

Abstract
Atonal homolog1 (Atoh1) encodes a basic helix–loop–helix protein that is the first transcription factor to be expressed in differentiating hair cells. Previous work suggests that expression of Atoh1 in prosensory precursors is necessary for the differentiation and survival of hair cells, but it is not clear whether Atoh1 is required exclusively for these processes, or whether it regulates other functions later during hair cell maturation. We used EGFP-tagged Atoh1 knock-in mice to demonstrate for the first time that Atoh1 protein is expressed in hair cell precursors several days before the appearance of differentiated markers, but not in the broad pattern expected of a proneural gene. We conditionally deleted Atoh1 at different points in hair cell development and observe a rapid onset of hair cell defects, suggesting that the Atoh1 protein is unstable in differentiating hair cells and is necessary through an extended phase of their differentiation. Conditional deletion of Atoh1 reveals multiple functions in hair cell survival, maturation of stereociliary bundles, and auditory function. We show the presence of distinct critical periods for Atoh1 in each of these functions, suggesting that Atoh1 may be directly regulating many aspects of hair cell function. Finally, we show that the supporting cell death that accompanies loss of Atoh1 in hair cells is likely caused by the abortive trans-differentiation of supporting cells into hair cells. Together our data suggest that Atoh1 regulates multiple aspects of hair cell development and function.
Introduction
The organ of Corti, the auditory sensory organ in mammals, comprises a precise and invariant pattern of mechanosensory hair cells and nonsensory supporting cells in the cochlea. Cochlear hair cells and supporting cells arise from a common postmitotic precursor population, the prosensory domain (Kelley, 2006). This differentiation follows a gradient, starting near the basal region of the cochlea and progressing toward the apex over a period of several days (Li and Ruben, 1979; Lim and Anniko, 1985; Chen et al., 2002; Lumpkin et al., 2003; Montcouquiol and Kelley, 2003; Kelley, 2006). Atoh1, the mouse homolog of the Drosophila proneural gene atonal, is a basic helix–loop–helix (bHLH) transcription factor that is the earliest known gene expressed in differentiating hair cells (Bermingham et al., 1999; Chen et al., 2002; Woods et al., 2004). In the cochlea, Atoh1 is both necessary and sufficient for hair cell development: the absence of Atoh1 results in a complete loss of hair cells (Bermingham et al., 1999), while ectopic expression of Atoh1 is sufficient to direct ectopic hair cell formation in the greater epithelial ridge, the nonsensory epithelium next to the organ of Corti (Zheng and Gao, 2000). In Atoh1-null mice, massive cell death is observed within the presumptive sensory epithelia at the base of the cochlea at embryonic day (E) 15.5. Later, the apoptosis progresses to the whole cochlear duct (Chen et al., 2002). Transient expression of Atoh1 in mice in which Atoh1 is conditionally deleted by an Atoh1-Cre transgene is not sufficient to prevent the majority of hair cells from dying and cannot support the proper function of the remaining hair cells (Pan et al., 2012), indicating the level and duration of Atoh1 expression is critical for maintaining the viability and differentiation of hair cells. However, it remains unclear whether there is a critical period for Atoh1 to keep hair cells alive and whether Atoh1 functions differently at later stages of hair cell development. A recent genome-wide survey of Atoh1 target genes in the cerebellum suggested that Atoh1 regulates a broad range of biological processes, including cell proliferation, differentiation, migration, and metabolism (Klisch et al., 2011). The variety of pathways Atoh1 might regulate during cerebellar development suggests that Atoh1 might be involved in multiple developmental processes in hair cells as well.
To dissect the function of Atoh1 during hair cell development, we established a conditional knockout (CKO) system to delete Atoh1 at specific developmental stages. By exposing pregnant or neonatal mice to tamoxifen to activate Cre-mediated recombination driven by an Atoh1 autoregulatory enhancer (Atoh1-CreERT2), we deleted Atoh1 from hair cells at different embryonic and neonatal stages. We found a critical time window, ∼2 d after initiation of Atoh1 expression, in which Atoh1 is absolutely required for hair cell survival. Atoh1 deletion within this time window also led to the loss of the surrounding supporting cells, and we used a Cre reporter allele to show that some of these supporting cells attempt to upregulate Atoh1 in response to hair cell loss. Significantly, later deletion of Atoh1 also revealed a time-dependent requirement for its activity in hair bundle maturation and auditory function. Together, our data suggest Atoh1 has multiple functions in the survival, differentiation, and maturation of hair cells during cochlear development.
Materials and Methods
Experimental animals.
Atoh1 −/− [Mouse Genome Informatics (MGI): Atoh1tm1Hzo], Atoh1A1GFP/A1GFP (MGI: Atoh1tm4.1Hzo), and Atoh1flox/flox (MGI: Atoh1tm3Hzo) mice were generated as previously described (Ben-Arie et al., 1997; Shroyer et al., 2007; Rose et al., 2009). Atoh1- CreERT2 [MGI: Tg(Atoh1-cre/Esr1*)14Fsh] (Machold and Fishell, 2005) and R26R-YFP [MGI: Gt(ROSA)26Sortm1(EYFP)Cos] (Srinivas et al., 2001) transgenic lines were obtained from Jackson Laboratories. Genotyping was performed by PCR using the following primers: for different Atoh1 alleles, Atoh1-forward (ACG CAC TTC ATC ACT GGC), Atoh1-reverse (GGC ACT GGC TTC TCT TGG), and Neo-forward (GCA TCG CCT TCT ATC GCC) yield a 600 bp wild-type allele band and a 400 bp null allele band. HA-forward (GCG ATG ATG GCA CAG AAG G) and HA-reverse (GAA GGG CAT TTG GTT GTC TCA G) yield a 1 kb Atoh1 EGFP-tagged allele band and a 350 bp floxed allele band. For Atoh1-CreERT2, Cre1F (GCC TGC ATT ACC GGT CGA TGC AAC GA) and Cre1R (GTG GCA GAT GGC GCG GCA ACA CCA TT) yield a 700 bp band. For R26R-YFP, oIMR0316 (GGA GCG GGA GAA ATG GAT ATG), oIMR0883 (AAA GTC GCT CTG AGT TGT TAT) and oIMR4982 (AAG ACC GCG AAG AGT TTG TC) yield a 320 bp yellow fluorescent protein-positive (YFP+) band. To generate the inducible Atoh1 CKO mice, Atoh1-CreERT2; Atoh1+/− males were crossed with Atoh1flox/flox; R26R-YFP homozygous females. One dose of 2 mg tamoxifen and 2 mg progesterone was administered to pregnant females at E15.5, E16.5, or E17.5 by oral gavage. Tamoxifen and progesterone were dissolved together in peanut oil, both at a concentration of 20 mg/ml. For delivering tamoxifen into newborn animals, tamoxifen was dissolved in peanut oil at a concentration of 10 mg/ml and subcutaneously injected into animals at a dose of 75–100 μg/g body weight. The genotypes of embryos or newborn pups from these crosses were determined as above. The Baylor College of Medicine Institutional Animal Care and Use Committee approved all animal experiments.
Cochlea isolation.
Cochleas from E13.5 to E16.5 embryos were dissected in PBS and then incubated in calcium-magnesium-free PBS containing dispase (1 mg/ml; Invitrogen) and collagenase (1 mg/ml; Worthington) for 8 min at room temperature. The enzyme solution was then replaced by DMEM containing 10% FBS and the mesenchymal tissue was removed by fine syringes to free the cochlear duct. For neonatal pups, cochleas were dissected in PBS, with the spiral ganglia and Reissner's membrane removed to expose the organ of Corti. To obtain adult cochlear whole-mount preparations, temporal bones were dissected and the cochleas were slowly perfused through the oval window with 4% paraformaldehyde (PFA) in PBS, postfixed for 2 h at room temperature, and decalcified in 150 mm EDTA for 5–7 d at 4°C before further dissection.
Immunohistochemistry.
Primary antibodies used in this study were anti-active Caspase 3 (1:500, rabbit; R&D Systems), anti-GFP (1:1000, chicken; Abcam; this detects GFP, EGFP, and YFP), anti-Hey2 (1:500, rabbit; Doetzlhofer et al., 2009), anti-Jag1 (1:200, rabbit; Santa Cruz Biotechnology), anti-Myosin6 (1:500, rabbit; Proteus), anti-Myosin7a (1:100, mouse; Developmental Studies Hybridoma Bank), anti-p27kip1 (1:250, mouse; NeoMarker), anti-Prox1 (1:500, rabbit; Millipore Bioscience Research Reagents), anti-Sox2 (1:500, rabbit; Millipore), anti-Sox10 (1:250, goat; Santa Cruz Biotechnology), anti-β tubulin (TuJ1, 1:1000, mouse; Covance), anti-TMHS (tetraspan transmembrane protein of hair cell stereocilia) (1:1000, rabbit; a gift from Kenneth Johnson; Longo-Guess et al., 2005), anti-espin (1:1000, rabbit; gift from Jim Bartles; Sekerkov á et al., 2004), and anti-Atoh1 (1:10,000, chick; a gift from Matthew Kelley and Thomas Coate; Driver et al., 2013). Secondary antibodies were Alexa 488, Alexa 594, or Alexa 647 (1:2000; Invitrogen). Actin in stereociliary bundles was labeled with Alexa 594-conjugated phalloidin (1:1000, Invitrogen). Cell nuclei were labeled by DAPI. For sections of ear tissue, animal heads were fixed 1–2 h in 4% PFA at room temperature, cryoprotected in 30% sucrose in PBS at 4°C, embedded in OCT compound, and cryosectioned at 12–14 μm. The immunohistochemistry procedure followed standard protocols with some minor modifications. For anti-Atoh1 staining, tissue was fixed in 4% paraformaldehyde for 30 min before sectioning, and sections were blocked in PBS containing 0.5% Triton X-100, 10% goat serum, and 10% Henblocker (Aves Labs). For anti-p27kip1, Jag1, and Sox2 staining, sections were boiled for 10 min in 10 mm citric acid, pH 6.0.
Uptake of FM1-43.
FM1-43 (Invitrogen) was prepared in water at a stock concentration of 5 mm. Cochleas were dissected from postnatal day (P) 5 mouse pups and briefly rinsed in HBSS (Invitrogen). Cochleas were exposed to a 5 μm solution of FM1-43 in HBSS for 15 s before being washed several times, mounted, and viewed with fluorescence microscopy.
Auditory brainstem response recording.
Before testing, mice were anesthetized by intraperitoneal injection of a ketamine–xylazine mix at a dose of 100 mg/kg ketamine and 10 mg/kg xylazine. Testing was performed in a soundproof booth. Normal body temperature was maintained throughout the procedure by placing the mice on a heating pad. Pure tone bursts (0.1 ms rise/fall, 2 ms duration, 21 presentations/s) from 4 to 48 kHz were generated using System 3 digital signal processing hardware and software (Tucker Davis Technologies). The intensity of the tone stimuli was calibrated using a type 4938 one-quarter inch pressure-field calibration microphone (Brüel & Kjær). EC1 ultrasonic, low-distortion electrostatic speakers were coupled to the ear canal to deliver stimuli within 3 mm of the tympanic membrane. Response signals were recorded with subcutaneous needle electrodes inserted at the vertex of the scalp (channel 1), the postauricular bulla region (reference), and the back leg (ground), and averaged over 500 presentations of the tone bursts. Electrode-recorded activity was filtered (high pass, 300 Hz; low pass, 3kHz; notch, 60Hz) before averaging to minimize background noise. Auditory thresholds were determined by decreasing the sound intensity of each stimulus to 10 dB from 90 dB in 5 dB steps until the lowest sound intensity with reproducible and recognizable waveforms was detected. Thresholds were determined to within 5 dB for each frequency by two raters to ensure reliability. Mean hearing thresholds ± SD (dB SPL) were plotted as a function of stimulus frequency (kilohertz) and analyzed for group differences at individual frequencies using two-tailed t tests accompanied by a one-way ANOVA to reveal overall trends. R (version 2.13) was used for all statistical analyses.
Results
Atoh1 protein marks hair cell precursors during mouse cochlear development
The organ of Corti differentiates from a band of prosensory cells that runs along the length of the cochlear duct (Kelley, 2007; Kelly and Chen, 2009). This prosensory domain begins to exit the cell cycle in mice at E12.5, starting at the apex of the cochlea. Over the next 2–3 d, cell cycle exit proceeds along the cochlea from the apex to the base (Ruben, 1967; Lee et al., 2006). The differentiation of this postmitotic precursor domain into hair cells and supporting cells begins between E13 and E14, starting close to the base of the cochlea and spreading along the length of the cochlea to the apex over the next 4 d (Li and Ruben, 1979; Lim and Anniko, 1985; Chen et al., 2002; Montcouquiol and Kelley, 2003). At intermediate times, different regions of the cochlea are therefore at different stages of differentiation. For example, at E16, the basal region of cochlea contains a full complement of one row of inner hair cells and three rows of outer hair cells as revealed by the hair cell marker Myosin6, while at the apex of the cochlea, no Myosin6-expressing hair cells can be detected (Fig. 1D). In the middle regions of the cochlea, the first inner hair cells can be observed differentiating, but their neighboring outer hair cells and associated supporting cells have yet to differentiate. Thus, different regions of the cochlea contain hair cells or their precursors at different stages of differentiation.

Atoh1 protein expression in the mouse cochlea. A, Schematic diagram of the Atoh1A1GFP/A1GFP knock-in mouse line, referred to in the figure as Atoh1-EGFP. An Atoh1-EGFP fusion gene was targeted to the Atoh1 genomic locus. B, Whole-mount Atoh1A1GFP/A1GFP cochleas from E13.5 to P0. The right two rows show higher magnification of the base and apex regions marked by box at the left. Hey2 (red) was used to mark the prosensory domain at E13.5 and E14.5. Scale bars, 200 μm. C, Sections of Atoh1A1GFP/A1GFP cochlea at E13.5. Scattered EGFP-labeled cells (arrows) were localized within the prosensory domain labeled by p27kip1 and Hey2. D, Sections through different regions of E16.5 Atoh1A1GFP/A1GFP cochlea. Hair cells were labeled by Myosin6 (red). Hair cell differentiation begins near the base of the cochlea and spreads down to the apex over a period of days. Sections through the base, middle turn, and apex of the cochlea at E16.5 therefore reveal different stages in the differentiation of hair cell precursors. In the base, most Atoh1-EGFP protein is localized in the nucleus of Myosin6-expressing hair cells. However, in the apex, much cytoplasmic Atoh1-EGFP protein can be observed in precursors that have not yet expressed Myosin6. E, An Atoh1 antibody also reveals nuclear staining in hair cells in the base of the cochlea, more cytoplasmic staining in the middle turn of the cochlea, but no detectable staining in the apex. Arrowhead, inner hair cells; bracket, outer hair cells.
Atoh1 is one of the first genes expressed in differentiating hair cells and is both necessary and in some circumstances sufficient for their differentiation (Bermingham et al., 1999; Zheng and Gao, 2000). However, the onset of Atoh1 expression in the cochlea has been hard to visualize accurately, with different techniques giving different results. For example, using a β-gal reporter knocked in to the Atoh1 locus, Woods et al. showed that Atoh1 promoter activity can be observed in a wide patch of cells along most of the cochlear duct at E13 (Woods et al., 2004), suggesting Atoh1 is a marker of hair cell and supporting cell precursors in the prosensory epithelium. Such a pattern of expression would be consistent with Atoh1 functioning as a proneural gene. To clarify the expression pattern of Atoh1 protein in the mouse cochlea, we made use of Atoh1A1GFP/A1GFP knock-in mice, in which the endogenous Atoh1 gene is tagged with EGFP (Fig. 1A; Rose et al., 2009). Homozygous Atoh1A1GFP/A1GFP mice are viable, breed normally, and exhibit no observable phenotype in any organ (Rose et al., 2009). We used these mice to visualize Atoh1 protein by staining with antibodies against EGFP together with markers of the prosensory domain and hair cells.
The earliest stage at which we could detect Atoh1-EGFP expression in the Atoh1A1GFP/A1GFP cochlea was E13.5 (Fig. 1B,C). In whole-mount preparations of the E13.5 cochlea, we observed expression of EGFP in a diffuse, salt-and-pepper patch of cells in the midbasal region, localized within the prosensory domain marked by the transcription factor Hey2 (Fig. 1B) (Hayashi et al., 2008; Doetzlhofer et al., 2009). We did not observe broad domains of Atoh1 protein expression in sections of the Atoh1A1GFP/A1GFP cochlea. Instead, cells expressing Atoh1-EGFP frequently lined up in columns in the sensory epithelium or existed as single cells (Fig. 1C). Costaining with p27kip1 and Hey2 indicated that these columns or isolated EGFP-labeled cells localized within the prosensory domain on its neural side (Fig. 1C). This arrangement of the earliest Atoh1-expressing cells in the Atoh1A1GFP/A1GFP cochlea was consistent with previous immunohistochemical studies using anti-Atoh1 antibodies (Chen et al., 2002; Driver et al., 2013). However, the higher sensitivity of the anti-EGFP antibody in our study suggests that the onset of Atoh1 protein expression in the mouse cochlea begins as early as E13.5.
At E16.5 in the basal cochlea, Atoh1-EGFP protein was restricted to hair cells that express the early hair cell differentiation marker Myosin6, forming one row of inner hair cells and three rows of outer hair cells (Fig. 1D). However, in the middle turn region, where Myosin6 is just beginning to be expressed in outer hair cells, Atoh1-EGFP was expressed in columns of cells spanning the sensory epithelium. This pattern of EGFP-expressing cells was also seen in the apex of E16.5 cochlea, where there were no signs of Myosin6 expression (Fig. 1D). This suggests that Atoh1 is expressed in a subset of postmitotic precursors within the cochlear sensory primordium, before the differentiation of hair cells. As differentiation proceeds, Atoh1 becomes restricted to Myosin6-labeled hair cells (Fig. 1D). Intriguingly, we observed that Atoh1-EGFP expression in hair cells in the basal third of the cochlea was largely restricted to the nucleus. However, in the middle turn and apical regions that contain Myosin6-negative hair cell precursors at earlier stages of differentiation, EGFP was diffusely localized throughout the cell cytoplasm (Fig. 1D). These changes in subcellular distribution of Atoh1 protein between precursors and differentiating hair cells suggests there is a redistribution of cytosolic Atoh1 or an active transport of Atoh1 protein from the cytoplasm into the nucleus during hair cell differentiation.
It is possible that the cytoplasmic localization of the Atoh1-EGFP fusion protein in hair cell precursors is a consequence of the EGFP tag. To test this, we stained E16.5 cochlear sections with a recently described chicken antibody to the Atoh1 protein (Driver et al., 2013). Although the staining we observed with this antibody was significantly fainter than that seen with the Atoh1-EGFP fusion protein and absent at the apex of the cochlea, we were able to observe a diffuse cytoplasmic staining in middle turn regions of the cochlea, with nuclear staining becoming more distinct in the most basal regions of the cochlea (Fig. 1E).
Cell death in the prosensory domain of Atoh1 mutant mice
Genetic inactivation of Atoh1 in mice results in the death of a subpopulation of cells in the prosensory domain and a consequent failure of hair cell formation (Chen et al., 2002; Pan et al., 2011, 2012). To examine the time of onset of the apoptosis in detail, we performed immunohistochemistry on whole-mount Atoh1-null (Atoh1−/−; Bermingham et al., 1999) cochleas at different stages, using antibody against the active form of Caspase3 (ActCasp3). No apoptotic cells were observed in the prosensory domain of the Atoh1-null cochlea at E14.5 (data not shown), but we observed many ActCasp3-labeling cells at the base of E15.5 cochleas (Fig. 2A–C'). Staining of the cochlear sections showed the dying cells were located within the sensory epithelial region labeled by p27kip1 (Fig. 2D). These data suggest there is an ∼48 h delay between the time at which Atoh1 protein expression would normally initiate in the cochlea and the death of hair cell precursors in the absence of Atoh1.

Precursor cells in the sensory epithelia start to undergo apoptosis at E15.5 in the Atoh1-null (−/−) mice. A–C, E–G, Whole-mount cochleas from E15.5 Atoh1-null (A–C) and Atoh1 heterozygous (E–G) littermates. p27kip1 (green) marks the prosensory epithelial region. ActCasp3 (red) labels apoptotic cells in the prosensory epithelium of the Atoh1-null cochlea. C', G', Higher-power magnifications of the midbasal region of the cochlea marked by box in C and G, respectively. The dotted lines highlight the prosensory epithelium. D, H, Sections of the basal cochlea at E15.5. ActCasp3 (red) staining was only observed in the Atoh1 mutants (D) but not their heterozygous littermates (H).
A transgenic mouse system to conditionally delete Atoh1
Our immunohistochemistry data from Atoh1A1GFP/A1GFP cochleas suggest Atoh1 expression is sustained in cochlear hair cells for at least a week after hair cell differentiation. We next asked whether Atoh1 is continuously required for the survival of hair cells after differentiation has begun. We established a CKO system to remove Atoh1 from hair cells at different stages using the inducible Cre-lox system. We crossed Atoh1+/−; Atoh1-CreERT2 males with Atoh1flox/flox; R26R-YFP female mice. By oral gavage of the pregnant females or injecting mouse pups subcutaneously with tamoxifen, we were able to generate Atoh1 CKO (Atoh1flox/−; Atoh1-CreERT2; R26R-YFP) and control (Atoh1flox/+; Atoh1-CreERT2; R26R-YFP) animals (Fig. 3A). One advantage of this system is that the R26R-YFP reporter provides a clear visualization of the Cre recombination efficiency in the cochlea and reveals cells in which Atoh1 has been deleted. The system displayed almost no recombination in the absence of tamoxifen. Figure 3B shows regions from a neonatal Atoh1flox/+; Atoh1-CreERT2; R26R-YFP mouse that had not been exposed to tamoxifen. No YFP can be seen in either apical or basal regions of the cochlea; we typically saw one YFP-labeled cell in every five cochleas from untreated control mice. Moreover, we saw no evidence of YFP-labeled supporting cells in control animals treated with tamoxifen at any age between E15.5 and P2 (data not shown).

A, Breeding scheme to generate Atoh1-CKO mice. Female Atoh1flox/flox; R26R-YFP mice were mated with Atoh1+/−; Atoh1-CreERT2 males to generate mice that carried the CreERT2 allele, one copy of the R26R-YFP Cre reporter, and either an Atoh1flox/+ (50%) or Atoh1flox/− (50%) allele. Use of the R26R-YFP Cre reporter provides a readout of the leakiness, speed, and efficiency of the system. B, The base and apex of a cochlea from a P3 Atoh1flox/−; Atoh1-CreERT2; R26R-YFP mouse whose mother did not receive tamoxifen during pregnancy. No YFP-labeled cells can be detected in either region of the cochlea, and we typically observed an average of 1 YFP+ cell in every five cochleas examined. C, The base and apex of cochleas from Atoh1flox/+; Atoh1-CreERT2; R26R-YFP mice whose mothers received a single dose of tamoxifen (TMX) at E17.5. The embryos were collected 8, 24, and 48 h after tamoxifen dosing. A small number of YFP cells can be observed in both base and apex after 8 h. After 24 h, significantly more labeled cells can be observed in the base, in which hair cells differentiate first, compared with the apex. After 48 h, similar numbers of YFP-labeled cells (80–85%) are present in both regions of the cochlea. D, The Atoh1-CreERT2 transgene remains active until at least 2 d after birth. Neonatal pups were injected with tamoxifen at either P0 or P2 and allowed to develop for 24 h. After birth, Atoh1 starts to be downregulated from the cochlea, starting at the base and proceeding down to the apex. Consequently, significantly fewer YFP-labeled cells can be seen in the base at both ages, but ∼50% YFP+ cells are seen in the apex.
Tamoxifen-mediated recombination occurred rapidly in our CKO system. Figure 3C shows examples of embryos in which tamoxifen was administered at E17.5 and the animals killed 8, 24, or 48 h later. We could readily detect YFP expression in the embryonic cochleas 8 h after tamoxifen treatment. After 24 h, >80% of hair cells in the base of the cochlea were labeled with YFP. At the apex, which contains less mature hair cells, fewer (50%) hair cells were labeled by Cre recombination, but by 48 h, similar numbers (80–85%) of YFP-labeled cells could be seen in both apical and basal regions.
Atoh1 expression is gradually downregulated in cochlear hair cells in the first few days after birth, commencing at the base of the cochlea and finally being extinguished at the apex several days later. For example, at P0, Atoh1 is being downregulated in the base of the cochlea and is strongly expressed in the apex. Accordingly, when tamoxifen was injected into P0 pups, we saw few YFP-labeled cells in the base of the cochlea but many more (∼50%) in the apex (Fig. 3D). We continued to see ∼50% labeled cells in the apical third of the cochlea when tamoxifen was administered at P2 (Fig. 3D), suggesting that the Atoh1-CreERT2 allele is still active at this time.
Identification of a critical period for Atoh1 function in hair cell survival
To test whether there is a critical time window for Atoh1 to maintain the survival of hair cells, we delivered one dose of tamoxifen to pregnant females at three stages, E15.5, E16.5, or E17.5 (Fig. 4A). Animals were allowed to develop to the day of birth (P0), when offspring were genotyped and the cochleas collected from both CKO and control animals (Fig. 4A). We labeled and counted hair cells in whole-mount cochlea preparations using anti-Myosin6 antibodies and detected cells in which Atoh1 had been deleted by visualizing the YFP reporter (Fig. 3A). As described above, hair cell differentiation in the cochlea follows a basal-to-apical gradient, in which differentiation at the apex starts ∼3 d later than at the base (Li and Ruben, 1979; Lim and Anniko, 1985; Chen et al., 2002; Montcouquiol and Kelley, 2003). We therefore divided each cochlea into three parts along the length of the cochlear duct (base, middle turn, and apex) and quantified the hair cells in each region separately. When tamoxifen was administered at E15.5, hair cell numbers were significantly reduced along the entire length of CKO cochleas (Fig. 4B) compared with controls. When tamoxifen was administered at E16.5, we observed significant loss of hair cells in the middle turn to apical parts of the CKO cochleas, but not at the base (Fig. 4C), where hair cells are more mature. When tamoxifen was administered at E17.5, significantly reduced numbers of hair cells were only observed in the apex of the CKO cochleas (Fig. 4D). We examined the timing of cell death in the sensory epithelia by labeling the cochleas with antibodies to ActCasp3. In the CKO cochleas, we first observed apoptotic cells 20 h after tamoxifen treatment, with large numbers of dying cells observed by 24 h (see Fig. 8A). Since we first detect recombination in our mice 8 h after tamoxifen administration (Fig. 3C), this suggests that Atoh1 protein is unstable and that loss of Atoh1 from differentiating hair cells causes rapid cell death. Moreover, since hair cells mature in a basal–apical gradient along the cochlea starting at E13.5, our data suggest that Atoh1 is required for the survival of hair cell precursors in the base of the cochlea for 72 h after the first appearance of Atoh1 protein, but that deletion of Atoh1 after this critical period (for example, at E16.5 or later in the base of the cochlea) does not compromise hair cell survival.

Atoh1 is required for the survival of hair cells in a time-dependent manner. A, Diagram showing the experimental design for the analysis of Atoh1-CKO cochleas in B–D. Female Atoh1flox/flox; R26R-YFP mice were mated with Atoh1+/−; Atoh1-CreERT2 males and received a single dose of tamoxifen (TMX) at E15.5, E16.5, or E17.5. Mouse pups were analyzed on the day of birth (P0). B–D, Different regions of whole-mount cochleas from P0 Atoh1-CKO and control littermates in which one dose of tamoxifen was administered at E15.5 (B), E16.5 (C), and E17.5 (D). YFP (green) was used as a reporter to show Cre-mediated recombination. Red, Myosin6-labeled hair cells. The number of hair cells in both CKO and control cochleas is quantified and shown in the graphs at the right (n ≥ 3, error bars show SD).
Identification of a second critical period for Atoh1 in hair cell function
We next addressed whether surviving hair cells in the Atoh1 CKO cochlea retain any aspects of normal structure or function. We tested the effect of Atoh1 deletion on hair bundle structure by staining the actin-rich stereocilia with fluorescently labeled phalloidin. Administration of tamoxifen at E17.5 causes hair cell loss in the apex, but not in the midregion or basal region (Fig. 4D). In these animals, we observed disorganized hair bundles in both in the middle turn (where hair cell numbers were normal) and in the apical regions of the cochlea (which show significant hair cell loss; Fig. 5A). We did not observe abnormal hair bundle structure in the basal, more mature hair cells of CKO cochleas (Fig. 5A). We saw similar results with antibodies to the hair bundle proteins Espin (Sekerkov á et al., 2004; Fig. 5B) and TMHS (Longo-Guess et al., 2005; data not shown). We also examined cochleas in which we delivered tamoxifen to neonatal pups at P0, and only observed very subtle defects at the apex of the CKO organs (Fig. 5A). These data suggest that Atoh1 plays an additional role in hair bundle development as well as in regulating hair cell survival. Since deletion of Atoh1 at E17.5 can cause hair cell defects without hair cell death, this suggests that there is a second delayed critical period for Atoh1 in the regulation of hair bundle morphology independent of cell survival.

Atoh1 is required for stereociliary bundle development in hair cells. A, Phalloidin staining of different regions of whole-mount cochlea preparations from P2 Atoh1-CKO and control littermates. One dose of tamoxifen administration at E17.5 resulted in disorganized hair bundles in the middle and apical turns of CKO cochleas, but no defects in the more mature hair cells in the basal region. However, only very subtle defects in hair bundles were observed in the apical cochlea of CKO animals that received one dose of tamoxifen at P0. This suggests that Atoh1 is initially required for hair cell survival, but later for other functions, including hair bundle morphology B, Whole-mount cochleas from Atoh1-CKO and control animals receiving one dose of tamoxifen at E17.5 and stained with anti-espin antibodies at P2. C, Whole-mount cochleas from Atoh1-CKO and control animals receiving one dose of tamoxifen at E17.5 and then dissected at P5 and exposed to 5 μm of the styryl dye FM1-43 for 15 s to detect dye uptake through mechanotransduction channels. Surviving hair cells from apical regions of the cochlea displaying abnormal bundle morphology (A, B) still take up FM1-43. Scale bars, 20 μm. TMX, tamoxifen.
To determine whether Atoh1 can influence assembly of the mechanotransduction machinery in hair cell stereociliary bundles, we labeled living cochleas with FM1-43, a fluorescent styryl dye that can rapidly enter hair cells through open mechanotransduction channels (Meyers et al., 2003; Lelli et al., 2009). We administered tamoxifen at E17.5 and let the mice develop for 5 d after birth. Dissected cochleas were then bath-exposed to FM1-43 for 15 s and then washed and examined for FM1-43 fluorescence. We detected dye uptake in hair cells all along the CKO cochleas regardless of bundle morphology, even at the apex, where significant hair cell loss had occurred (Fig. 5C). Similar results were observed by injecting mouse pups with the fixable dye AM1-43 (data not shown). This suggests that deletion of Atoh1 does not affect the correct expression of mechanotransduction channels in hair cells over the time period examined, and that the assembly of mechanotransduction apparatus does not require morphologically normal hair bundles.
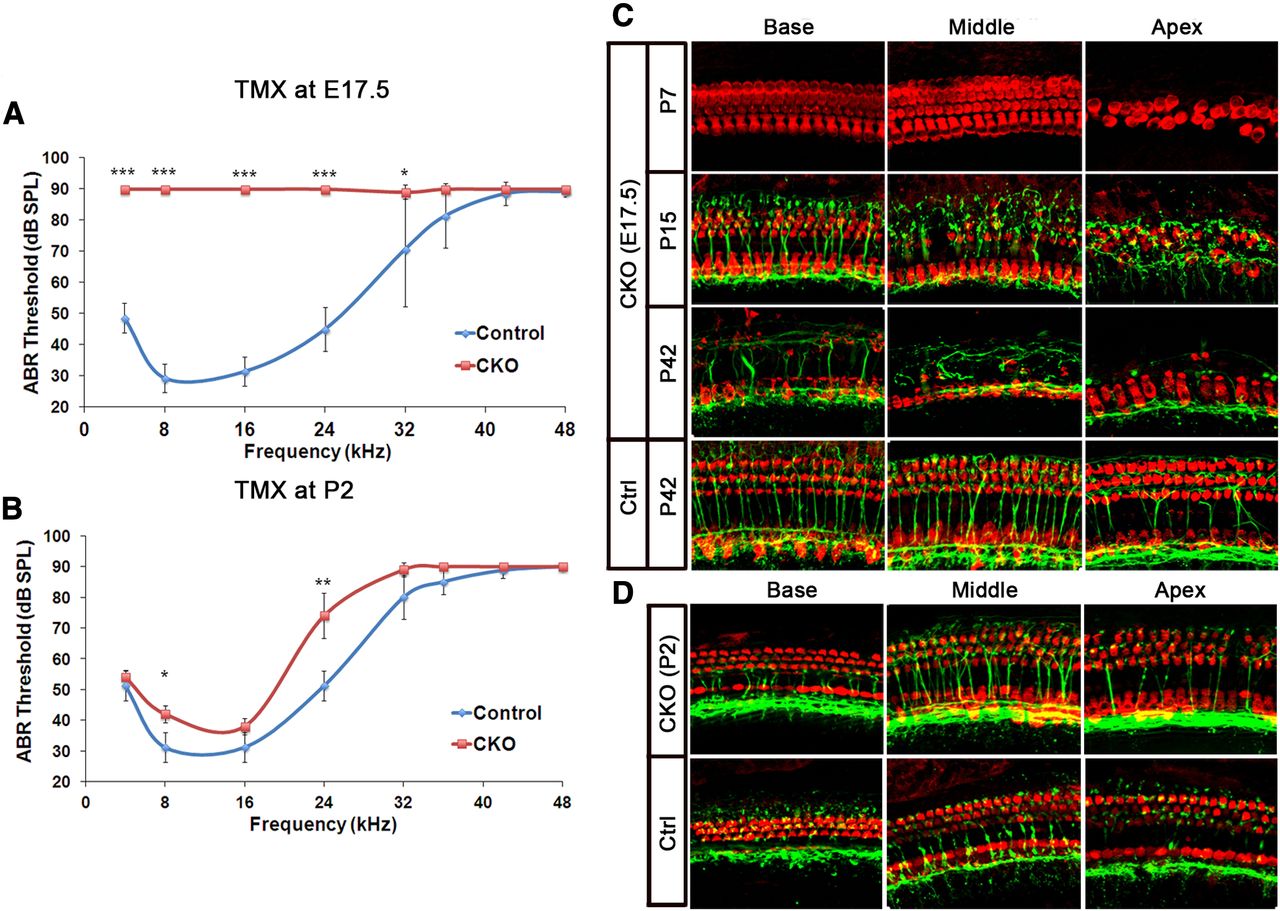
To determine the degree of hearing loss caused by deletion of Atoh1, we performed auditory brainstem response (ABR) measurements on 6-week-old Atoh1-CKO animals in which tamoxifen was administered at E17.5. We were surprised to see that these mice had a severe hearing loss at all frequencies tested (Fig. 6A), despite the fact that mice given tamoxifen at E17.5 only display apical hair cell loss at birth (Fig. 4D). This suggested that the loss of Atoh1 might lead to further hair cell loss or dysfunction as the animals age. We therefore treated mice with tamoxifen at E17.5 and isolated cochleas from these CKO at different postnatal stages and performed immunohistochemistry for Myosin6 to label hair cells and the TuJ1 anti-tubulin antibody to label neuronal fibers. At P7, no hair cells loss was observed in basal or middle turn regions of the CKO cochleas (Fig. 6C), although significant hair cell loss was seen in the apical regions, as we had observed at P0 (Fig. 4D). However, by P15, we started to observe loss of outer hair cells in the middle turn region of the cochlea (Fig. 6C). In 6-week-old adult mice, most outer hair cells were missing along the entire length of the cochlea (Fig. 6C) and efferent innervation of the outer hair cell region was severely disrupted. These data suggest that although outer hair cells in the basal and middle turns of the cochlea initially survive following loss of Atoh1, it is required for their integrity and survival in the week before the onset of hearing.

Timing of Atoh1 deletion affects extent of hair cell survival and preservation of hearing function. A, B, Atoh1 CKO mice have profound hearing loss when tamoxifen is administered at E17.5 (ANOVA p = 7.96e–11) (A), but only mild hearing loss when tamoxifen administration is delayed until P2 (ANOVA p = 0.1849) (B). Error bars represent SD. The Student's t test with Benjamini–Hochberg adjustment of p values for multiple comparisons was used to estimate statistical significance between genotypes at individual frequencies. *p < 0.05, **p < 0.02, ***p < 0.001. C, When tamoxifen is administered at E17.5, hair cells, visualized with Myosin6 immunostaining (red), are only missing from the apex of the cochlea at P7. However, by P15, hair cell loss is seen in the middle turn of the cochlea and, by P42, at the base. Control animals show no hair cell loss. Regions lacking hair cells also have defects in afferent and efferent innervation, revealed by staining with the TuJ1 antibody (green). D, When tamoxifen is administered at P2, no significant defects are seen in either hair cell numbers or innervation.
To determine whether this delayed effect of Atoh1 loss on hair cell survival also occurred within a critical time window, we administered tamoxifen to CKO pups at 2 d after birth and again measured hearing and cochlear morphology at 6 weeks of age. At P2, Atoh1 is still expressed in the apex of the cochlea, and Atoh1-CKO mice still show recombination in ∼50% of hair cells (Fig. 3D). In contrast to mice in which Atoh1 was deleted at E17.5, P2 CKO mice had ABR thresholds only marginally higher than those of controls (Fig. 6B). Moreover, the morphology and innervation of the cochlea of 6-week-old CKO was similar to controls along the length of the cochlea, and only very few missing outer hair cells were observed at the apex (Fig. 6D). These data suggest that although deletion of Atoh1 hair cells at E17.5 can cause delayed, severe hair cell death, hair cells in P2 mice no longer require Atoh1 for their short-term or long-term survival.
Atoh1 is indirectly required for differentiation and survival of cochlear supporting cells
Previous studies have suggested Atoh1 expression in hair cells might play an indirect role in the development and maturation of the neighboring supporting cells (Woods et al., 2004). Consistent with this idea, immunostaining of cochleas from Atoh1-null E19 mice or cochleas from Atoh1-CKO E19 mice that received tamoxifen between E13.5 and E15.5 showed significantly decreased expression or loss of several supporting cell markers, including Prox1, Sox2, and Jag1 (Fig. 7A). To examine whether deleting Atoh1 from hair cells affects the development or survival of the surrounding supporting cells, we collected neonatal CKO cochleas after tamoxifen treatment at E15.5, E16.5, or E17.5 (an identical schedule to that shown in Fig. 4A) and performed immunohistochemistry for Prox1 as a marker of Deiters' and pillar cells (Fig. 7B–D). For quantification, each cochlea was divided into three regions: base, middle turn, and apex. When tamoxifen was administered at E15.5, there was a significant loss of supporting cells from basal to middle turn regions of the CKO cochleas (Fig. 7B). However, when tamoxifen was administered at E17.5, supporting cell numbers were comparable between CKO and control organs at the base. Significant decrease of supporting cells was only observed in the middle turn to apical regions (Fig. 7D). The loss of supporting cells in the Atoh1-CKO cochlea closely followed the loss of hair cells (Fig. 4B–D). These data suggest that the critical period of Atoh1 requirement for the survival of hair cells is approximately the same as that which indirectly regulates the survival of surrounding supporting cells.

Atoh1 is required for the survival of the surrounding supporting cells in a time-dependent manner. A, Sections of middle turn regions of the cochlea from Atoh1-null and heterozygous embryos at E19. The expression of supporting markers Prox1, Sox2, and Jag1 was abolished in the knock-out mutants. Similar, albeit slightly milder phenotypes were also seen in Atoh1-CKO mice that received three daily doses of tamoxifen between E13.5 and E15.5 and were killed at E19. B–D, Whole-mount cochleas from P0 Atoh1-CKO and control littermates receiving one dose of tamoxifen at E15.5 (B), E16.5 (C), and E17.5 (D). This is the same experimental regimen shown in Figure 4A. Supporting cells were visualized by staining with a Prox1 antibody (red). The number of Prox1-labeled cells in both CKO and control cochleas on each injection day is quantified and shown in the graph.
To gain more insight into the mechanism of supporting cell death, we used anti-ActCasp3 antibodies to identify cells undergoing apoptosis in the cochlear sections of CKO mice that we harvested at 24 h after tamoxifen treatment at E17.5. Within the sensory epithelial region, we only observed ActCasp3 staining in the apex of the CKO cochleas, consistent with the restriction of hair cell and supporting cell loss to this region (Figs. 4D, 7D). Although many of the apoptotic cells were present in the upper hair cell layer, significant numbers of ActCasp3-labeled cells were located in the supporting cell layer close to the basilar membrane, suggesting that Atoh1 deletion in hair cells results in the death of surrounding supporting cells as well (Fig. 8A). Interestingly, when we examined Atoh1-CreER-mediated recombination in these mice, we observed some cells expressing the supporting cell markers Prox1 and Sox10 that were also labeled by YFP in the apical region of the CKO cochleas (Fig. 8C,D). However, we did not observe coexpression of YFP and Prox1 in any region of the control cochleas (Fig. 8C), nor at the base of the CKO organs. This suggested that the loss of hair cells leads to activation of Atoh1, and hence the Atoh1-CreERT2 transgene in supporting cells. We quantified the YFP-labeled cells within the hair cell layer and supporting cell layer (Fig. 8E). In the apex of the CKO cochleas, about half of the YFP+ cells were localized in the supporting cell layer (79 cells of a total of 170 YFP+ cells counted from 3 different cochleas; Fig. 8E). In contrast, we observed only two YFP+ cells in total within the supporting cell layer in the apical regions of three control cochleas (Fig. 8E). Since the Atoh1 autoregulatory enhancer used to drive CreERT2 expression requires Atoh1 expression (Helms et al., 2000; Machold and Fishell, 2005; Raft et al., 2007), the presence of YFP in the supporting cell layer suggests that loss of hair cells induces the upregulation of Atoh1 in the surrounding supporting cells, followed by Cre-mediated deletion of Atoh1 in these cells and their rapid death.

Atoh1 is indirectly required for differentiation and survival of cochlear supporting cells. A–D, Sections of apical regions of the cochlea from Atoh1-CKO and control animals harvested 24 h after tamoxifen treatment at E17.5. YFP shows cells that underwent recombination after tamoxifen administration. ActCasp3 (red) labels apoptotic cells in the apex of CKO organ (A). Myosin6 labels differentiated hair cells (B). Prox1 (C) and Sox10 (D) were used to examine supporting cell development. YFP+ cells (arrow) in the supporting cell layer were only observed in the apical region of CKO cochlea. This suggested that cells in the supporting cell layer of the CKO, but not wild-type cochlea, were activating the Atoh1-CreERT2 transgene. In the apex of control cochlea, we often saw inner hair cells labeled by Sox10 (D, arrowhead). E, Quantification of YFP+ cells in the hair cell and supporting cell layers of Atoh1-CKO and control mice harvested 24 h after tamoxifen treatment at E17.5. The total number of YFP+ cells was quantified in regions approximating to the basal and apical thirds of the cochlea from three different specimens of each genotype. Cells were assigned based on their position in the hair cell or supporting cell layers. Significant numbers of YFP-labeled cells (79) were observed in the supporting cell layer from the apical regions of three Atoh1-CKO cochleas. In contrast, only two YFP-labeled supporting cells from three cochleas could be observed in the apical turn of control cochleas. HC, hair cell; SC, supporting cell.
Discussion
Atoh1 is first expressed in a subset of precursors before hair cell differentiation
In Drosophila, atonal acts as a proneural gene: it is expressed in proneural clusters and the sensory precursors that give rise to the chordotonal organs, and is both necessary and sufficient for the differentiation of sensory organ precursors (Jarman et al., 1993). However, different techniques to reveal Atoh1 mRNA or protein expression in the mouse cochlea have yielded inconsistent results (Chen et al., 2002; Woods et al., 2004). We attempted to resolve this issue by using an Atoh1-EGFP fusion construct knocked into the Atoh1 locus. The Atoh1-EGFP allele is fully functional (Rose et al., 2009) and displays no observable phenotypes, being able to breed normally and survive to adulthood, unlike Atoh1-null mice. Our data show that Atoh1 protein is expressed before the expression of hair cell and supporting cell markers, but its expression is restricted to a subset of precursors in a salt-and-pepper pattern close to the border of the prosensory domain with the greater epithelial ridge (Fig. 1B,C). This region will give rise to inner hair cells, the first hair cell type to differentiate in the organ of Corti (Lim and Anniko, 1985; Chen et al., 2002; Lumpkin et al., 2003; Jacques et al., 2007). The restricted expression of Atoh1 suggests that, unlike atonal in Drosophila, Atoh1 is not expressed in a manner consistent with a proneural gene in the mammalian cochlea.
We observed a striking change in the subcellular localization of Atoh1-EGFP protein as hair cell precursors differentiated. Before hair cell differentiation, Atoh1-EGFP is ubiquitously expressed in the nucleus and cytoplasm of hair cell precursors (Fig. 1D). However, as cells started expressing the hair cell differentiation marker Myosin6, Atoh1-EGFP becomes restricted to the nucleus. We observed similar results with a recently described polyclonal antibody to Atoh1 (Driver et al., 2013), although the antibody appeared to be less sensitive than the Atoh1-EGFP fusion protein (compare Fig. 1D and 1E). The change in Atoh1 subcellular localization might be caused by the nuclear transport of Atoh1 when precursors differentiate into hair cells. Although nuclear-cytoplasmic shuttling has been reported for many transcription factors, there are few examples of this process regulating bHLH genes (Ghosh and Baltimore, 1990; Van Der Heide et al., 2004; Reich and Liu, 2006; MacDonald et al., 2009). At present, we do not know whether this translocation is regulated by post-translational modification of Atoh1 itself, or by accessory proteins that actively import Atoh1 into the nucleus. Regardless of the mechanism, it is intriguing to consider that the onset of nuclear restriction of Atoh1 coincides with the first appearance of differentiated hair cell markers, such as Myosin6 (Fig. 1D,E), and with the timing of hair cell precursor death in Atoh1-null embryos (see below). We are currently exploring the significance of the altered subcellular localization of Atoh1 in more detail.
Recent studies using Cre recombinase-mediated lineage tracing of Atoh1-expressing precursors suggests that at least some cells expressing Atoh1 will ultimately differentiate as supporting cells (Yang et al., 2010; Driver et al., 2013). We therefore cannot be certain that all of the Atoh1-EGFP-labeled cells we observe are destined to become hair cells. The random and variable distribution of cochlear supporting cells labeled by Atoh1 lineage tracing in these studies suggests that some Atoh1-expressing cells may be diverted to a supporting cell fate, possibly through Notch-mediated lateral inhibition (Jarman and Groves, 2013).
Atoh1 regulates the survival of hair cells and hair cell precursors
We and others have observed the first signs of apoptosis in the Atoh1-null cochlea at E15.5, 2 d after Atoh1 protein can be visualized in the wild-type organ (Chen et al., 2002; Pan et al., 2012). However, when we conditionally removed Atoh1 at stages between E15.5 and E17.5, conspicuous cell death was observed much more rapidly, starting at 20 h after tamoxifen treatment. The rapid cell death in our Atoh1 CKO mice is especially striking because we first observe recombination of the R26R-YFP locus in these mice 8 h after tamoxifen gavage. The fact that only 12 h elapses between the first cessation of Atoh1 transcription and the onset of cell death suggests that Atoh1 protein is quite unstable and that its absence rapidly leads to hair cell death. However, this critical period in which hair cells are vulnerable to loss of Atoh1 lasts for only 72 h in the base of the cochlea, as deletion of Atoh1 after E16.5, 72 h after the first expression of Atoh1 protein, does not cause cell death. It is likely that hair cells begin to express additional factors that promote hair cell survival toward the end of this critical period. Although we currently do not know the identity of these factors, two possible candidates are the transcription factors Gfi1 and Pou4f3, as mutant mice for either gene show significant hair cell death in the cochlea at later developmental stages (Xiang et al., 1998; Wallis et al., 2003). Atonal also participates in cell fate decisions mediating survival and death in the Drosophila antenna, where only sensory lineages specified by Atonal can respond to EGF signaling and survive (Sekerkov á et al., 2004). This suggests Atoh1 might also regulate cell survival in the cochlea by modulating hair cell responses to trophic factors.
The role of Atoh1 in hair bundle development and function
Previous studies on Atoh1 function in the cochlea focused mainly on the regulation of hair cell differentiation (Mulvaney and Dabdoub, 2012). However, the sustained expression of Atoh1 in differentiating hair cells until after birth suggests Atoh1 might also participate in other biological programs involved in the maturation and function of hair cells. We observed disorganized hair bundles in Atoh1-CKO cochleas when Atoh1 was deleted at E17.5, but not when Atoh1 was removed at P0, suggesting Atoh1 is required for the formation and orientation of stereocilia in a time-dependent manner. The fact that disorganized hair bundles can be observed in regions of Atoh1-CKO cochleas that have a normal complement of hair cells and supporting cells (Fig. 5A,B) suggests that this effect is a direct result of Atoh1 loss, rather than morphological rearrangements as an indirect consequence of cell death. Our FM1-43 labeling data suggests that mechanotransduction channels are still present in surviving Atoh1-CKO hair cells bearing disorganized stereociliary bundles (Fig. 5C). However, our ABR measurements of Atoh1-CKO mice suggest that animals in which Atoh1 is deleted at E17.5 have severe hearing loss at 6 weeks. Our whole-mount data suggests the hearing loss results from the degeneration of hair cells, particularly outer hair cells, and that this degeneration is already apparent by the onset of hearing at 2 weeks. We suggest that one factor contributing to the delayed loss of hair cells in the CKO animals seen at adult stages might be disrupted formation of stereocilia. It has been previously shown that loss of genes involved in stereocilia development, such as Whirlin, Cadherin23 and Pcdh15, can cause hair cell degeneration with age (Alagramam et al., 2001; Holme et al., 2002; Mustapha et al., 2007; Kane et al., 2012), suggesting healthy stereociliary bundles are essential for the long-term survival of hair cells. Once again, we observe another critical period in which Atoh1 is required for hair bundle formation and long-term hair cell survival, as we observed far less severe hearing loss and hair cell loss in CKO mice treated with tamoxifen at P2. Our histology data show that despite significant (>50%) recombination in the apex of the P2 cochlea, these mice lack only a few outer hair cells in this region, with no obvious signs of hair cell degeneration in basal and middle turn regions. The slight loss of hair cells in the apex may explain why these CKO mice show some degree of hearing loss at lower frequencies (<16 kHz).
Hair cell loss leads to trans-differentiation and death of surrounding supporting cells
Previous studies have shown that Atoh1-null mice lack both hair cells and supporting cells (Woods et al., 2004; Pan et al., 2012). It has been proposed that nascent hair cells may regulate the differentiation of supporting cells, but the ongoing role of hair cells in supporting cell survival is more controversial (Woods et al., 2004). We show here that conditional deletion of Atoh1 in cochlear hair cells leads to supporting cell death, but only in regions in which hair cells are also missing. This suggests that hair cells are indirectly regulating the survival of supporting cells. A possible mechanism for this death is suggested by our observation of Cre-mediated recombination in some supporting cells after tamoxifen treatment. We see strong expression of EGFP in cells in the lower layer of the CKO cochlea, colocalizing with both Prox1 and Sox10. We have never observed this expression in control cochleas or in regions of the CKO cochlea that show no hair cell death, suggesting that Atoh1 is upregulated in supporting cells in regions of the cochlea where hair cells are dying. It has been proposed that nascent hair cells normally send inhibitory signals (likely through the Notch pathway) to neighboring supporting cells, preventing their differentiation into hair cells (Lanford et al., 1999; Kiernan et al., 2005). Moreover, a number of previous studies in embryonic or neonatal mammals have shown that loss of hair cells following trauma can cause supporting cells to trans-differentiate into hair cells (Kelley et al., 1995; Burns et al., 2012). Atoh1 upregulation in the supporting cells of CKO cochleas might therefore be the consequence of loss of lateral inhibition following hair cell death. This would suggest that supporting cells might undergo an ill-fated attempt to trans-differentiate into hair cells, which then undergo Atoh1 deletion due to the continuing presence of circulating tamoxifen (Reinert et al., 2012).
In conclusion, our data suggest that Atoh1 fulfills several separate functions during the development and maturation of hair cells. In addition to being required for the initial differentiation of hair cells, conditional deletion of Atoh1 has also revealed the existence of distinct critical periods for hair cell survival, stereociliary bundle maturation, and the long-term viability of hair cells. By analogy to the recent finding that Atoh1 can directly regulate a wide variety of cellular functions in the developing cerebellum (Klisch et al., 2011), we suggest that Atoh1 may have a similarly diverse set of functions during the maturation of hair cells. A systematic survey of Atoh1 targets in hair cells, similar to that performed in the cerebellum, is likely to reveal further candidates and pathways that promote hair cell survival and function.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants DC006185 (A.K.G.), Department of Defense Grant W81XWH1020116 (A.K.G.), a training fellowship from the Houston Laboratory and Population Sciences Training Program in Gene–Environment Interaction (T.C.), National Research Service Award Grant F31DC012503 (M.L.S.), and NIH Grant DC000354 (F.A.P.). We thank Alyssa Faught and Huiling Li for excellent technical assistance, members of the Groves laboratory for helpful discussions, and Kurt Chonko and Steven Maricich for discussing data and exchanging ideas before publication. We thank Matt Kelley and Tom Coate for a gift of Atoh1 antibody, Jim Bartles for a gift of anti-espin antibody, and Kenneth Johnson for a gift of anti-TMHS antibody. The Myo7a 138-1 monoclonal antibody developed by Dana Orten and Tama Hasson was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biology. We are extremely grateful to Huda Zoghbi for providing mouse lines, for her suggestions for experimental design, and her comments on the manuscript.
- Correspondence should be addressed to Andrew Groves, Baylor College of Medicine, BCM295, 1 Baylor Plaza, Houston, TX 77030. akgroves{at}bcm.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}