

Abstract
Emerging evidence implicates serotonergic descending facilitatory pathways from the brainstem to the spinal cord in the maintenance of pathologic pain. Upregulation of the serotonin receptor 2A (5-HT2AR) in dorsal horn neurons promotes spinal hyperexcitation and impairs spinal μ-opioid mechanisms during neuropathic pain. We investigated the involvement of spinal glutamate receptors, including metabotropic receptors (mGluRs) and NMDA, in 5-HT2AR-induced hyperexcitability after spinal nerve ligation (SNL) in rat. High-affinity 5-HT2AR agonist (4-bromo-3,6-dimethoxybenzocyclobuten-1-yl)methylamine hydrobromide (TCB-2) enhanced C-fiber-evoked dorsal horn potentials after SNL, which was prevented by mGluR1 antagonist AIDA [(RS)-1-aminoindan-1,5-dicarboxylic acid] but not by group II mGluR antagonist LY 341495 [(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl)propanoic acid] or NMDA antagonist d-AP5 [d-(−)-2-amino-5-phosphonopentanoic acid]. 5-HT2AR and mGluR1 were found to be coexpressed in postsynaptic densities in dorsal horn neurons. In the absence of SNL, pharmacological stimulation of 5-HT2AR with TCB-2 both induced rapid bilateral upregulation of mGluR1 expression in cytoplasmic and synaptic fractions of spinal cord homogenates, which was attenuated by PKC inhibitor chelerythrine, and enhanced evoked potentials during costimulation of mGluR1 with 3,5-DHPG [(RS)-3,5-dihydroxyphenylglycine]. SNL was followed by bilateral upregulation of mGluR1 in 5-HT2AR-containing postsynaptic densities. Upregulation of mGluR1 in synaptic compartments was partially prevented by chronic administration of selective 5-HT2AR antagonist M100907 [(R)-(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenyl)ethyl]-4-pipidinemethanol], confirming 5-HT2AR-mediated control of mGluR1 upregulation triggered by SNL. Changes in thermal and mechanical pain thresholds following SNL were increasingly reversed over the days after injury by chronic 5-HT2AR blockade. These results emphasize a role for 5-HT2AR in hyperexcitation and pain after nerve injury and support mGluR1 upregulation as a novel feedforward activation mechanism contributing to 5-HT2AR-mediated facilitation.
Introduction
Activity-dependent plasticity of glutamatergic neurotransmission to dorsal horn neurons initiated by burst of primary afferent activity following peripheral injury or inflammation is widely accepted as a major mechanism of central sensitization and the shift from acute to chronic pain (Willis, 2001, 2002; Moore et al., 2002; Latremoliere and Woolf, 2009). In addition, aminergic pathways descending to the dorsal horn from supraspinal sites can exert control over centripetal progression of nociceptive impulses through the dorsal horn (Millan, 2002), and mounting evidence now supports the importance of facilitatory influences exerted via descending projections arising largely from the rostral ventromedial medulla in the maintenance of pathologic pain in animal models (Dubner, 2004; Bannister et al., 2009; Heinricher et al., 2009; Ossipov et al., 2010).
Neurotransmitter serotonin [5-hydroxytryptamine (5-HT)] is instrumental in descending regulation of pain (Oliveras et al., 1979; Millan, 2002). Of the seven receptor families (5-HT1 through 5-HT7) so far identified (Hoyer et al., 1994; Barnes and Sharp, 1999), all but 5-HT3 are G-protein-coupled receptors modulating ligand- and voltage-gated channels (Barnes and Sharp, 1999). The involvement of 5-HT receptors in pain neurotransmission and modulation is still ill understood. Our previous work has shown that subtypes 2A (5-HT2AR) and 2B (5-HT2BR) become upregulated and tonically active in the dorsal horn in the spinal nerve ligation (SNL) model of neuropathic pain, and both promote spinal hyperexcitation and impair spinal μ-opioid mechanisms (Aira et al., 2010, 2012), suggesting a major mediator role for these receptors in descending facilitation.
Early evidence implicates 5-HT in the modulation of glutamate-evoked neuronal excitation (McCall and Aghajanian, 1979; White and Neuman, 1983), and cross talk mechanisms between glutamate- and 5-HT receptors have recently been reported. For example, serotonin signaling can regulate both AMPA receptor function in pyramidal neurons of the prefrontal cortex (PFC) (Cai et al., 2002) and expression of GluR1 receptor in the amygdala (Tran and Keele, 2011). Reciprocally, serotonin facilitation of long-term depression (LTD) in PFC neurons depends on concurrent activation of group I mGluRs (mGluR1/5) (Zhong et al., 2008). In PFC neurons, serotonin and glutamate receptors may cooperatively induce LTD through converging activation of postsynaptic p38 MAPK (Zhong et al., 2008). The spinal dorsal horn is highly enriched in these two receptor classes, and both are prominent in spinal nociception. This led us to hypothesize that, in inducing spinal hyperexcitability after SNL, one important target of the spinal 5-HT2AR and 5-HT2BR could be glutamate receptors expressed by dorsal horn neurons, and that dysregulation of glutamatergic transmission and plasticity by the altered serotonin system may contribute to the development of pain disorders after nerve injury. Here, we report that activation of 5-HT2AR rapidly upregulates mGluR1 expression via PKC in dorsal horn neurons after SNL, a plastic change that in turn enables 5-HT2AR to promote hyperexcitation to noxious input and pain. Given the critical role of glutamatergic transmission in conveying noxious afferent input to dorsal horn neurons and pain pathways, these results provide a molecular and cellular mechanism for 5-HT2A/PKC regulation of spinal nociception and pain.
Materials and Methods
Animal experiments were performed according to the European Communities Council Directive (86/609/ECC) on adult male Sprague Dawley rats (250–350 g; OF1; Iffa Credo). The protocols for animal care and use were approved by the appropriate committee at the University of the Basque Country. Efforts were made to keep the number of animals used to a minimum as well as to minimize animal suffering.
SNL surgery.
The left spinal L5 nerve root was ligated under 4% chloral hydrate anesthesia (0.4 g/kg, i.p.), as described by Kim and Chung (1992). Sham-operated animals received no nerve ligation, being otherwise identically prepared and assessed. After surgery, the animals were allocated to individual cages to recover, with food and water available ad libitum. Successful nerve ligation was confirmed 9 d later by evaluation of mechanical paw withdrawal thresholds (see below, Behavioral evaluation).
Electrophysiology.
Procedures were performed under urethane anesthesia (1.5 g/kg, i.p.). A tracheotomy was performed to maintain an open, low-resistance airway, and cannulae were inserted into the left common carotid artery and the right internal jugular vein for arterial blood pressure monitoring (mean 80–100 mmHg) and continuous infusion of Tyrode's solution (in mm: 137 NaCl, 2.7 KCl, 1.4 CaCl2, 1 MgCl2, 6 NaHCO3, 2.1 NaH2PO4, 6.5 d-(+)-glucose; pH 7.4) at 0.8–1 ml/h, respectively. Colorectal temperature was continuously monitored and euthermia (37–38°C) was maintained via a feedback-controlled under-body heating pad for the duration of the experimental procedure. The left sciatic nerve was exposed, gently freed from connective tissue, and placed onto platinum hook electrodes for bipolar electrical stimulation. Bilateral dorsal laminectomies were performed at vertebrae T13–L1, the vertebral column was immobilized to a rigid frame, and the dura mater overlaying lumbosacral spinal segments was carefully removed.
The electrophysiological setup was essentially as described previously (Azkue et al., 2003). Tungsten microelectrodes (impedance, 5 MΩ) were inserted 1 mm lateral to the spinal midline at a depth of 100–300 μm from the dorsal surface of the cord (i.e., into laminae I–II) by means of a hydraulic microdrive. The position of the tip of the recording electrode in the spinal cord was marked with a small electrolytic lesion by delivery of an anodal current through the recording electrode (50 μA anodal current for 10 s) and histologically verified. Single monophasic, square-wave electrical pulses were delivered as test stimuli to the sciatic nerve trunk at a midthigh level on a per-minute basis by means of a computer-controlled stimulus isolator, and the elicited spinal field potentials were amplified (analog bandpass set at 1–550 Hz), displayed on an oscilloscope, and digitized to a PC-based computer at a 10 kHz sampling rate via an A/D converter card (MIO16; National Instruments). Electrical stimulation of the sciatic nerve trunk did not permit us to discern the relative contribution of cutaneous, muscle, and joint fibers to the recorded signals. Care was taken to avoid stimulus repetition. Field potentials were evoked in superficial laminae of the spinal dorsal horn by suprathreshold, electrical C-fiber stimulation (3–3.5 mA pulses of 0.5 ms duration) and quantified as described previously (Buesa et al., 2006).
To confirm that the potentials considered here to be mediated by C-fibers were actually attributable to this afferent input, we examined the effect of shifting the position of the stimulation electrodes along the proximal–distal axis of the sciatic nerve on the latencies of potentials evoked in the dorsal horn. C-fiber-evoked field potentials elicited by positioning the stimulation electrodes (10 mm apart) at the most proximal location possible (i.e., ∼100 mm away from the dorsal root entry zone) exhibited an average peak latency of 104.49 ms (SD, 0.80; average conduction velocity, 0.95 m/s). Displacement of the stimulation electrode pair 10 or 20 mm distally increased the average peak latency of the evoked potentials linearly to 114.97 ms (SD, 0.55) or 125 ms (SD, 0.88), respectively (Fig. 1), with Spearman's rank correlation coefficient of 0.996 (p < 0.001), providing strong support for C-fibers as the actual mediators of evoked field potentials measured here.

Effect of proximal–distal displacement of stimulation electrodes over the sciatic trunk on the peak latencies of C-fiber-evoked field potentials. Shifting of the stimulation electrode pair 10 or 20 mm distally from the reference position located at the most proximal location possible (distant ∼100 mm from the dorsal root entry zone) results in a linear increase in the average peak latencies, confirming that the field potentials considered here to be mediated by C-fibers are actually attributable to this afferent input. A typical location of recording electrodes is indicated by a solid circle in the schematic on the right. Error bars indicate SEM.
To measure the ability of 5-HT2AR to enhance C-fiber-evoked spinal field potentials, these were recorded during spinal superfusion with successively increasing, cumulative concentrations of selective agonist (4-bromo-3,6-dimethoxybenzocyclobuten-1-yl)methylamine hydrobromide (TCB-2). TCB-2-induced hyperexcitation is 5-HT2A receptor specific, as shown previously by blockade with the selective 5-HT2AR antagonist 4-(4-fluorobenzoyl)-1-(4-phenylbutyl)piperidine (4F 4PP) (Aira et al., 2010). Each drug concentration change lasted for 20 min, and only the last 10 evoked field potentials were extracted for analysis from the baseline control period and from each treatment period. The areas of field potentials evoked during each treatment period were compared with those recorded during a control, aCSF superfusion period, by using univariant ANOVA and post hoc Bonferroni's or Tamhane's multiple-comparison tests. To evaluate the influence of mGluR1, mGluR2/3, or NMDA receptor blockade on field potential enhancement by TCB-2, the agonist was administered in combination with subclinical concentrations of selective receptor-specific antagonists (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA) (10 μm), (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl)propanoic acid (LY 341495) (10 μm), or d-(−)-2-amino-5-phosphonopentanoic acid (d-AP5) (10 μm). Antagonist concentrations were selected on the basis of preliminary experiments (see Fig. 4).
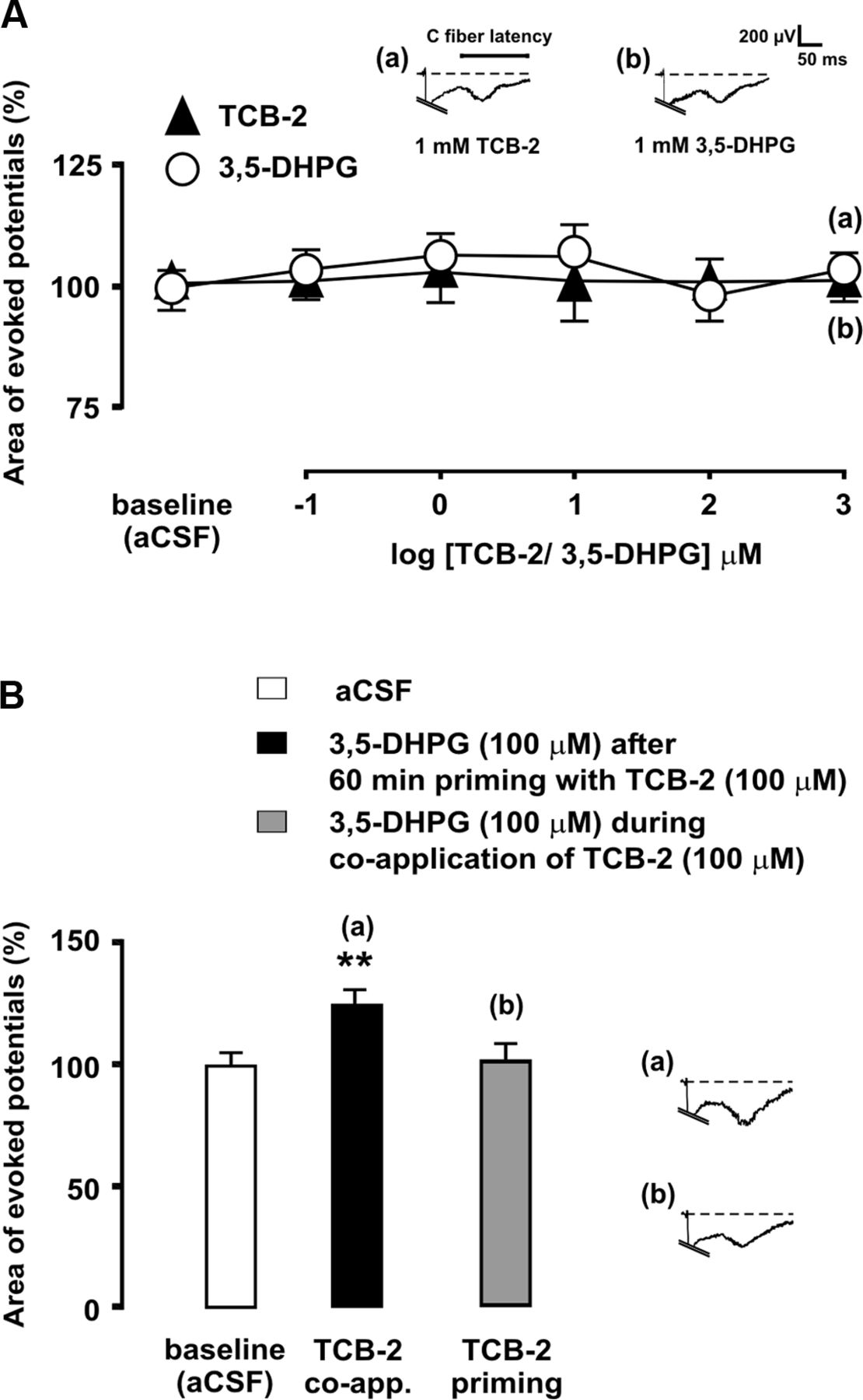
In a separate set of experiments, we tested the ability of increasing, cumulative concentrations of the 5-HT2AR agonist TCB-2 to enhance C-fiber-evoked potentials in sham-operated rats, both alone and in the presence of simultaneously administered mGluR1 agonist (RS)-3,5-dihydroxyphenylglycine (3,5-DHPG) at a concentration (100 μm) that would not itself affect evoked potentials if administered alone.
Drug preparation and delivery.
Drugs used for in vivo electrophysiological recordings included TCB-2 (5-HT2A agonist) (McLean et al., 2006), AIDA (mGluR1 antagonist) (Moroni et al., 1997), 3,5-DHPG (group I mGluR agonist) (Ito et al., 1992), LY 341495 (mGluR2/3 antagonist) (Sakagami et al., 2008), and d-AP5 (NMDA antagonist) (Davies et al., 1981), all five from Tocris, as well as PKC inhibitor chelerythrine (Sigma-Aldrich). Stock solutions were obtained by diluting drug powder in double-distilled water, and working solutions were prepared in artificial CSF (aCSF) (in mm: 130 NaCl, 3.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 1.2 CaCl2, 1.2 MgSO4, 10 d-(±) glucose; pH 7.4) immediately before delivery. Drugs were applied in small volumes (10–15 μl) by controlled superfusion via a silicon, 40–50 mm2 pool attached to the dorsal surface of the spinal cord (Beck et al., 1995).
In experiments of chronic blockade of 5-HT2AR, antagonist (R)-(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenyl)ethyl]-4-pipidinemethanol (M100907) (Sigma-Aldrich) (Sorensen et al., 1993) was administered by daily intraperitoneal injections at 0.4 mg/kg (McMahon and Cunningham, 2001; Minabe et al., 2001; Boothman et al., 2006). We confirmed that intraperitoneally delivered M100907 could depress C-fiber-evoked spinal field potentials as above (Fig. 2), which served as confirmation of a spinal locus of action of M100907 also when using a systemic route of administration.

Intraperitoneally delivered 5-HT2AR antagonist M100907 depresses C-fiber-evoked dorsal horn potentials. The effect of acute intraperitoneal administration of M100907 (0.4 mg/kg) on evoked potentials is shown 9 d after SNL. C-fiber-evoked potentials were decreased significantly 40 min after M100907 delivery, and depression increased with time at least until recordings were discontinued 100 min after administration. The asterisks indicate statistical significance (p < 0.01; n = 5) at Bonferroni test's when comparing mean potential areas to before drug treatment. Error bars indicate SEM.
Subcellular fractionation.
Biochemical fractionation of dorsal horn proteins was performed with minor variations according to previous studies (Yang et al., 2009; Cao et al., 2011). Briefly, rats were deeply anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and killed by decapitation. L4–L5 segments were quickly extracted into ice-cold aCSF. Tissue was separated and homogenized mechanically with a motor-driven glass/glass tissue homogenizer in ice-cold lysis buffer (10 mm Tris, pH 7.6, 320 mm sucrose, 5 mm EDTA) containing protease inhibitors (5 mm EGTA, 1 mm PMSF, 10 U/ml aprotinin, 0.0001% chymostatin, 0.0001% leupeptin, and 0.0001% pepstatin). Dorsal horn samples ipsilateral and contralateral to surgery were taken and processed separately. Homogenates were centrifuged at 1000 × g for 10 min to remove cell nuclei (P1) from the low supernatant (S1). S1 was collected and centrifuged at 10,000 × g during 15 min to separate a P2 pellet containing the crude synaptosomal fraction and a cytoplasmic fraction S2 with microsomes. The P2 pellet was incubated in the lysis buffer containing 0.5% Triton and centrifuged at 32,000 × g for 20 min to obtain the crude synaptic vesicle fraction (S3) and the final pellet containing the synaptic fraction (P3). The latter was solubilized in resuspension buffer (10 mm Tris, pH 8.0, 1 mm EDTA, 2% SDS). All fractions were stored at −80°C. We confirmed that only P3 fraction was enriched with the postsynaptic density protein PSD-95 (see Fig. 7).
Western blot.
BCA protein assay kit (Pierce) was used for determining protein concentration. Identical amounts of protein (50 μg) were loaded to SDS-PAGE using 8% running gels and transferred to nitrocellulose membranes (GE Healthcare). After a blocking step with 5% nonfat milk in PBST for 1 h at room temperature (RT), membranes were incubated overnight at 4°C with rabbit antibody to mGluR1 (1:1000; Millipore). Membranes were then washed three times in PBST for 10 min and incubated with HRP-conjugated donkey anti-rabbit antiserum (1:5000; GE Healthcare) for 1 h at RT. Thermo Fisher Scientific SuperSignal Chemiluminescent Substrate was used to detect HRP on the blots. Reversible validated Ponceau staining was used to check equal loading of gels (Aldridge et al., 2008; Romero-Calvo et al., 2010). Protein load, antiserum dilution, and film development parameters were adjusted to optimize visualization of both strongly and weakly stained bands. For quantitation, protein band densities were analyzed by using NIH ImageJ software. The dorsal horn contralateral to surgery site in sham condition was used as the reference to normalize protein band densities in relative density analyses. Student's t test was used for comparison to the reference condition.
Cell culture.
HEK293 cells were plated either in 25 mm2 culture flasks or in 12-well plates onto poly-d-lysine-coated 18 mm glass coverslips (both at a density of 5 × 104 cells/mm2) and maintained in DMEM (Invitrogen) supplemented with 10% FBS and 1% penicillin–streptomycin mixture. When they had reached 60–70% confluence, cells from two 25 mm2 flasks were homogenized with 2 ml of the above lysis buffer, and protein concentration was determined using BCA protein assay kit (Pierce). Cells plated onto poly-d-lysine-coated coverslips were fixed by incubation for 10 min at RT with 4% paraformaldehyde in PB and subjected to double immunofluorescence staining.
Immunofluorescence.
Deeply anesthetized rats (sodium pentobarbital; 50 mg/kg, i.p.) were perfused transcardially with 250 ml of 0.9% saline followed by 900 ml of 4% paraformaldehyde in phosphate buffer (PB) (0.1 m), pH 7.4. Fifteen animals were used for immunofluorescence experiments. L4–L5 spinal segments were removed, postfixed with 4% paraformaldehyde in PB for 4 h, and then cryoprotected for 48 h with 30% sucrose in PBS at 4°C. Coronal, 40-μm-thick cryotome sections were serially collected in PBS and preincubated with 1% bovine serum albumin (Sigma-Aldrich) and 1% normal serum (1 h; RT). In double immunolabelings for PSD-95/mGluR1, sections were incubated overnight at 4°C with rabbit antiserum against mGluR1 (1:500; Millipore) and mouse antibody to PSD-95 (1:400; Thermo Fisher Scientific). In triple immunostainings, sections were incubated with antibodies to mGluR1, with goat polyclonal H-18 against 5-HT2AR (1:400; sc-15074; Santa Cruz Biotechnology), and with mouse antibodies to either PSD-95 as above or to Pan neuronal marker (1:400; Millipore). After preincubations with normal serum of species other than those in which the secondary antibodies were raised, sections were sequentially incubated with Cy5 650-conjugated donkey anti-rabbit, Dylight 549-conjugated donkey anti-mouse, and Alexa 488-conjugated donkey anti-goat fluorescent antibodies (1:200; Jackson ImmunoResearch) and mounted in Mowiol (Vector Laboratories). Double- and triple-labeled sections were viewed in a Fluoview FV500 Olympus confocal microscope, and digital photomicrographs were acquired sequentially to avoid overlapping of fluorescent emission spectra. ImageJ software (intensity correlation plug-in) (Li et al., 2004) was used to adjust brightness and contrast, to obtain image colocalization overlays, as well as to perform intensity correlation-based analyses of confocal photomicrographs of labeled tissue. Pearson's correlation coefficient and Fisher's exact test were used to determine and contrast colocalization values, respectively.
Specificity of this antibody to mGluR1 has been documented previously in transfected HEK293 cells expressing splice variants of mGluR1, synaptosomal and membrane preparations of cortical and subcortical regions, and mGluR1-deficient mice (Ferraguti et al., 1998). Specificity of the affinity-purified goat polyclonal H-18 antibody (Santa Cruz Biotechnology; sc-15074) against 5-HT2AR was tested here by preabsorption with an excess of antigen (3:1 mass ratio of peptide to IgG) for 2 h at RT followed by immunofluorescence analysis in spinal cord tissue sections (Fig. 3). To further confirm its specificity, the H-18 antibody was combined with the mouse monoclonal A-4 antibody (Santa Cruz Biotechnology; sc-166775) and double-immunofluorescence experiments were conducted in the embryonic kidney 293 (HEK293) cell line (Fig. 3), which can be used as a positive control for expression of 5-HT2AR as indicated by the A-4 antibody manufacturer and confirmed in our laboratory (Fig. 3). The A-4 antibody has been raised against a different epitope of the 5-HT2AR from that of H-18, and detects a single protein band consistent with the theoretical molecular weight of ∼53 kDa of the 5-HT2AR in both nervous tissue (Aira et al., 2012) and lysates from HEK293 cells (present study) (Fig. 3).

Specificity of the 5-HT2AR H-18 antibody. Confocal scanning fluorescent micrographs of the spinal cord dorsal horn immunolabeled with the goat polyclonal H-18 antibody are shown before (A) and after (B) preabsorption with the immunizing peptide at a 3:1 ratio of peptide to antibody protein. The insets depict higher magnification images taken at the level of the lamina II of the dorsal horn. Dense 5-HT2AR immunostaining in the neuropil was almost abolished by preabsorption of the antibody with the antigen. Double immunofluorescence experiments in HEK293A cells using the mouse monoclonal A-4 (C) and goat polyclonal H-18 (D) antibodies generated against different epitopes of the 5-HT2AR revealed an almost identical distribution of the immunofluorescence signals (E), further supporting the specificity of the goat polyclonal H-18 antibody. Western immunoblot analysis of total lysate (20 μg) using the mouse monoclonal A-4 antibody confirmed the endogenous expression of 5-HT2AR in HEK293 cells (F). Scale bars: B, 100 μm; insets, 10 μm; E, 50 μm.
For double immunofluorescence, paraformaldehyde-fixed HEK293 cells were treated for 1 h at RT with blocking buffer (0.066% saponin, 0.22% gelatin in PB) containing 1% bovine serum albumin and 1% normal donkey serum followed by overnight incubation at 4°C with mouse monoclonal A-4 (1:400) and goat polyclonal H-18 (1:100) primary antibodies in blocking buffer. Thereafter, cells were incubated for 1 h at RT in fluorescent conjugated secondary antibodies DyLight 549 donkey anti-mouse F(ab′)2 fragment (1:400; Jackson ImmunoResearch) and Alexa Fluor 488 donkey anti-goat IgG (1:400; Invitrogen). After extensive washing with PBS, coverslips were mounted in Mowiol and subjected to confocal laser-scanning analysis. Negative controls for double and triple immunofluorescence experiments included omission of primary antibodies and incubations with species-mismatched secondary antibodies. No labeling above background was detected in any of these controls.
Behavioral evaluation.
Mechanical and thermal pain thresholds were evaluated before surgery and 2, 5, 7, and 9 d after ligation in three experimental groups, namely in sham-operated rats, nerve-ligated rats thereafter chronically treated with M100907 (0.4 mg/kg, i.p.), and nerve-ligated rats receiving vehicle treatment. For mechanical pain threshold evaluation, we determined 50% mechanical paw withdrawal thresholds for the right and left hindpaws to plantar stimulation with von Frey monofilaments (North Coast Medical) according to an up–down algorithm (Chaplan et al., 1994). We assessed thermal pain thresholds on a hot-plate device (World Precision Instruments) according to Almási et al. (2003). Briefly, rats were placed in a Plexiglas chamber on the plate at 30°C for 5 min for habituation, and plate temperature was raised at a rate of 6°C per minute until the animal exhibited paw licking or withdrawal behaviors (cutoff temperature, 50°C). The heat pain threshold was calculated as the mean temperature eliciting response from two repeated procedures, 30 min apart. The evaluator was blind to received treatments. Nonparametric Kruskal–Wallis one-way ANOVA and Mann–Whitney rank test were used to compare thresholds with those before surgery and across time points (SPSS, version 15.0).
Results
Spinal hyperexcitability induced by 5-HT2A after SNL depends on endogenous coactivation of mGluR1
In addition to the well established role of spinal glutamatergic mechanisms in central sensitization (Schmidtko et al., 2008; Li et al., 2010), there is recent evidence that 5-HT2A is upregulated and plays a proexcitatory role in dorsal horn neurons after SNL (Aira et al., 2010, 2012). Previous evidence of cross talk mechanisms between glutamate and serotonin receptors in cerebral cortex (Gewirtz and Marek, 2000; Molinaro et al., 2009) led us to hypothesize the existence of an analogous interplay in dorsal horn neurons during sustained pain. Spinal superfusion with high-affinity 5-HT2AR agonist TCB-2 at 100 nm concentration significantly and concentration-dependently increased C-fiber-evoked potentials by 35.23 ± 0.30%, with an EC50 of 0.26 ± 1.32 μm, in nerve-injured rats (Fig. 4). Coadministration of the selective mGluR1 antagonist AIDA at subclinical concentration (10 μm) significantly raised the EC50 value of the agonist to 37.86 ± 1.09 μm (p < 0.01), and elevated by 3 orders of magnitude the lowest significant concentration of TCB-2 to enhance evoked potentials (14.64 ± 0.98% increase of mean potential areas at 100 μm drug concentration; p < 0.01; n = 6; Fig. 4).

Enhancement of C-fiber-evoked spinal field potentials that is promoted by 5-HT2AR after SNL is mediated by mGluR1, but not by mGluR2/3 or NMDA. Preliminary experiments to determine the effects of pharmacological blockade of mGluR1, mGluR2/3, and the NMDA receptor in nerve-ligated rats are shown in A. Administration of NMDA receptor antagonist d-AP5 at 100 μm significantly decreased mean evoked potential areas, and thus a 10 μm concentration was considered as subclinical for further experiments, whereas 10 and 100 μm concentrations were selected for mGluR1 antagonist AIDA and for group II mGluR antagonist LY 341495, respectively. Mean areas of C-fiber-evoked potentials are shown in B during spinal superfusion with increasing, cumulative concentrations of 5-HT2AR agonist TCB-2 (100 μm) either alone (open triangles) or in combination with subclinical concentrations of AIDA (solid triangles), LY341495 (open circles), or d-AP5 (solid circles). Coadministration of neither LY341495 nor d-AP5 altered the ability of TCB-2 to augment evoked field potentials in nerve-ligated rats. In contrast, coadministration of AIDA increased the minimal effective concentration of TCB-2 by 3 orders of magnitude and dramatically increased the EC50 of TCB-2. Statistical significance (p < 0.01 at Bonferroni's test) of increases in field potential areas by effect of TCB-2 relative to baseline controls in the absence or presence of AIDA is denoted by asterisks or section signs, respectively. Representative recordings shown at the top illustrate the enhancing effect of 100 μm TCB-2 on C-fiber-evoked field potentials in the absence (a) or concurrent presence of 10 μm AIDA (b). Error bars indicate SEM.
In contrast, the ability of TCB-2 (100 nm) to increase C-fiber-evoked field potentials was not altered by the coadministration of subclinical concentrations of either the group II mGluR antagonist LY 341495 (100 μm) or the NMDA antagonist d-AP5 (10 μm) (35.00 ± 6.90 and 27.75 ± 2.34%, increases by TCB-2, respectively; p < 0.01; n = 6; Fig. 4).
5-HT2AR and mGluR1 are coexpressed in postsynaptic sites in dorsal horn neurons
The question as to whether mGluR1 acts at the cellular or supracellular level to enable 5-HT2AR-mediated facilitation as above has not yet been addressed. If interaction between mGluR1 and 5-HT2AR takes place at the cellular level, then a requisite is that both receptor proteins are jointly expressed in dorsal horn neurons. To investigate for this possibility, we performed colocalization studies in the superficial laminae I–II of the dorsal horn, since synaptic axon terminals of descending serotonergic fibers and glutamatergic primary afferents both are concentrated in this region (Ruda et al., 1981; Alvarez et al., 1992; Todd, 2010; Wei et al., 2010). At low magnification, mGluR1 and 5-HT2A immunoreactivities were found to be coexpressed in laminae I–II (Fig. 5). At higher magnification, 5-HT2AR/mGluR1 coexpression was circumscribed to neuropilar elements reminiscent of dendritic processes, consistent with previous studies locating both receptors at postsynaptic sites in dorsal horn (Alvarez et al., 1993; Pitcher et al., 2007). The neuronal location of coexpressed 5-HT2AR/mGluR1 was confirmed by triple immunofluorescence with the neuronal marker Pan (Fig. 5). We further confirmed that 5-HT2AR/mGluR1 were coexpressed in postsynaptic sites in dorsal horn neurons by using triple immunofluorescence with the postsynaptic marker PSD-95 (Fig. 5).

Enrichment in mGluR1 in 5-HT2AR-containing postsynaptic sites in superficial laminae of the spinal dorsal horn after SNL. Micrographs of double- and triple-immunofluorescence labelings for confocal laser-scanning microscopy of a transverse section of the L5 segment are shown. Lower magnification micrographs in A provide an overview of the dorsal horn, where approximate boundaries of superficial laminae are shown as dashed lines and the region from which high-power micrograph below was taken is indicated by a box. Scale bar, 100 μm. At higher magnification, mGluR1/5-HT2AR colocalization overlay was found to be located in neuropilar elements reminiscent of dendritic process. Scale bar, 3 μm. The neuronal location of 5-HT2AR was confirmed by colocalization with the Pan neuronal marker. The schematic drawing at the top provides a spatial clue as to the convergence of descending serotonergic axons and primary C-fiber afferents in the dorsal horn. In B, coexistence of mGluR1 and 5-HT2AR is shown in postsynaptic densities in dorsal horn neurons ipsilateral and contralateral to injury, as identified by the postsynaptic marker PSD-95, as well as coexpression changes at day 9 after SNL. Scale bar, 3 μm. The asterisks in correlation coefficient graphs indicate statistical significance (p < 0.01) at Fisher's exact test for comparison of Pearson's correlation coefficients. Error bars indicate SEM.
Facilitation of evoked potentials by coactivation of 5-HT2A and mGluR1 in the absence of nerve injury
Having shown that 5-HT2AR-mediated hyperexcitability following SNL was dependent on concurrent mGluR1 activation, it remained to be established whether simultaneous activation of mGluR1 and 5-HT2AR would be sufficient itself to enhance C-fiber-evoked potentials in the absence of SNL. We first determined that neither the mGluR1 agonist 3,5-DHPG nor TCB-2 alone, at concentrations of up to 1 mm, was able to alter C-fiber-evoked potentials in sham-operated rats (Fig. 6). In contrast, superfusion of the spinal cord simultaneously with 100 μm 3,5-DHPG and 100 μm TCB-2 significantly increased evoked potentials (16.08 ± 3.13% increase; p < 0.01; n = 6; Fig. 6). These observations strongly supported a regulatory role of mGluR1 on 5-HT2AR function. Since pharmacological activation of 5-HT2AR results in rapid upregulation of mGluR1 (see below), we wanted to determine whether mGluR1 activation by 3,5-DHPG would become capable of enhancing evoked potentials after transient stimulation of 5-HT2AR. The results showed that superfusion with 100 μm TCB-2 for 60 min and subsequent washout of the drug did not result in the enhancement of evoked field potentials by 100 μm DHPG application (Fig. 6).

Coactivation of 5-HT2AR and mGluR1 augments C-fiber-evoked dorsal horn potentials in sham-operated rats. In A, mean areas of C-fiber-evoked field potentials are shown during spinal superfusion with increasing, cumulative concentrations the mGluR1 agonist 3,5-DHPG (open circles) or the 5-HT2AR agonist TCB-2 (solid triangles). The bar diagram in B shows the magnitudes of evoked potentials during administration of 100 μm 3,5-DHPG, either during simultaneous application of 100 μm TCB-2 or after prior priming with TCB-2, which consisted in administration of 100 μm TCB-2 for 60 min and subsequent washout. The asterisks indicate statistical significance (p < 0.01 at Bonferroni's test) of mean evoked potential area comparisons to baseline (aCSF) controls. Representative recordings illustrate the effects of drug administration on evoked field potentials. Error bars indicate SEM.
SNL leads to rapid, bilateral upregulation of mGluR1
In light of the observed dependence of 5-HT2A-mediated hyperexcitability on a mGluR1-based mechanism, we hypothesized that SNL should result in upregulation of mGluR1 in spinal neurons as an essential change to enable 5-HT2AR-mediated facilitation. Previous studies had suggested synaptic plasticity of mGluR1 expression to occur in dorsal horn neurons during sustained inflammatory pain (Pitcher et al., 2007). We addressed this issue by assessing mGluR1 expression in dorsal horn homogenates at several time points after L5 ligation, starting at 60 min after injury to up to 9 d after ligation. Western blot analyses showed a significant (p < 0.01 in protein density analysis) ipsilateral increase in mGluR1 expression in synaptic fraction 1 h after injury followed by a dramatic bilateral increase in expression from day 5 onward (Fig. 7). Confocal immunofluorescence experiments confirmed that 5-HT2AR-containing postsynaptic densities in dorsal horn neurons become highly enriched in mGluR1 bilaterally by day 9 after SNL (Fig. 5).

Western blot analysis of the time course of upregulation of mGluR1 in synaptic fraction (P3) of dorsal horn homogenates in rats subjected to SNL, ipsilateral and contralateral to injury site. Relative density analyses from before injury revealed a significant ipsilateral increase of mGluR1 protein density 1 h after ligation, whereas increase in mGluR1 expression in dorsal horn contralateral to injury became significant at day 5 after ligation. The asterisks indicate statistical significance (p < 0.01) at Student's t test for density comparisons to before injury (total n was 20). As shown at the top, only the synaptic fraction but not cytoplasmic fraction S2 or crude synaptic vesicle fraction S3, was enriched in postsynaptic density protein PSD-95. Ponceau staining was used to confirm equal protein loading. Error bars indicate SEM.
Tonic activation of 5-HT2AR that is induced by SNL is responsible for upregulation of mGluR1 at postsynaptic sites
Mechanisms potentially leading to upregulation of mGluR1 following SNL are unknown. Since our previous work had shown that the spinal 5-HT2AR was upregulated and tonically active following SNL (Aira et al., 2010, 2012), an intriguing possibility was that 5-HT2AR-mediated serotonergic input itself might contribute to mGluR plasticity (Li et al., 1999). We thus sought to determine whether chronic blockade of 5-HT2AR by daily intraperitoneal administration of the selective 5-HT2AR antagonist M100907 could prevent mGluR1 upregulation 9 d after SNL. As a positive control, we confirmed that an acute intraperitoneal dose of M100907 (0.4 mg/kg) could decrease C-fiber-evoked spinal field potentials 9 d after SNL (Fig. 2) and thus could block the spinal 5-HT2AR as effectively as the selective antagonist 4F 4PP applied directly to the dorsal horn by spinal superfusion (Aira et al., 2010). Western blot analyses revealed that chronic treatment by daily intraperitoneal injections of M100907 (0.4 mg/kg), but not vehicle (isotonic saline), was able to markedly and bilaterally attenuate upregulation of mGluR1 both in cytoplasmic (S2) and synaptic (P3) fractions of dorsal horn homogenates by day 9 after SNL (Fig. 8).

Chronic blockade of 5-HT2AR attenuates SNL-induced upregulation of mGluR1 in dorsal horn neurons. Western blot analyses are shown illustrating significant bilateral increases in mGluR1 protein in S2 (A) and P3 fractions (B) from dorsal horn homogenates ipsilateral and contralateral to injury site after SNL. Daily intraperitoneal injections of 5-HT2AR antagonist M100907 (0.4 mg/kg) prevented and attenuated in mGluR1 upregulation in S2 fraction contralateral and ipsilateral to injury site, respectively. In synaptosomal (P3) fraction, mGluR1 upregulation was bilaterally and markedly attenuated by chronic M100907 treatment. As revealed by relative density analyses normalized to contralateral side in sham-operated rats, chronic 5-HT2AR blockade significantly attenuated SNL-induced upregulation of mGluR1 in both subcellular fractions and both sides of the body (asterisks indicate significance at p < 0.01 at Student's t test; total n was 12). Error bars indicate SEM.
To confirm that upregulation of mGluR1 after SNL involved an increased presence of the receptor at postsynaptic sites, we made use of PSD-95 as a postsynaptic marker. PSD-95 is closely related to synaptic plasticity during maintained pain (LeBlanc et al., 2010; D'Mello et al., 2011) and is known to form interaction complexes with glutamate receptors (D'Mello et al., 2011). We predicted that colocalization of mGluR1 and PSD-95 would be increased in nerve-ligated rats. Double immunofluorescence experiments confirmed this increase on the injury side, and colocalization also was significantly increased contralaterally (Fig. 9). Correlation analyses of double-labeled photomicrographs revealed significant bilateral increases in mGluR1/PSD-95 colocalizations, yielding Rr values of 0.86 after SNL vs 0.21 in sham-operated rats (z = −131.82; p < 0.01) on the side of injury and 0.81 versus 0.24 (z = −107.66; p < 0.01) contralaterally (Fig. 9).

Bilateral upregulation of mGluR1 in postsynaptic densities after SNL is dependent on 5-HT2AR activation. Micrographs of double-immunofluorescence labelings for mGluR1 and the postsynaptic density marker PSD-95 are shown of a transverse section of L5. All pictures were taken from superficial layers of the dorsal horn as indicated in Figure 2. Scale bar, 5 μm. Coexpression of mGluR1 and PSD-95, as shown by white overlay and confirmed by correlation coefficient analyses, is increased after SNL both ipsilaterally (A) and contralaterally (B) to injury, indicating increased presence of mGluR1 in postsynaptic densities. Chronic blockade of 5-HT2AR by daily injections of antagonist M100907 significantly and bilaterally reduced SNL-induced upregulation of mGluR1. The asterisks in correlation coefficient graphs (C) indicate statistical significance (p < 0.01) at Fisher's exact test for comparison of Pearson's correlation coefficients. Error bars indicate SEM.
In light of the preventative effect of chronic 5-HT2AR blockade on mGluR1 upregulation in synaptic fraction of tissue homogenates, we further examined whether intraperitoneal M100907 treatment could disrupt PSD-95/mGluR1 colocalization in nerve-ligated rats. High-power magnification images confirmed that 5-HT2AR blockade could at least partially block upregulation of mGluR1 9 d after SNL. Correlation analyses revealed Rr values of 0.44 after 5-HT2AR blockade on the side of injury (z = −100.20 vs absence of treatment; p < 0.01) and 0.42 contralateral to surgery (z = −82.90 vs absence of treatment; p < 0.01) (Fig. 9).
Upregulation of mGluR1 is inducible by 5-HT2AR activation and dependent on PKC
To gain insight into intracellular mechanisms leading to upregulation of mGluR1 following SNL, we first sought to determine whether 5-HT2AR activation was sufficient to upregulate mGluR1 in dorsal horn neurons. Therefore, the lumbosacral enlargement of the spinal cord was superfused with the 5-HT2AR agonist TCB-2 (100 μm) for 60 min and then removed for assessing mGluR1 protein density in tissue homogenates. The results showed that 5-HT2AR activation, but not superfusion with NaCl, effectively triggered upregulation of mGluR1 both in cytoplasmic (S2) and synaptic (P3) compartments in dorsal horn neurons (Fig. 10).

Upregulation of mGluR1 is induced by 5-HT2AR via activation of PKC. As shown by Western blot analyses, 5-HT2AR stimulation by spinal superfusion with agonist TCB-2 can upregulate mGluR1 protein in the spinal dorsal horn both in S2 and P3 fractions. For quantification, Western blot bands were normalized to S2 fraction band density in the control condition (superfusion with NaCl). Coadministration of PKC inhibitor chelerythrine markedly reduced TCB-2-induced upregulation of mGluR1. The asterisks indicate statistical significance at p < 0.01 at Student's t test. Ponceau staining was used to confirm equal protein loading. Error bars indicate SEM.
Since activation of 5-HT2AR, whether synaptic or pharmacological, could upregulate mGluR1 via an intracellular mechanism, a candidate pathway may be PKC activation, a major signaling route coupled to subtype 2 5-HT receptors (Hoyer et a., 1994; Hori et al., 1996). Indeed PKC is involved in many aspects of regulation of mGluR function (Gereau and Heinemann, 1998; Francesconi and Duvoisin, 2000; Ferguson et al., 2008; Lee et al., 2008). We wanted to determine whether the plasticity of mGluR1 in the dorsal horn that is triggered by activation of 5-HT2AR is dependent on mediation by PKC. In parallel with the preceding set of experiments, we tested the effect of PKC inhibitor chelerythrine on TCB-2-induced increase in mGluR1 expression. The results showed that coadministration of chelerythrine visibly reduced mGluR1 upregulation led to by TCB-2 application (p < 0.01; Fig. 10).
Time-dependent, 5-HT2AR mediation of thermal and mechanical allodynia after SNL
To better understand the involvement of 5-HT2AR-mediated serotonergic input in central sensitization and pain following SNL, we evaluated the occurrence of thermal and mechanical allodynia over several time points in nerve-injured rats that were chronically treated for the entire postoperative period with daily intraperitoneal injections of either vehicle or the 5-HT2AR antagonist M100907 (0.4 mg/kg). In vehicle-treated rats, mechanical allodynia to plantar stimulation with von Frey monofilaments was already present at the side of injury at day 2 after SNL, as shown by the dramatic decrease in paw withdrawal thresholds (1.31 ± 0.59 vs 11.23 ± 0.42 g before surgery; p < 0.01; n = 6), and lasted for the entire evaluation period. Similarly, heat allodynia measured by the hot-plate test was found from the second postsurgery day onward (38.29 ± 0.97 vs 48.42 ± 0.58°C before surgery; p < 0.01; n = 6). Lesser yet significant changes were found contralaterally at day 2 after SNL (mechanical threshold at 8.99 ± 0.76 g and heat pain threshold at 46.48 ± 0.48°C; both p < 0.01 relative to preinjury measures), which progressively increased until reaching maximal expression at day 9 (2.64 ± 0.54 g and 42.89 ± 0.64°C, respectively) (Fig. 11). No changes in mechanical or thermal thresholds were detected in sham-operated rats.

5-HT2AR mediates late-onset thermal and mechanical allodynia after SNL. Heat pain thresholds assessed in the hot-plate test (A), as well as the 50% paw withdrawal threshold to a static mechanical stimulus with von Frey monofilaments (B) are shown over multiple time points after SNL. After ligation of L5, both thermal and mechanical threshold were significantly lowered both ipsilaterally and contralaterally to injury site in vehicle-treated animals. The latter were mild and evolved over a few days after surgery, whereas profound ipsilateral changes were already present at day 2 after surgery and lasted for the entire evaluation period (9 d). No changes in mechanical and thermal thresholds were found in sham-operated animals. The asterisks indicate statistical significance at p < 0.01 at the nonparametric Kruskal–Wallis one-way ANOVA and Mann–Whitney rank tests when comparing mean threshold values to before surgery in vehicle-treated animals (for the sake of clarity, only the earliest time point demonstrating significant changes is labeled). Chronic 5-HT2AR blockade with M100907 progressively decreased SNL-induced mechanical and thermal allodynia over the days following injury. Antiallodynic effects of M100907 were conspicuous from day 5 onward and mechanical and thermal thresholds were reverted by M100907 to near-basal levels by day 9 after injury. The section signs and pound signs denote statistical significance (same α and statistical test as above) when comparing mean thresholds values in M100907-treated rats to values in vehicle-treated rats and to values in sham-operated rats, respectively (for clarity, only data from day 9 after injury are labeled for statistical significance). Error bars indicate SEM.
Chronic 5-HT2AR blockade with M100907 progressively decreased SNL-induced mechanical and thermal allodynia over the days following injury. Thus, both types of allodynia were little, if any, affected by M100907 relative to vehicle-treated animals at day 2 after injury, and only at day 5 were they attenuated by M100907 (Fig. 11). Mechanical and thermal thresholds were reverted by M100907 to near-basal levels at day 9 after injury. Mean heat pain thresholds contralateral to injury and mechanical pain thresholds ipsilateral to injury 9 d after ligation were not significantly different from those in sham-operated rats (47.81 ± 0.46 vs 48.13 ± 0.64°C, and 10.11 ± 0.54 vs 10.90 ± 0.47 g, respectively), whereas mean heat pain thresholds ipsilateral and mechanical pain thresholds contralateral to injury were markedly reverted by M100907 treatment by this time point, compared with vehicle-treated animals (Fig. 11), yet still significantly lower than in sham-operated animals [47.80 ± 0.56 vs 48.62 ± 0.44°C (p < 0.001) and 11.09 ± 0.61 vs 9.48 ± 0.49 g (p < 0.01), respectively].
Discussion
The results of this work provide evidence for a cross talk between receptors 5-HT2A and mGluR1 in dorsal horn neurons that is critical to the development of spinal hyperexcitability and thermal and mechanical allodynia after SNL. This interplay entails (1) upregulation of mGluR1 in dorsal horn synapses induced by 5-HT2AR recruitment and activation of downstream PKC, and (2) establishment of mGluR1-dependent, 5-HT2AR-promoted spinal hyperexcitation and pain.
5-HT2AR-induced increase in mGluR1 expression in dorsal horn synapses
Previous studies have shown that spinal mGluR1/5 promote central sensitization and pain (Neugebauer et al., 1999; Azkue et al., 2003), and contribute to maintaining pain behaviors after peripheral neuropathy (Yashpal et al., 2001; Fisher et al., 2002). Colocalization of mGluR1 with postsynaptic marker PSD-95 (Fig. 9) is in agreement with previous ultrastructural data (Alvarez et al., 1993) and supports a postsynaptic mechanism of action for mGluR1. Plasma membrane-associated mGluR1 has been shown to move significantly closer to the synaptic cleft during inflammatory pain, which has been proposed to increase probability of receptor activation (Pitcher et al., 2007). Here, we provide evidence that mGluR1 is coexpressed with 5-HT2AR in postsynaptic densities in dorsal horn neurons, and that these synapses become highly enriched in mGluR1 following SNL (Fig. 5). Rapid changes in mGluR1 expression are detectable in the synaptic compartment 60 min after SNL and increase over several days after ligation (Fig. 7). Less prominent changes in cytoplasmic fraction are consistent with increased de novo mGluR1 protein synthesis that may be required for the maintenance of pain (Fundytus et al., 2001, 2002; Yashpal et al., 2001).
We show that chronic blockade of 5-HT2AR by daily intraperitoneal injections of antagonist M100907 (0.4 mg/kg) impedes upregulation of mGluR1 (Figs. 8, 9), suggesting that increased activity of 5-HT2AR secondary to SNL (Aira et al., 2010) is responsible for mGluR1 plasticity in dorsal horn neurons. In further support of this is our finding that pharmacological stimulation of 5-HT2AR by direct spinal superfusion with TCB-2 is sufficient to initiate upregulation of mGluR1, as well as spinal hyperexcitation upon coactivation of mGluR1 (Fig. 6). In addition, we show that mGluR1 stimulation with DHPG fails to enhance C-fiber-evoked potentials after transient administration of TCB-2 (Fig. 6), suggesting that 5-HT2AR activation may be required not only for initiating but also for maintaining increased responses to peripheral C-fiber input.
A salient feature of 5-HT2AR-induced plasticity of mGluR1 here was its bilateral nature. This finding is relevant because contralateral sensory abnormalities are a common characteristic of central sensitization not only in animal models of sustained pain (Ossipov et al., 2000) (see below) but also in experimental (Shenker et al., 2008) and clinical pain in the human being (Burstein et al., 2000; Fernández-de-las-Peñas, 2009, 2010; La Touche et al., 2010). Most if not all 5-HT-containing fibers arise from supraspinal sites, where major serotonergic sources such as the raphe magnus nucleus give rise to bilateral spinopetal projections (Skagerberg and Björklund, 1985). Consistently, 5-HT release increases bilaterally after peripheral nerve injury (Satoh and Omote, 1996). Moreover, we have recently shown that 5-HT2AR is upregulated bilaterally in dorsal horn neurons after SNL (Aira et al., 2012), an event that is crucial to plasticity of mGluR1 as well as to bilateral development of pathologic pain as found here. In further support for mGluR1 mediation of the 5-HT2AR-mediated hyperexcitability and pathologic pain that follow SNL, the time course of mGluR1 upregulation triggered by SNL paralleled that of thermal and mechanical allodynia attenuated here by pharmacological blockade by M100907 (i.e., allodynia assumed to be mediated by 5-HT2AR) (Fig. 11).
A PKC-based mechanism in mGluR1 plasticity
We show here that 5-HT2AR and mGluR1 are coexpressed in postsynaptic densities in superficial I–II layers, a region where afferent C-fibers and descending serotonergic axon terminals converge (Alvarez et al., 1993; Doly et al., 2004; Van Steenwinckel et al., 2008; Todd, 2010; Wei et al., 2010). Both receptors share PKC as a common downstream transduction pathway that is known to promote neuronal excitation (Houamed et al., 1991; Hoyer et al., 1994; Conn and Pin, 1997; Shum et al., 2002) and to contribute to allodynia and neuropathic pain (Hua et al., 1999; Honda et al., 2007; Miyoshi et al., 2007). The impeding effect of chelerythrine on upregulation of mGluR1 that is otherwise induced by TCB-2 in cytoplasmic and synaptic fractions (Fig. 10) supports a mediator role for PKC in mGluR1 plasticity. A putative intracellular mechanism that may link 5-HT2AR to de novo expression of mGluR1 might include transcription factor CREB, whose phosphorylation can be promoted by PKC (Wu et al., 2005). For example, 5-HT2AR and CREB both can regulate μ- and δ-opioid receptor expression (Lee and Lee, 2003; Aira et al., 2012).
Activation of mGluR1 is essential to 5-HT2AR facilitation of C-fiber-evoked spinal excitation
There is evidence of cross talk mechanisms between mGluRs and 5-HT receptors in the cerebral cortex (Gewirtz and Marek, 2000; Molinaro et al., 2009). We show that activation of mGluR1 in dorsal horn neurons, but not that of group II mGluRs or the NMDA receptor, is essential for 5-HT2AR to generate spinal hyperexcitation following SNL (Fig. 4). Furthermore, pharmacological stimulation of mGluR1 by 3,5-DHPG in the absence of SNL was sufficient to enable the 5-HT2AR agonist TCB-2 to effectively enhance C-fiber-evoked potentials (Fig. 6). This is suggestive that the activity of mGluR1, which is low or absent in normal physiological conditions (Azkue et al., 2003), may be insufficient to effectively interact with 5-HT2AR (Aira et al., 2010) but becomes upregulated by 5-HT2AR after SNL and thus may act as a feedforward activation mechanism for the latter. Increased central glutamate release over the days following nerve injury (Kawamata and Omote, 1996) may additionally contribute to mGluR1 activation, and the lack of contralateral changes in glutamate release (Kawamata and Omote, 1996) is consistent with the milder behavioral and biochemical changes observed here contralaterally.
Time-specific contribution of 5-HT2AR to neuropathic pain
Full development of thermal and mechanical allodynia ipsilateral to ligation site by day 2 after surgery (Fig. 9) is in keeping with other studies (Röyttä et al., 1999; Sinnott et al., 1999; Clayton et al., 2007; Kaku et al., 2007; Kobayashi et al., 2008; Pérez-Severiano et al., 2008; Wei et al., 2010). In addition, the development of milder contralateral changes over >1 week as found here (Fig. 11) is in agreement with previous reports (Sinnott et al., 1999; Araujo et al., 2003; Clayton et al., 2007; Arguis et al., 2008). Here, chronic blockade of 5-HT2AR after ligation resulted in a dramatic decrease of thermal and mechanical allodynia, demonstrating a crucial role for 5-HT2AR in promoting sustained pain and sensory abnormalities after SNL. A key role for 5-HT2AR in mechanical hypersensitivity has also been reported in other models of neuropathic pain, such as those induced by 2′,3′-dideoxycytidine or vincristine (Thibault et al., 2008; Van Steenwinckel et al., 2008).
The contribution of 5-HT2AR to facilitation of nociceptive responses may initiate by day 2 after injury, and continues building up until days 7–9, as revealed here by chronic blockade experiments (Fig. 11). This increasing involvement of 5-HT2AR roughly parallels the progressive dependence of spinal hyperexcitation and pain on descending facilitatory influences after nerve injury. Although the precise timing of this transition process remains to be established, the dependence of mechanical allodynia on increased primary afferent input may start to decline approximately by day 1 after injury (Sun et al., 2005), a time by which serotonin concentration is already significantly increased (Satoh and Omote, 1996). Mechanical allodynia may be largely dependent on serotonergic influence by day 2, in view that serotonin depletion by 5,7-DHT (5,7-dihydroxytryptamine) can effectively attenuate SNL-induced mechanical allodynia by this time, and importantly this dependence further increases from approximately day 4 onward (Rahman et al., 2006). Consistent with this progression, allodynia appears to become dependent on integrity of descending pathways to the dorsal horn by days 5–7 after injury (Porreca et al., 2001; Burgess et al., 2002; Rahman et al., 2006; Vera-Portocarrero et al., 2006; Mase et al., 2011) and largely independent from increased activity of primary afferents (Li et al., 2000; Jang et al., 2010). The observed time course of the 5-HT2AR contribution to thermal and mechanical allodynia here broadly coincides with that of serotonergic descending facilitatory influences on spinal nociception, thus reinforcing the view that 5-HT2AR may act as a mediator in this phenomenon. Additional support to this notion comes from our findings contralateral to injury site, where central sensitization was entirely dependent on descending serotonergic mechanisms subserved by a spinal-bulbo-spinal loop, rather than on increased primary afferent input. It is thus likely that, in the absence of primary afferent-mediated induced excitation, 5-HT2AR blockade could suppress allodynia contralaterally at a much earlier stage than on the injury side (Fig. 11).
In conclusion, the present findings emphasize the role of 5-HT2AR in facilitation of spinal nociceptive transmission and pain following SNL, and suggest the upregulation of mGluR1 in dorsal horn neurons as a novel feedforward activation mechanism critically contributing to hyperexcitation generated by 5-HT2AR.
Footnotes
This work was supported by the Government of the Basque Country under Program SAIOTEK (SA-2010/00110) and Ayudas a Grupos de Investigación del Sistema Universitario Vasco.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Jon Jatsu Azkue, Department of Neurosciences, School of Medicine and Dentistry, University of the Basque Country, P.O. Box 699, 48080 Bilbao, Spain. jonjatsu.azkue{at}ehu.es





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}