]. The findings are in agreement, however, with those of Rehens et al. (2001), who found a comparable level of hyperploidy in their analysis. Others have reported even higher levels (Yurov et al., 2005; Mosch et al., 2007)
We have shown that the ectopic expression of cell cycle proteins persists in the E2F1-deficient neurons for a very long period, up to 1 year. It would seem that, although they may be stressed initially, neurons can tolerate this cycling status for a long period. Furthermore, despite the fact that the levels of calbindin expression appear to retreat somewhat with age in the E2f1−/− Purkinje cells, other markers such as Map2 and synapsin suggest little structural atrophy in the older animals. Others have noted a mild but significant cerebellar atrophy in E2F1-deficient mice (Cooper-Kuhn et al., 2002), but in this case it is unclear whether the degenerative process is induced directly by aberrant cell cycle events or other factors.
Cell cycle reentry in cultured E2f1−/− primary neurons indicates that the ectopic cell cycles are a cell-autonomous response to the deficiency of E2F1. However, the earlier initiation of the CCEs in vitro argues that E2F1 collaborates with some extrinsic factor(s) that normally holds the cell cycle in check for almost a week longer in vivo than in vitro. We conclude from this that an ectopic cell cycle event is regulated by both intrinsic and extrinsic mechanisms, but the extrinsic factors cannot compensate for intrinsic failure in the end.
In summary, our findings indicate that E2F1 functions as a cell cycle suppressor in CNS neurons. These findings help explain why, despite the predictions of the previous tissue culture experiments, the absence of E2F1 does not protect granule cell neurons from target-related cell death in staggerer and lurcher mice. Tissue culture results suggest that, although the timing of the cell cycle initiation might be advanced slightly, the failure to suppress the cycle is intrinsic to the mutant neurons. Cell cycle reentry in E2f1−/− neurons is begun in the second and third postnatal weeks in vivo but then stalls, and several proteins, typically found only in actively cycling cells, continue to be expressed for many months with few harmful affects apparent in neuronal structure or function.
Footnotes
-
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (R01-NS20591), the National Institute on Aging (P50-AG08012; R01-AG24494), and the Blanchette Hooker Rockefeller Fund. We thank Xiaoying Tang for technical help.
- Correspondence should be addressed to Karl Herrup, Department of Cell Biology and Neuroscience, Rutgers, The State University of New Jersey, 604 Allison Road, Piscataway, NJ 08854. herrup{at}biology.rutgers.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}