

Abstract
Cerebellar deficit contributes significantly to disability in multiple sclerosis (MS). Several clinical and experimental studies have investigated the pathophysiology of cerebellar dysfunction in this neuroinflammatory disorder, but the cellular and molecular mechanisms are still unclear. In experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, proinflammatory cytokines, together with a degeneration of inhibitory neurons, contribute to impair GABAergic transmission at Purkinje cells (PCs). Here, we investigated glutamatergic transmission to gain insight into the pathophysiology of cerebellar dysfunction in EAE. Electrophysiological recordings from PCs showed increased duration of spontaneous excitatory postsynaptic currents (EPSCs) during the symptomatic phase of EAE, suggesting an alteration of glutamate uptake played by Bergmann glia. We indeed observed an impaired functioning of the glutamate-aspartate transporter/excitatory amino acid transporter 1 (GLAST/EAAT1) in EAE cerebellum caused by protein downregulation and in correlation with prominent astroglia activation. We have also demonstrated that the proinflammatory cytokine interleukin-1β (IL-1β), released by a subset of activated microglia/macrophages and infiltrating lymphocytes, was involved directly in such synaptic alteration. In fact, brief incubation of IL-1β in normal cerebellar slices replicated EAE modifications through a rapid GLAST/EAAT1 downregulation, whereas incubation of an IL-1 receptor antagonist (IL-1ra) in EAE slices reduced spontaneous EPSC alterations. Finally, EAE mice treated with intracerebroventricular IL-1ra showed normal glutamatergic and GABAergic transmissions, along with GLAST/EAAT1 normalization, milder inflammation, and reduced motor deficits. These results highlight the crucial role played by the proinflammatory IL-1β in triggering molecular and synaptic events involved in neurodegenerative processes that characterize neuroinflammatory diseases such as MS.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the CNS, for which the myelin sheath has been considered to be the primary target for many years. However, an increasing number of clinical (Plaut, 1987; Werner et al., 2001; Pitt et al., 2003) and experimental studies (Pitt et al., 2000; Smith et al., 2000; Centonze et al., 2010) have pointed out neurodegenerative aspects of the disease pathogenesis occurring in early phases and independently of demyelination (Trapp and Nave, 2008; Calabrese et al., 2010). It has been suggested that an imbalance between glutamatergic and GABAergic transmission likely represents a possible cause of excitotoxic damage observed in MS and experimental autoimmune encephalomyelitis (EAE; Gottesfeld et al., 1976; Pitt et al., 2000; Sarchielli et al., 2003; Clements et al., 2008; Centonze et al., 2009; Rossi et al., 2011).
We have demonstrated that EAE causes a dramatic potentiation of excitatory transmission by altering both presynaptic and postsynaptic sites of glutamate synapses in the striatum (Centonze et al., 2009; Grasselli et al., 2013; Rossi et al., 2012). This alteration is responsible for dendritic spine degeneration and severe motor deficits (Centonze et al., 2009). Furthermore, a decrease in GABAergic signal gives a relevant contribution to the enhancement of neuronal excitability in striatum during EAE, likely representing a further cause of excitotoxic damage (Centonze et al., 2009; Rossi et al., 2011). These data are consistent with in vivo observations showing that imbalanced glutamate homeostasis contributes to neuronal and oligodendroglial pathology in MS and that blockade of glutamate receptors ameliorates the clinical course of both MS (Plaut, 1987) and EAE (Smith et al., 2000; Pitt et al., 2000; Centonze et al., 2009).
A growing body of evidence indicates that proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and IL-1β are responsible for such synaptic and neuronal pathology in EAE (Lai et al., 2006; Centonze et al., 2009; Froger et al., 2010; Tolosa et al., 2011) and in MS. During an acute MS attack, inflammation increases brain IL-1β signaling, enhancing in turn glutamate-mediated synaptic excitability and neurotoxicity (Rossi et al., 2012). The potential role of other proinflammatory cytokines such as IL-17 (Chisholm et al., 2012; Miao et al., 2013) or chemokines needs further investigation.
Due to the strong impact that enhanced glutamate transmission seems to have in the inflammation-driven neurodegenerative process of MS, we extended our electrophysiological and morphological investigations to the cerebellum, a brain region largely affected in both MS and EAE (Kumar and Timperley, 1988; Waxman, 2005; Kutzelnigg et al., 2007; Chin et al., 2009; MacKenzie-Graham et al., 2009, 2012; Crespy et al., 2011). Recently, we reported an impairment of the inhibitory transmission at Purkinje cells (PCs) of EAE mice during the symptomatic phase of the disease associated with a degeneration of inhibitory interneurons. We proposed IL-1β as a potential player of the electrophysiological alterations observed in EAE (Mandolesi et al., 2012).
Here, we investigated cerebellar glutamatergic transmission in EAE and the cellular and molecular mechanisms underlying the synaptic alterations caused by IL-1β.
Materials and Methods
EAE induction and clinical evaluation.
Female C57BL/6 mice (The Jackson Laboratory) were used for all the experiments. EAE was induced in 7- to 8-week-old animals as described previously (Centonze et al., 2009; Mandolesi et al., 2012). Mice were injected subcutaneously at the flanks with 200 μg of myelin oligodendrocyte glycoprotein peptide 35-55 (MOG35-55) emulsion to induce EAE by active immunization. The emulsion was prepared under sterile conditions using MOG35-55 (>85% purity; Espikem) in 300 μl of complete Freund's adjuvant (CFA; Difco) containing Mycobacterium tuberculosis (8 mg/ml, strain H37Ra; Difco) and emulsified with PBS. All animals were injected with 500 ng of pertussis toxin (Sigma) intravenously on the day of immunization and 2 days later. Control animals received the same treatment as EAE mice without the immunogen MOG peptide, including complete CFA and pertussis toxin (referred to as hereafter as “CFA”). Animals were scored daily for clinical symptoms of EAE according to the following scale: 0 = no clinical signs, 1 = flaccid tail, 2 = hindlimb weakness, 3 = hindlimb paresis, 4 = complete bilateral hindlimb paralysis, and 5 = death due to EAE; intermediate clinical signs were scored by adding 0.5 (Aktas and Zipp, 2003; Centonze et al., 2009; Mandolesi et al., 2012). For each animal, the onset day was recorded as the day postimmunization (dpi) when it showed the first clinical manifestations (score >0). In all of the experiments performed at the symptomatic phase, the animal scores were ≥2, whereas in those performed at the presymptomatic phase, the score was zero. All efforts were made to minimize animal suffering and to reduce the number of mice used in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC).
Minipump implantation and continuous intracranial infusion.
One week before immunization, some mice were implanted with a minipump to allow continuous intracerebroventricular (icv) infusion of either vehicle (n = 16) or IL-1 receptor antagonist (IL-1ra, 150 ng/day; R&D Systems; n = 17) for 4 weeks (2 sets of immunization). Alzet osmotic minipumps (model 1004; Durect) connected via catheter tube to an intracranial cannula (Brain Infusion Kit 3; Alzet), delivered vehicle or IL-1ra into the right lateral ventricle at a continuous rate of 0.11 μl/h. The coordinates used for icv minipump implantation were as follows: anteroposterior= −0.4 mm from bregma; lateral= −1 mm; depth: 2.5 mm from the skull (Haji et al., 2012).
Electrophysiology.
Mice were killed by cervical dislocation under halothane anesthesia, and cerebellar parasagittal slices (210 μm) were prepared from fresh tissue blocks of the brain using a vibratome (Mandolesi et al., 2012). After 1 h of recovery time in a chamber containing oxygenated artificial cerebrospinal fluid (ACSF), single slices were transferred to a recording chamber and submerged in a continuously flowing ACSF at 2–3 ml/min gassed with 95% O2- 5% CO2. PCs could be easily identified using an Olympus BX50WI upright microscope with a 40× water-immersion objective combined with an infrared filter. Whole-cell patch-clamp recordings were made with borosilicate glass pipettes (1.8 mm outer diameter; 2–5.5 MΩ) in voltage-clamp mode at the holding potential of −70 mV. To detect spontaneous EPSCs (sEPSCs), the recording pipettes were filled with internal solution containing the following (in mm): 125 K+-gluconate, 10 NaCl, 1.0 CaCl2, 2.0 MgCl2, 0.5 BAPTA, 10 HEPES, 0.3 GTP, 3.0 Mg-ATP, adjusted to pH 7.3 with KOH. Bicuculline (10 μm) was added to the external solution to block GABAA-mediated transmission. To detect spontaneous GABAA-mediated IPSCs, intraelectrode solution was used containing the following (in mm): 110 CsCl, 30 K+-gluconate, 1.1 EGTA, 10 HEPES, 0.1 CaCl2, 4 Na-ATP, and 0.3 Na-GTP, adjusted to pH 7.3 with CsOH. MK-801 (30 μm; Tocris Bioscience) and CNQX (10 μm; Tocris) were added to the external solution to block NMDA and non-NMDA glutamate receptors, respectively. IL-1β (30 ng/ml; Rossi et al., 2012) and threo-β-benzyloxyaspartate (TFB-TBOA), a blocker (0.1 μm; Tsukada et al., 2005), were applied by dissolving them in the bathing ACSF. In some experiments, cerebellar slices were incubated in the presence of CD3+ cells or a drug in a total volume of 1 ml of oxygenated ACSF before the electrophysiological recordings. The incubation time and substance concentrations were as follows: CD3+ cells for 30–60 min (5000 cells/ml; Centonze et al., 2009), TNF-α 2 h and 30 min (0.6 μm; Musumeci et al., 2011), IL-1β 2 h and 10 min (30 ng/ml), and IL-1ra 1 h (100 ng/ml; Rossi et al., 2012).
Spontaneous synaptic events were stored using P-CLAMP 9 (Molecular Devices) and analyzed offline on a personal computer with Mini Analysis Version 5.1 software (Synaptosoft). The detection threshold of spontaneous and miniature excitatory events was set at twice the baseline noise. Positive events were confirmed by visual inspection for each experiment. Analysis was performed on spontaneous synaptic events recorded during a fixed time epoch (1–2 min) sampled every 2 or 3 min. Only cells that exhibited stable frequencies and amplitudes were taken into account. For sEPSC kinetic analysis, events with peak amplitude between 5 and 40 pA were grouped, aligned by half-rise time, and normalized by peak amplitude. In each cell, all events between 5 and 40 pA were averaged to obtain rise times, decay times, and half-widths. One to six cells per animal were recorded.
CD3+ cell isolation and culture.
Mice from both the EAE (n = 3; 19–21 dpi) and CFA (n = 3) groups were killed through cervical dislocation and the spleens were quickly removed and stored in sterile phosphate-buffered saline (PBS). After mechanical dissociation of the tissue, the cell suspension was passed through a 40 μm cell strainer (BD Biosciences) to remove cell debris and centrifuged. The cell suspension obtained was subjected to magnetic cell sorting separation (CD3 microbeads kit; Miltenyi Biotec) to obtain a pure lymphocyte population. For electrophysiological experiments, 5 × 103 pure cells were incubated with cerebellar slices for 30–60 min before electrophysiological recordings. To assess the level of IL-1β released by lymphocytes, freshly isolated pure CD3+ cells from both EAE (n = 3) and CFA (n = 3) mice were plated at 5 × 105 cells/well in RPMI 1640 medium (Invitrogen) containing 1% pen/strep (Invitrogen) and 10% FBS. Cells were maintained at 37°C for 1 or 24 h and then supernatants were collected, centrifuged, and stored in −80 until use.
IL-1β ELISA assay.
Both lymphocyte medium (1 and 24 h incubation) and cerebella from EAE (n = 5; 21 dpi) and CFA (n = 5) mice were assayed for IL-1β content through a direct IL-1β ELISA (Quantikine ELISA; R&D Systems) performed according to the manufacturer's instructions. Each sample was loaded in duplicate in the ELISA plate and actual cytokine concentrations were determined by reference to a standard curve generated using highly purified recombinant cytokine at various concentrations performed simultaneously with each assay. Each measurement was made in the linearity range of the standard curve (12.5–400 pg/ml). For lymphocytes, before loading into the ELISA plate, equal volumes (1.35 ml) of both 1 h and 24 h lymphocyte medium were defrosted and concentrated through centrifugal filters with a nominal molecular weight limit of 3 kDa (Amicon Ultra; Millipore). IL-1β level was expressed as picograms per milliliter. For cerebella, mice were killed through cervical dislocation and cerebella were quickly removed and frozen. Tissues were lysed in a 1:3 ratio of lysing buffer (Cell Lysis Buffer 2; R&D Systems). Protein content was determined according to the Bradford method. Equal amounts of proteins were loaded (1 mg per well per sample) into the ELISA plate. The amount of IL-1β in cerebellar extracts was expressed as picograms per milligram of total proteins.
RNA extraction and quantitative real-time PCR.
Cerebella of EAE (n = 6; 21 dpi) and CFA (n = 4) mice were dissected and total RNA was extracted according to the standard TRIzol protocol (Life Technologies). The quantity and the quality were analyzed on a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific) and by visual inspection of the agarose gel electrophoresis images. Next, 2 μg of total RNA was reversetranscribed using Superscript III Reverse Transcriptase (Life Technologies) according to the manufacturer's instructions. Then, 10 ng of cDNA was amplified with SensiMix SYBR Hi-Rox Kit (Bioline; Meridian Life Science) in triplicate using the Applied Biosystem 7900HT Fast Real Time PCR system. Relative quantification was performed using the ΔΔCT method. Actin and GAPDH were used as internal controls. Primer sequences were as follows: IL1β (NM_008361): 5′-GGACCTTCCAGGATGAGGACAT-3′ (sense), 5′-GCTCATGGAGAATATCACTTGTTGG-3′ (antisense); ACTIN (NM_007393): 5′-CCTAGCACCATGAAGATCAAGATCA-3′ (sense), 5′-AAGCCATGCCAATGTTGTCTCT-3′ (antisense); and GAPDH (NM_008084): 5′-GCCTGGAGAAACCTGCCAA-3′ (sense), 5′-TCCACCACCCTGTTGCTGTAG-3′ (antisense).
Cerebellar total extract preparation and Western blot.
For standard Western blot (WB), EAE (n = 4; 21 dpi) and CFA (n = 4) mice were killed through cervical dislocation and cerebella were quickly removed and frozen until use. For WB experiments performed on cerebellar slices, both the cutting procedure and IL-1β or IL-1ra incubation were performed as described in the “Electrophysiology” section, and then slices were removed from the recording chamber and frozen.
Tissues were homogenized in RIPA buffer plus protease inhibitor mixture (Sigma). After sonication, the homogenates were centrifuged at 16000 × g for 15 min and the supernatant was collected. Protein content was quantified according to the Bradford method. Next, 5–20 μg of proteins were loaded onto 10% polyacrylamide gel, blotted onto nitrocellulose membrane (Protran; Whatman), and then blocked for 1 h at room temperature (RT) by 5% nonfat dry milk in 0.1% Tween 20-PBS. All of the following incubations were performed in Tween 20-PBS. The following primary antibodies were used: mouse anti-β-actin (1:10000; Sigma) for 1 h at RT; rabbit anti-GLAST/EAAT1 (1:20000; Abcam) overnight at +4°C; mouse anti-glial fibrillary acidic protein (GFAP) (1:1000; Immunological Science) overnight at +4°C. Membrane was incubated with secondary HRP-conjugated IgG (1:10000; Millipore) in 5% milk for 1 h at RT. After washing, immunodetection was performed by ECL-Plus reagent (GE Healthcare) and membrane was exposed to film (Sigma). Densitometric analysis of protein levels was performed with ImageJ software (http://rsb.info.nih.gov/ij/). WB results were presented as data normalized to control CFA values.
Bromodeoxyuridine labeling.
To visualize proliferating cells in vivo, EAE (n = 3; 15 dpi) and CFA (n = 3) mice were intraperitoneally injected once a day for 4 consecutive days with the thymidine analog bromodeoxyuridine (BrdU) (100 mg/kg; Serotec). Forty-eight hours after the last injection, mice were anesthetized and transcardially perfused with PBS and 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer. The brains were removed and postfixed overnight in 4% PFA. Cerebellar sagittal serial sections (30 μm) were processed for immunohistochemistry.
Immunohistochemistry and confocal microscopy.
The immunofluorescence (IF) experiments were performed similarly to a method described previously (Mandolesi et al., 2012). Briefly, mice at least from 2 to 3 different immunization experiments were killed at the peak of the symptomatic phase (21 dpi, score ≥2), deeply anesthetized, and intracardially perfused with ice-cold 4% PFA. Brains were postfixed for 2 h and equilibrated with 30% sucrose for at least one night. Thirty-micrometer-thick sagittal sections were permeabilized in PBS with Triton X-100 0.25%. Electrophysiological slices (210 μm) from several experimental groups were fixed in 4% PFA for 24 h, washed in PBS, and permeabilized in Triton X-100 0.25% for 2–3 d to allow good penetration of antibodies. All of the following incubations were performed in Triton X-100 0.25%. Sections were preincubated with 10% normal donkey serum solution for 1 h at RT, incubated with the primary antibody overnight at +4°C, and then, after washing, incubated with secondary antibodies for 2 h at RT and rinsed. The following primary antibodies were used: rabbit anti GLAST/EAAT1 (1:10000; Abcam), rat anti CD3 (1:250 AbD Serotec), rabbit anti-calbindin (1:1000; Swant), mouse anti-calbindin (1:2000; Swant), goat anti-IL-1β (1:200; R&D Systems), rabbit anti-GFAP (1:500; DAKO), rabbit anti-Iba1 (1:750; Wako), and mouse anti TNF-α (1:500; Abcam). These were used in combination with the following secondary antibodies: Alexa Fluor-488 or Alexa Fluor-647-conjugated donkey anti-mouse (1:200; Invitrogen) and Alexa Fluor-488- or Cy3-conjugated donkey anti-rabbit or anti-rat (1:200; Jackson Laboratories). To exclude aspecific signals derived from the secondary antibody, immunostainings were also performed on slices incubated only with secondary antibodies (data not shown). The specificity of GLAST/EAAT1 and IL-1β antibodies was assessed by immunoblot analysis (data not shown): the positive control for GLAST/EAAT1 was a cerebellar lysate; negative controls were activated and nonactivated BV2 microglial cell lysates. For IL-1β, the specificity of the antibody was determined either by WB (the recombinant IL-1β protein) or by immunostaining on activated (positive control) and nonactivated (negative control) BV2 microglia cells. A positive signal was detectable only on activated cells (data not shown).
For BrdU immunolabeling, sagittal sections of the cerebellum were incubated in 2 n HCl at 37°C for 30 min for DNA denaturation, washed in 0.1 m borate buffer, pH 8.5, and then washed in PBS. Sections were then incubated with 10% normal donkey serum for 1 h at RT. Double immunostaining for BrdU-GFAP was performed using rat anti-BrdU (1:150; Abd Serotec) and rabbit anti-GFAP (1:500; DAKO) antibodies overnight at 4°C. Subsequently, the sections were exposed for 2 h at RT to an Alexa Fluor 488-conjugated goat anti-rat IgG (1:200) and Alexa Fluor 594 goat anti-rabbit (1:200).
Images from immunelabeled samples were acquired using a model LSM7 Zeiss confocal laser-scanner microscope with 20× (zoom 0.7×) or 63× oil objective (numerical aperture: 1.4; zoom: 0.5× or 1.5×, z-step: 1 μm). The images had a pixel resolution of 1024 × 1024. The confocal pinhole was kept at 1.0, the gain and the offset were lowered to prevent saturation in the brightest signals, and sequential scanning for each channel was performed. Z-stack images were acquired, z-projected, and exported in TIFF file format and adjusted for brightness and contrast as needed using ImageJ software. Smooth filters were used to reduce noise on stacks and z-projections. All qualitative analyses were performed on at least four images acquired from at least four serial sections per animal from at least two independent experiments.
Statistical analysis.
For each type of experiment and time point, at least five mice of each group were used unless otherwise specified. For electrophysiological data throughout the text, “n” refers to the number of cells unless otherwise specified. Data are presented as means ± SEM. The significance level was established at p < 0.05. Statistical analysis was performed using a paired or unpaired Student's t test. Multiple comparisons were analyzed by one-way ANOVA, followed by Tukey's HSD. Throughout the text, “df” refers to degrees of freedom. Differences between groups in clinical score analysis were tested by Mann–Whitney test and by log-rank test.
Results
sEPSC decay time and half-width are increased at PC synapses of EAE mice
To study the contribution of EAE to the modulation of glutamatergic synaptic transmission in the cerebellum, we made whole-cell voltage-clamp recordings from PCs and monitored spontaneous glutamatergic sEPSCs in MOG(35-55)-EAE mice compared with control animals (CFA; n = 10 mice). We killed EAE mice and relative controls at two different stages of the disease before clinical onset (presymptomatic: 7–9 dpi, n = 5 EAE mice) and during the acute phase of the disease (symptomatic phase: 20–25 dpi; n = 11 EAE mice). The mean values and SEM of sEPSC kinetics are reported for each experimental group in Table 1.
sEPSC kinetics of PC synapses recorded from CFA and EAE mice
PCs receive excitatory inputs from parallel fibers and climbing fibers and inhibitory inputs from basket and stellate interneurons; the isolation of sEPSCs was thus obtained by adding bicuculline to the bathing fluid. The sEPSCs were mediated by AMPA receptor activation because they were totally and reversibly blocked by CNQX. We found that in EAE mice, the mean sEPSC frequency (data not shown) was indistinguishable from that of control mice (0.85 ± 0.12 Hz; n = 25) at both the presymptomatic (0.66 ± 0.07 Hz; n = 12) and the symptomatic phase of the disease (0.73 ± 0.08 Hz; n = 18; one-way ANOVA, p = 0.32), as well as the sEPSC amplitude (one-way ANOVA, p = 37, df = 53; Table 1). On the contrary, we observed an increase of the duration of sEPSCs in symptomatic EAE mice in comparison to both CFA and EAE-presymptomatic mice (Fig. 1; Table 1). In particular, a slower decay phase (Fig. 1A) and half-width (Fig. 1B) accounted for increased sEPSC duration (one-way ANOVA, p = 0.0003 for decay and p = 0.0009 for half-width, df = 38). A trend of a slower sEPSC rise time turned out to be not statistically different (one-way ANOVA, p = 0.28; Fig. 1C).

The duration of spontaneous glutamatergic transmission is increased in the cerebellum of EAE mice. Whole-cell patch-clamp recordings from PCs show a slower decay phase (A) and half-width (B) of the sEPSCs in EAE symptomatic mice (21–25 dpi; score ≥2) relative to CFA (p < 0.001) and presymptomaptic (p < 0.05) mice, whereas the rise time was not significantly different (C). The kinetics of glutamate-mediated sEPSCs was normal in the presymptomatic phase (7–9 dpi) of EAE (p > 0.05 versus CFA). D, Electrophysiological traces are examples of sEPSCs recorded from PCs in control conditions (CFA) and in symptomatic EAE. Data are presented as means ± SEM. *p < 0.05, ***p < 0.001, one-way ANOVA.
The kinetic properties of the postsynaptic current at glutamatergic synapses is shaped by postsynaptic factors and by the rate of glutamate removal from the synaptic cleft by diffusion and uptake (Barbour et al., 1994; Takahashi et al., 1995, 1996). In particular, AMPA receptor desensitization could potentially modify the kinetic properties of the synaptic currents (Nakazawa et al., 1997; Koike et al., 2000). Here, we focused our attention on alterations of glutamate uptake and therefore GluT functioning because they have been extensively characterized at PC synapses (Takahashi et al., 1995; Takayasu et al., 2004, 2009). In addition, alteration of GluTs have been reported in MS patients and EAE rodents in association with glutamate excitotoxicity (Ohgoh et al., 2002; Vercellino et al., 2007; Mitosek-Szewczyk et al., 2008, Sulkowski et al., 2009).
Astrogliosis and downregulation of GLAST/EAAT1 in EAE cerebellum
The clearing of the majority of glutamate that floods out of the synaptic cleft immediately after transmitter release from parallel fibers and climbing fibers is mainly performed by the Bergmann glia (BG; Palay and Chan-Palay, 1974; Spacek, 1985) through a very high density of GluTs (Barbour et al., 1994; Chaudhry et al., 1995; Lehre and Danbolt, 1998). Among the five GluTs expressed in the cerebellum, the GLAST/EAAT1 is the main transporter expressed specifically by BG (Rothstein et al., 1994; Takayasu et al., 2004, 2009).
Therefore, to gain insight into the cellular and molecular mechanisms responsible for the electrophysiological alterations observed in EAE cerebellum, we first investigated the level of astroglia activation (Fig. 2A–B′,G–G′) and then the expression of GLAST/EAAT1 (Fig. 2C–F′′,G–G′′) in EAE and CFA mice by means of both IF and WB analysis. Using the antibody anti-GFAP, a marker for astroglia, we observed a prominent astroglia activation that was evident in the molecular layer (ML), where BG processes associate closely with PC synaptic contacts, and in the granular layer (GL) of the cerebellar cortex (Fig. 2A,B, red signal). Accordingly, using WB, we observed an upregulation of ∼2-fold of GFAP in EAE cerebellar extract relative to CFA control mice (GFAP/actin ratio: CFA 1 ± 0.073 vs EAE 1.97 ± 0.151, unpaired t test: p = 0.009; n = 4 mice each group; df = 6; Fig. 2G,G′). In addition, we verified the proliferative state of astroglia in EAE cerebellum by BrdU incorporation in EAE and CFA mice. After 4 d of BrdU injection, we killed the mice at 21 dpi and performed immunolabeling for BrdU and GFAP. In the cerebellar cortex of CFA mice, we did not detect any BrdU-positive nuclei (Fig. 2A, green), whereas in EAE mice, GFAP-positive cells (Fig. 2B,B′, red) showed BrdU-positive nuclei (Fig. 2B,B′, green), indicating proliferation of these cells during the symptomatic phase of the disease. The GFAP-negative cells bearing BrdU-positive nuclei in the ML of EAE mice are likely microglia/macrophage cells, which are strongly activated in EAE mice (Mandolesi et al., 2012).

Astrogliosis and downregulation of GLAST/EAAT1 in EAE cerebellum. A, B′, Confocal images of GFAP immunostaining (red) of cerebellar sagittal slices show morphological changes typical of activated astroglia in EAE mice (B; 21–25 dpi; score ≥2) relative to CFA control mice (A). BrdU-positive nuclei (B,B′, green) were evident all over the cerebellar cortex (ML, GL, Purkinje cell layer [PCl]) of EAE mice but not in CFA mice (A, green), indicating a prominent cellular proliferation in the symptomatic mice. (B′), High magnification of the double fluorescence staining shows BrdU-positive nuclei surrounded by GFAP labeling (arrowheads), indicating a strong astroglia proliferation in EAE cerebellum. Immunofluorescence analysis of GLAST/EAAT1 expression in CFA (C,D′′) and EAE (E,F′′) cerebella. To distinguish the cerebellar layers we stained calbindin (Calb) to visualize PCl and ML (green), and DAPI+ cell nuclei (gray) in the GL. A specific labeling of the anti-GLAST antibody (red) characterizes the ML of both CFA (C′,D, high magnification) and EAE cerebellar slices (E′,F, high magnification), reflecting localization of the protein in the processes associated closely with PC spines and dendrites (Calb, green; D′,D′′,F′,F′′). GLAST/EAAT1 expression was less prominent in the ML of EAE symptomatic mice (E′,F). G, G′′, WB analysis of the expression level of GFAP and of GLAST/EAAT1. The quantitative analysis reported in the graph shows an ∼2-fold upregulation of GFAP in EAE cerebellar extract relative to CFA control mice (G,G′). On the contrary, GLAST/EAAT1 was less abundant in EAE compared with CFA (G,G′). Quantification of GLAST/EAAT1 relative to GFAP protein levels emphasizes the differences between CFA and EAE cerebella (G,G′′). Data are presented as means ± SEM and are normalized to the CFA group. Scale bars, 50 μm in A,B,C,C′,E,E′; 10 μm in B′,D,D′′,F,F′′. *p < 0.05, **p < 0.01, unpaired t test.
We next verified the expression of GLAST/EAAT1 in the two experimental groups (Fig. 2C–G″). Using IF experiments, we observed a strong and specific labeling of GLAST/EAAT1 in the ML of CFA mice (Fig. 2C,C′, red), reflecting the expression of the protein in the processes associated closely with PC spines and dendrites (Fig. 2 D,D′′, green). In EAE symptomatic mice, the labeling appeared less prominent and variable (Fig. 2E,F′′). Using WB analysis, we quantified the GLAST/EAAT1 expression, which turned out to be less abundant in EAE mice (n = 4) compared with CFA mice (n = 4) cerebellar extracts (GLAST/actin ratio: CFA 1 ± 0.08 vs EAE 0.686 ± 0.072; unpaired t test: p = 0.044; df = 6; Fig. 2G,G′). This observation indicates that, despite an increase in the content of GFAP and therefore astroglia, GLAST/EAAT1 expression was reduced. Quantification of GLAST/EAAT1 relative to GFAP protein levels indeed emphasizes the differences between CFA and EAE mice (Fig. 2G′′; GLAST/GFAP ratio: CFA 1 ± 0.118 vs EAE 0.345 ± 0.030; unpaired t test: p = 0.0001; df = 6), suggesting that the prolonged sEPSC at PC synapses of EAE mice could be mediated in part by a downregulation of GLAST/EAAT1 correlated with astrogliosis.
Alteration of GLAST/EAAT1 functioning in EAE cerebellum
To clarify whether GLAST/EAAT1 was involved in the alterations of the glutamatergic transmission in EAE cerebellum, we addressed the effect of its inhibitor, TFB-TBOA (Takayasu et al., 2004). We first recorded the sEPSCs from cerebellar control slices (CFA) before and after incubation of TFB-TBOA (Tsukada et al., 2005; Bozzo and Chatton, 2009). After 5–10 min of incubation, we observed a statistically significant increase of the decay time (12.52 ± 1.18 ms; percentage of predrug: 124.59 ± 9.09%; paired t test: p = 0.02; n = 14) and half-width (11.58 ± 0.90 ms; percentage of predrug: 116.99 ± 8.79%; paired t test: p = 0.04; n = 13; Fig. 3A,B) of the EPSCs compared with the predrug basal level. Such an effect, as expected with a GLAST/EAAT1 inhibitor (Takayasu et al., 2004), seemed to resemble the alterations of glutamatergic synapses observed in symptomatic EAE mice. On the contrary, recording from cerebellar EAE slices in the presence of TFB-TBOA did not reveal any effect on the EPSC duration (decay time after drug: 14.78 ± 1.42 ms, percentage of predrug 103.46 ± 7. 9%; half-width after drug: 13.69 ± 0.87 ms, percentage of predrug 101.20 ± 4.93%; paired t test: p = 0.09 and p = 0.13 respectively; n = 10; Fig. 3A), suggesting an apparent ineffectiveness of the treatment on kinetic already compromised. These experiments support the hypothesis that GLAST/EAAT1 functioning and glutamate uptake were affected in EAE cerebellum.

The GLAST/EAAT1 inhibitor TBOA affects sEPSCs in normal cerebellar slices but not in EAE. A, Bath application of TBOA (10 min) in cerebellar slices of normal mice significantly increased the decay time and half-width of PC sEPSCs, as expected. In EAE symptomatic mice (21–25 dpi; score ≥2), the effect was completely abolished, suggesting a compromised glutamate uptake dependent on GLAST/EAAT1. B, Examples of electrophysiological traces (sEPSCs) recorded from PCs of CFA mice before and after TBOA application. Data are presented as the means ± SEM; *p < 0.05, paired t test.
Rapid and direct effect of IL-1β on glutamatergic synaptic transmission through GLAST/EAAT1 downregulation
It has been shown that IL-1β and other inflammatory mediators can precipitate elevations in extracellular glutamate and/or exacerbate excitotoxic insults to nervous tissue (Casamenti et al., 1999; Rothwell and Luheshi, 2000; Fogal et al., 2007) through GluT regulation. In addition, we have observed previously that TNF-α and IL-1β are responsible for significant changing of glutamate transmission in EAE striatum by altering the duration and the frequency of the sEPSCs, respectively (Centonze et al., 2009; Haji et al., 2012; Rossi et al., 2012). Such effects were evident both in vitro after brief incubation of the cytokine on normal slices (Centonze et al., 2009; Rossi et al., 2012) and in vivo after a prolonged infusion of an antagonist in EAE mice (Haji et al., 2012).
Therefore, we first investigated whether each cytokine incubated on cerebellar slices from untreated mice could replicate the glutamatergic alterations observed in the EAE cerebellum. We first tested TNF-α using two different incubation times and the treatments did not affect the sEPSCs recorded from PCs (Fig. 4A, Table 2). Both decay time and half-width were not significantly different after 30 minutes (n = 7; unpaired t test: p = 0.77 and p = 0.89 respectively; Table 2) or 2 h of incubation (n = 8; unpaired t test: p = 0.44 and p = 0.15, respectively, Fig. 4A, Table 2) compared with control conditions (n = 10). On the contrary, we observed an effect in the presence of IL-1β, which induced after 10 min of incubation an increase of synaptic events for both decay time (percentage of predrug: 153.22 ± 18.5%; paired t test: p = 0.01; n = 7) and half-width (percentage of predrug: 149.27 ± 19.2%; paired t test: p = 0.03; n = 7; Fig. 3B) relative to the baseline (Table 2). The same effect was observed after 2 h of IL-1β incubation (n = 13; unpaired t test: p = 0.04 for both parameters Table 2).

IL-1β affects cerebellar glutamatergic transmission by inhibiting GLAST/EAAT1 expression. A, B, Graphs showing that TNFα incubation (2 h) of control cerebellar slices did not affect sEPSC duration (A); conversely, application of IL-1β (10 min) in control slices enhanced decay time and half-width of sEPSCs (B), mimicking the synaptic alterations obtained in symptomatic EAE mice. C, The protein level of GLAST/EAAT1 was quantified by WB analysis in cerebellar slices after IL-1β incubation (10 min) and in control conditions (ACSF). The treatment induced a significant reduction of GLAST/EAAT1 expression. WB data were normalized to β-actin. D, E, Graphs showing that incubation of IL-1ra was able to rescue the EAE-mediated effect on sEPSCs. D, Left: Decay time was faster in EAE-IL-1ra slices compared with EAE-untreated slices (p < 0.01) and indistinguishable from CFA-IL-1ra slices. D, Right: The half-width was also faster but did not recover completely (p > 0.05 vs EAE and CFA-IL-1ra). Dotted lines represent the mean values obtained in CFA-untreated slices. E, Examples of sEPSC events recorded from PCs in symptomatic EAE slices, after IL-1ra incubation in EAE slices and in control conditions (CFA-IL-1ra). Data are presented as means ± SEM. Unpaired t test in A and paired t test in B, C; one-way ANOVA in D: *p < 0.05, **p < 0.01, ***p < 0.001.
Effect of TNF-α and IL-1β on sEPSCs recorded from untreated mice
GluTs are highly sensitive to neuroinflammatory signaling and undergo downregulation in response to either IL-1β or TNF-α (Wang et al., 2003; Prow and Irani, 2008). Therefore, we performed WB analysis on protein extracts from cerebellar slices incubated with or without IL-1β (10 min, n = 9 slices for each group) to correlate the electrophysiological result to a potential GLAST/EAAT1 downregulation. As shown in Figure 4C, we observed a prominent reduction of GLAST/EAAT1 level after incubation of IL-1β compared with control slices (GLAST/actin ratio: CFA 1 ± 0.103 vs EAE 0.451 ± 0.031; unpaired t test p = 6,1 E-05; df = 16).
Such rapid and prominent effect of IL-1βon GLAST/EAAT1 expression prompted us to perform a complementary set of experiments by incubating EAE slices with IL-1ra to block the activity of endogenous IL-1β. Interestingly, we observed that, despite the presence of a prominent inflammatory reaction and a brief blocking of IL-1β signaling (30–60 min), the kinetic defects induced by EAE were partially rescued (Fig. 4D,E). The decay time was faster in EAE-IL-1ra slices (10.70 ± 0.75 ms, n = 18) compared with EAE untreated slices (14.61 ± 0.84 ms; n = 18), reaching values indistinguishable from CFA control slices (CFA-IL-1ra: 8.60 ± 0.50, n = 12; one-way ANOVA, p < 0,01; df = 61; Fig. 4D,E). The effect on the half-width was less remarkable: the values of EAE-IL-1ra (9.99 ± 0.73 ms) were not significantly different from EAE (12.11 ± 0.60 ms) or from CFA control slices (CFA- IL-1ra = 8.00 ± 0.41; one-way ANOVA, p > 0.05 for both groups; df = 61; Fig. 4D,E). IL-1ra did not affect half-width or decay time of the sEPSCs recorded from CFA slices compared with CFA untreated slices (one-way ANOVA, p > 0.05, df = 61 for both comparisons; Fig. 4D,E).
WB analysis did not reveal a significant modulation of GLAST/EAAT1 expression in EAE slices treated or not with IL-1ra (data not shown), suggesting that IL-1β could have a GLAST/EAAT1-independent synaptic effect. However, based on the present observation of a modulation of GLAST/EAAT1 by IL-1β in normal conditions, it might be possible that WB analysis was not as sensitive at detecting small variations of a protein as electrophysiology in detecting partial alterations of protein functioning in EAE conditions.
These results demonstrate that activation of the IL-1β signaling quickly downregulates GLAST/EAAT1 expression and that this effect likely contributes to inducing the prolonged sEPSCs observed in EAE PCs.
IL-1β is expressed by CD3+ lymphocytes and microglia/macrophage in EAE cerebellum
These data, together with our previous observations showing an effect of IL-1β on cerebellar GABAergic transmission (Mandolesi et al., 2012), prompted us to investigate the expression of the cytokine in the cerebellum of EAE mice at the peak of the disease. CFA and EAE cerebella in the symptomatic phase of the disease were first processed to perform quantification of both mRNA and protein levels of IL-1β by means of real-time PCR and ELISA assay, respectively (Fig. 5A). As shown in Figure 5A, we detected ∼7 times more IL-1β mRNA in EAE cerebella compared with the CFA group (n = 4 CFA, n = 6 EAE; unpaired t test: p = 0.019, df = 8). Accordingly, EAE cerebellum showed an abundant expression of IL-1β relative to the CFA group (n = 5 each group; CFA 4.33 ± 0.30 pg/mg, EAE 12.07 ± 2.55 pg/mg; p = 0.016, df = 8, Fig. 5A). The next step was to identify which cells of the immune system released IL-1β during the symptomatic phase (21 dpi) of the disease. During this phase, the cerebellum is characterized by extensive lesions in the white matter (WM), where CD3+ lymphocytes reside, and by a strong microglia/macrophage activation not only in the WM and GL, but also in the ML, where PC dendrites receive most of their inputs (Mandolesi et al., 2012). Therefore, we performed double IF experiments and confocal imaging on cerebellar sagittal slices derived from EAE and CFA mice to verify the expression of IL-1β in microglia/macrophage, astroglia, and CD3+ lymphocytes, all potential sources of proinflammatory cytokines.

Quantification of IL-1β expression in EAE cerebellum and its expression in microglia/macrophage. A, Quantification of both mRNA and protein of IL-1β at the symptomatic phase of the disease. Left: Upregulation of IL-1β mRNA in EAE versus CFA cerebella. A quantitative real-time PCR was performed using actin as internal control. Right: Upregulation of IL-1β expression in EAE cerebellar extract relative to CFA control mice evaluated by ELISA assay. B–E, Double immunostaining of cerebellar sagittal sections showing expression of IL-1β (red) in Iba1-positive microglia/macrophage cells (green) that are activated in EAE mice (B,B′′,D,D′′, 21 dpi; score ≥2), but not in CFA mice (C,E). B,B′′, In the WM and GL of EAE, a colocalization between the two makers (B′, merge image: IL-1β-Iba1) is evident. B′′ is an high magnification of the white box in B′. D, E, In the ML, activated microglial cells seem not to express detectable levels of IL-1β (D,D′′). D′′ is an high magnification of the white box in D′. These results show that IL-1β is expressed by subsets of microglia/macrophages populations of cells. Arrowheads in B′ and D′ show IL-1β-positive cells that are Iba1-negative and are likely lymphocytes (Fig. 7). Data are presented as the means ± SEM; unpaired t test in A: *p < 0.05, **p < 0.01. Scale bars in B,B′,C,D,D′,E: 25 μm; in B′′,D′′: 10 μm.
Colocalization analysis with Iba1, a specific marker of microglia/macrophages (Fig. 5B–E, green), revealed the expression of IL-1β (Fig. 5B–E, red) in these cell types, but not in all of them. In particular, we observed a differential expression between the WM/GL and ML of EAE cerebellum; in the former, most of the cells were IL-1β-positive (Fig. 5B,B′′, EAE; Fig. 5C, CFA), whereas in the latter, the expression was restricted to Iba1-negative cells (Fig. 5D,D′′, EAE; Fig. 5E, CFA). Microglia/macrophage cells in both WM/GL and ML were positive for TNF-α staining (data not shown). A diffuse and puntacted labeling of IL-1β was observed in the ML of both EAE and CFA mice, suggesting an endogenous expression of the cytokine. By performing the same colocalization analysis with a specific astroglia marker (GFAP; Fig. 6, green), a detectable expression of the cytokine (red) in these cell types was not observed in the WM/GL (Fig. 6A,A′, EAE; Fig. 6B,B′, CFA) or ML (Fig. 6C,C′, EAE; Fig. 6D,D′, CFA). Finally, we investigated whether lymphocytes infiltrating the WM could represent a potential source of IL-1β. As shown in Figure 7, the cytokine was indeed highly expressed in CD3+ cells at the level of WM lesion (Fig. 7A,A′′′, EAE; Fig. 7B,B′, CFA). Interestingly, we observed in the ML numerous lymphocytes that were IL-1β-positive (Fig. 7C,D, EAE; CFA not shown).

IL-1β is not detectable in activated astroglia in EAE cerebellum. Double immunostaining of cerebellar sagittal sections showing absence of colocalization signal between IL-1β (A,A′,C,C′, red) and GFAP (A′,C′, green), which is highly expressed by activated astroglia in the WM, GL (A,A′) and ML (C,C′) of EAE cerebellum (21 dpi; score ≥2) compared with CFA mice (B,B′,D,D′). Arrowheads in C, C′ show IL-1β-positive cells in the ML that are likely lymphocytes (Fig. 7). Scale bars, 25 μm.

IL-1β is highly expressed in CD3+ lymphocytes infiltrating EAE cerebellum. Double immunostaining of cerebellar sagittal sections showing in the WM/GL (A,A′′′, in gray DAPI nuclei) and ML (C,C′′) of EAE mice (21 dpi; score ≥2) a strong colocalization signal (A′′,A′′′ in WM/GL and C′′ in ML) between IL-1β (red, A in WM/GL and C in ML) and CD3+ infiltrating lymphocytes. D, High magnification of the white boxes in C,C′-C″ showing CD3+ lymphocytes as substantial source of IL-1β (C, arrowheads) in the ML of EAE cerebellum. No signal was present in CFA cerebellum except for DAPI staining of the cell nuclei (B,B′ GL and ML not shown). Scale bars in A–C′′: 25 μm; in D: 10 μm.
These results indicate that a prominent source of IL-1β in the cerebellum of EAE mice is represented by CD3+ lymphocytes in both the WM and gray matter. Microglia and macrophages contributed also to IL-1β production even if the expression was restricted to those microglia/macrophage cells localized in the WM/GL.
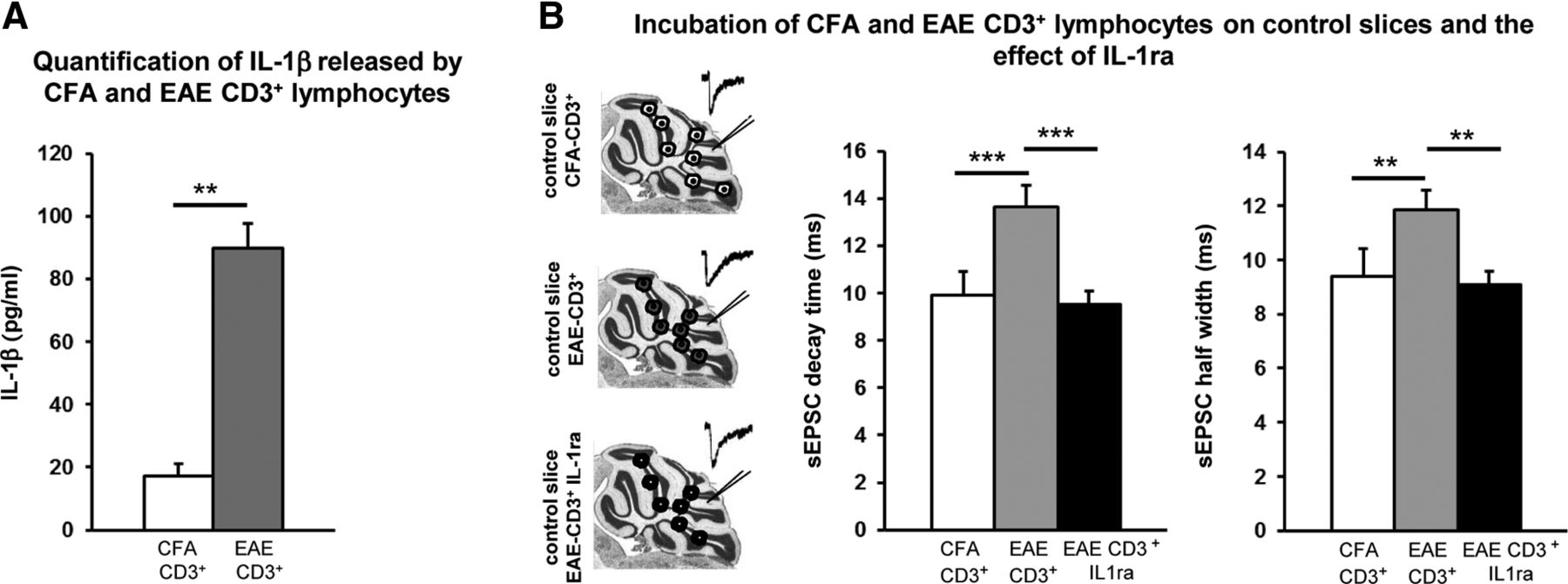
Incubation of EAE-CD3+ lymphocytes on control slices reproduces the IL-1β synaptic defects observed in EAE mice
Due to the characteristic expression of IL-1β in the CD3+ lymphocytes infiltrating the WM and ML of EAE cerebellum, we wanted to study their potential direct effect on cerebellar synaptic alterations. CD3+ lymphocytes were isolated from the spleens of EAE (20–30 dpi, EAE-CD3+) and CFA (CFA-CD3+) mice and put in culture to determine the amount of IL-1β released in the medium by ELISA assay. As shown in Figure 8A, the amount of IL-1β released by EAE-CD3+ lymphocytes after 24 h (n = 3 spleens; 89.86 ± 7.75 pg/ml) was 5 times that released by the CFA-CD3+cells (n = 3 spleens; 17.21 ± 3.87; p < 0.01; df = 4). Such a difference was appreciable already after 1 h of incubation, but was not statistically significant due to the sensitivity of the system (n = 3 spleens each group; CFA 0.03 ± 0.03 and EAE 6,75 ± 3,29; unpaired t test, p > 0.05). The next step was to perform electrophysiological experiments on cerebellar slices derived from untreated animals in the presence of EAE-CD3+ and CFA-CD3+ lymphocytes (Fig. 8B). The latter did not produce any effect on the sEPSCs (decay time: 9.89 ± 0.41 ms; half-width: 9.41 ± 0.32 ms; n = 17), whereas in slices incubated with EAE-CD3+ cells, we observed a relevant alteration of sEPSC kinetics (decay time: 13.62 ± 0.91 ms; half-width ms: 11.85 ± 0.73; n = 14; one-way ANOVA p < 0.0001, number of group = 4, df = 61 for both parameters; Fig. 8B), reminiscent of the defects seen in EAE (one-way ANOVA: CD3+-EAE vs EAE and CD3+-CFA vs CFA p > 0.05 for decay and half-width; CD3+-EAE vs CFA p < 0.01, df = 61 for both decay and half-width). These data suggested that EAE-infiltrating T cells induce changes in glutamate transmission in EAE cerebellum that possibly involve IL-1β signaling.

Incubation of EAE-CD3+ lymphocytes on normal slices reproduces the IL-1β synaptic defects observed in EAE mice. A, Protein quantification by ELISA assay of IL-1β released by CD3+ lymphocytes isolated from the spleens of EAE (EAE-CD3+) and CFA (CFA-CD3+) mice. Histogram shows an upregulation of IL-1β released by EAE-lymphocytes (24 h) relative to the protein released by CFA lymphocytes. B, The diagram on the left is a schematic representation of the experimental procedure; cerebellar slices from normal mice were incubated for 1 h with CFA-CD3+ or EAE-CD3+ lymphocytes by layering them on the top of the slices. After the incubation, electrophysiological recordings were performed (insets represent examples of sEPSCs). Circles represent CD3+ lymphocytes from CFA (white) or EAE (gray) mice and EAE-CD3+ lymphocytes in the presence of the IL-1ra (black circle). Graphs on the right show a slower decay time and half-width of PC sEPSCs in control slices incubated with EAE-CD3+ compared with CFA-CD3+ control conditions. EAE lymphocytes failed to affect sEPSCs after the blockade of IL-1β, suggesting that these inflammatory cells induce changes of glutamate transmission in EAE cerebellum through IL-1β signaling. Data are presented as the means ± SEM; unpaired t test in A; one-way ANOVA in B: **p < 0.01, ***p < 0.001.
To test this hypothesis, we performed recordings in slices incubated with both EAE-CD3+ cells and IL-1ra (30–60 min) to block the activity of released IL-1β. In this set of experiments, sEPSC kinetic properties (decay time: 9.51 ± 0.59 ms; half-width: 9.10 ± 0.48 ms; n = 25; one-way ANOVA: p = 0.61, number of group = 5, df = 86; Fig. 8B) were indistinguishable from those recorded in control conditions, whereas they were statistically different from the EAE and EAE-CD3+ groups (one-way ANOVA: p < 0.001, df = 86 for both comparisons).
These results suggest that during the symptomatic phase of the disease, infiltrating CD3+ T cells might contribute to EAE synaptic alterations in mice through the release of IL-1β.
In vivo modulation of IL-1β signaling ameliorates the clinical score and prevents inflammation-dependent synaptic alterations
This evidence and our previous work (Mandolesi et al., 2012) suggest that IL-1β is a key mediator of the abnormal spontaneous glutamatergic and GABAergic transmission observed in EAE cerebellum. To further support these observations, we investigated whether in vivo modulation of IL-1β could interfere with EAE alterations. To address this issue, icv IL-1ra or vehicle were delivered by osmotic minipump for 4 weeks in both EAE and CFA mice. One week after implantation, mice were immunized. We followed the score until the peak of the disease and then killed the mice to study inflammatory and electrophysiological status of the cerebellum.
Similarly to our previous observations (Furlan et al., 2007), intracerebroventricular delivery of IL-1ra (150 ng/d) ameliorated the EAE course. In particular, the disease severity was significantly reduced during the symptomatic phase (19–22 dpi; Mann–Whitney test, p < 0.05; Fig. 9A) in IL-1ra-treated mice (n = 17) compared with EAE-vehicle (n = 16). Regarding the clinical symptoms, the number of mice without motor deficits (score 0) was higher in IL-1ra-treated mice compared with the EAE vehicle, although it did not reach a significant difference (log-rank test, p = 0.10; Fig. 9B). Such observations indicate an important and broadened effect of the icv treatment.

Clinical course in EAE mice receiving IL-1ra or vehicle by intracerebroventricular delivery. A, Time course of EAE clinical score (mean ± SEM) in mice treated with IL-1ra (n = 17, black) or vehicle (n = 16, gray) until the acute phase of the disease. The severity of EAE was milder in EAE-IL-1ra-treated mice compared with EAE-vehicle-treated mice (19–22 dpi, Mann–Whitney test, *p < 0.05). B, Survival curve for disease onset in EAE mice preventively treated with intracerebroventricular IL-1ra (black line) or vehicle (gray line). The incidence of mice with a score of zero was higher in EAE-Il-1ra-treated mice relative to vehicle mice, although it did not reach a significant difference (p = 0.10, log-rank test), suggesting a protective effect of IL-1ra delivered by intracerebroventricular infusion.
We next explored the impact of IL-1ra delivery on EAE cerebellum by first recording sEPSCs from PCs of cerebellar slices derived from the four experimental groups (EAE-vehicle, EAE-IL-1ra, CFA-vehicle, CFA-IL-1ra). The IL-1ra-treated mice with score ≥2 were also recorded. As shown in Figure 10, both sEPSC decay time and half-width (Fig. 10A,B) were significantly reduced in EAE-IL-1ra mice (decay time = 10.84 ± 0.66 ms; half-width = 9.83 ± 0.62 ms; n = 19) compared with EAE-vehicle animals (decay time = 13.51 ± 0.64 ms; half-width = 11.61 ± 0.48 ms; n = 15; one-way ANOVA: p < 0.01 for decay time and p < 0.05 for half-width; df = 61) and not significantly different from those recorded in CFA-IL-1ra animals (decay time = 9.37 ± 0.27 ms; half-width = 8.83 ± 0.22 ms; n = 28; p > 0.05). The control group CFA-IL-1ra and CFA-vehicle mice were pooled together because their values were not significantly different. In support of such results, we performed WB analysis to correlate the electrophysiological observations with the degree of astrogliosis and of GLAST/EAAT1 expression in the recorded slices. We observed a modest astrogliosis in slices derived from EAE-IL-1ra-treated mice (n = 4, GFAP/actin ratio: 2.22 ± 0.47) relative to EAE-vehicle (n = 5, GFAP/actin ratio: 2.9 ± 0.31, p > 0.05) that was not significantly different from the CFA group (n = 5; GFAP/actin ratio: 1 ± 0.13; EAE-IL-1ra vs CFA-vehicle p > 0.05; CFA-vehicle vs EAE-vehicle p < 0.01; one-way ANOVA p = 0.0075; df = 12; Fig. 10C,C′). Such a trend was likely due to variability in the effectiveness of the treatment in a group of mice also including those with a score ≥2. Interestingly, despite a mild astrogliosis, the expression of GLAST/EAAT1 normalized to the GFAP level reached almost normal values after in vivo treatment of IL-1ra (EAE-IL-1ra, n = 4, GLAST/GFAP ratio: 0.894 ± 0.107; CFA-vehicle, n = 5, GLAST/GFAP ratio: 1 ± 0.061; EAE-IL-1ra vs CFA-vehicle, p > 0,05, Fig. 10C,C′′). Accordingly, GLAST/EAAT1 expression in the EAE-vehicle group (n = 5, GLAST/GFAP ratio: 0.457 ± 0.081) was significantly different from both the CFA-vehicle and EAE-IL-1ra groups (EAE-vehicle vs EAE-IL-1ra, p < 0.05; CFA-vehicle vs EAE-vehicle, p < 0.01; ANOVA p = 0.0012; df = 12; Fig. 10C,C′′). These results demonstrate that IL-1ra in vivo treatment was able to recover GLAST/EAAT1 functioning by acting on GLAST/EAAT1 expression and by attenuating astrogliosis.

In vivo modulation of IL-1β signaling prevents the synaptic, molecular, and inflammatory events mediated by EAE. A, Histograms showing that pharmacological inhibition of IL-1β signaling by means of IL-1ra icv treatment prevented the effect of EAE on glutamatergic sEPSC kinetic properties. B, Examples of electrophysiological traces (sEPSCs) recorded from PCs in the different experimental conditions. C, C′′, WB analysis of the expression levels of GFAP and GLAST/EAAT1 in the cerebella of CFA-vehicle, EAE-vehicle, and EAE IL-1ra mice. The quantitative analysis reported in the graph shows a mild reduction of GFAP in EAE IL-1ra cerebellar extract (C, C′, normalized to actin) but a significant recovery of GLAST/EAAT1 (C′′, normalized to GFAP) relative to EAE mice (C′′). D–I, Double immunostaining of cerebellar sagittal sections showing expression of Iba1-positive microglia/macrophage cells (green) and DAPI staining of the cell nuclei. In EAE IL-1ra slices (F,F′) microglia proliferation was less prominent (F) compared with EAE mice (D) and occasionally almost absent (F′), similar to CFA (E). G–I, Images showing the morphology of Iba-1-positive cells, which in EAE IL-1ra (I) resembled that of control mice (H). J, Whole-cell patch-clamp recordings from PCs show a slower frequency of the spontaneous GABAergic transmission (sIPSCs) in EAE mice relative to CFA mice. The strong reduction of the frequency was efficiently recovered in EAE-IL1ra mice. Data are presented as the means ± SEM; one-way ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars in D–F′: 200 μm; in G–I: 10 μm.
The next step was to verify the effect of the IL-1ra treatment on microglia activation on the three experimental groups. As shown in Figure 10D–F′, by means of an immunostaining for Iba-1 we observed a modest microglia activation in EAE IL-1ra cerebellar slices (n = 4–5 slices for each group; F-, F′). In some areas, microglia proliferation was less prominent (Fig. 10F′) relative to EAE mice (Fig. 10D) and the morphology of Iba-1 positive cells resembled that of CFA mice (Fig. 10G–I). Accordingly, quantification of Iba-1 by WB analysis confirmed a trend of a reduced expression of the protein in EAE-IL-1ra (n = 4, Iba1/actin ratio: 5.43 ± 3.84) relative to EAE-vehicle (n = 5, Iba1/actin ratio: 11.7 ± 2.29; CFA n = 5, Iba1/actin ratio: 1 ± 0.375) which did not reach statistical significance in comparison with both CAF and EAE groups (ANOVA p = 0.049; EAE-IL-1ra vs CFA and EAE, p > 0.05; EAE vs CFA, p < 0.05; df = 12; data not shown).
Due to such a relevant effect of the IL-1ra treatment, we also explored the spontaneous GABAergic transmission that we previously demonstrated to be impaired in EAE cerebellum mice (Mandolesi et al., 2012). By recording sIPSCs from PCs of all experimental groups at the peak of the disease, we observed that the strong reduction of the frequency observed in the EAE-vehicle mice (n = 14; 0.88 ± 0.183 Hz) was efficiently recovered in the EAE-IL-1ra group (n = 17; 3.79 ± 0.925 Hz) compared with the CFA-control group (n = 14; 5.36 ± 1 Hz; one-way ANOVA, p = 0.0021; CFA-vehicle vs EAE-vehicle, p < 0.01; CFA-vehicle vs EAE-IL-1ra p > 0.05; EAE vs EAE-IL-1ra, p < 0.05; df = 44; Fig. 10J). This result strongly suggests that the IL-1ra treatment was effective at halting synaptic neurodegenerative events that characterize the EAE cerebellum (Mandolesi et al., 2012).
These observations clearly demonstrate that IL-1β triggers a cascade of deleterious molecular and inflammatory events that cause important synaptic alterations and likely neurodegenerative damage in the EAE brain.
Discussion
The present investigation provides a molecular link between inflammation and altered glutamate transmission in the cerebellum of EAE mice. First, we observed an enhancement of glutamate transmission at PC synapses caused by a reduced expression and functioning of the GLAST/EAAT1, the most abundant GluT expressed by BG. Second, we demonstrated that IL-1β, highly expressed in EAE cerebellum and released by infiltrating lymphocytes, is one of the molecular players responsible for such synaptic alteration having an effect on GLAST/EAAT1 expression. Finally, intracerebroventricular inhibition of IL-1β signaling in EAE mice was able to ameliorate inflammatory reaction, electrophysiological response, and clinical disability, indicating a pivotal role of IL-1β in EAE disease.
A growing body of evidence shows that glutamate excitotoxicity is evoked by altered glutamate homeostasis and contributes to neuronal and oligodendroglia pathology in MS (Werner et al., 2001; Pitt et al., 2003; Srinivasan et al., 2005). In addition to an elevated concentration of glutamate in cerebrospinal fluid of patients with acute MS (Stover et al., 1997) and in acute myelin lesions (Srinivasan et al., 2005), an altered expression of specific GluTs in MS and EAE has been reported (Ohgoh et al., 2002; Pitt et al., 2003; Vallejo-Illarramendi et al., 2006; Mitosek-Szewczyk et al., 2008). However, a clear correlation between glutamate excitotoxicity and GluT alterations has not yet been drawn. In MS patients, an upregulation of GluTs has been interpreted as a protective mechanism to prevent cellular damage caused by enhanced glutamate level (Vallejo-Illarramendi et al., 2006). Conversely, a reduced expression of GluTs by oligodendroglia in WM of MS patients has also been found (Pitt et al., 2003; Korn et al., 2005). In the EAE rodent model, a dramatic decrease in GluT expression, including GLAST/EAAT1, was observed in rat spinal cord, forebrain, and cerebellum (Ohgoh et al., 2002; Mitosek-Szewczyk et al., 2008). However, the decreased GluT expression was accompanied by upregulation of their relative mRNAs, especially in forebrain and cerebellum, suggesting a dysregulation of posttranscriptional processes in pathological conditions.
It has been shown that downregulation of GluTs may be mediated by proinflammatory cytokines such as TNF-α (Pitt et al., 2003) or IL-1β (Ye and Sontheimer, 1996; Hu et al., 2000; Takahashi et al., 2003; Sama et al., 2008), as demonstrated by experiments on oligodendrocytes, astrocyte cultures, and spinal cord tissue (Prow and Irani, 2008). Despite this in vitro evidence (Ye and Sontheimer 1996; Szymocha et al., 2000; Wang et al., 2003; Korn et al., 2005), direct proof that proinflammatory cytokines contribute to MS and EAE defects in glutamate uptake and the development of excitotoxicity in vivo has thus far not been provided.
Here, we found that sEPSCs at PC synapses were enhanced in EAE cerebellum. Both posttranscriptional modifications of the GluR subunits and glutamate uptake alterations can potentially shape the sEPSC kinetics. We highlighted the crucial contribution played by GLAST/EAAT1 expressed by BG (Takayasu et al., 2009) in dysregulating glutamate homeostasis during EAE. We indeed observed that incubation of TBOA on EAE cerebellar slices did not affect the sEPSC kinetics that occurred in control conditions, indicating a compromised functioning of GLAST/EAAT1. Interestingly, we observed that in EAE cerebellum, the expression of GLAST/EAAT1 was reduced and inversely correlated with the level of astrogliosis/inflammation and therefore of IL-1β. We indeed provided electrophysiological evidence of an IL-1β-dependent enhancement of the sEPSC at PC synapses by means of both in vitro and in vivo experiments. A recovery of the synaptic alterations was observed, not only in cerebellar slices of EAE mice treated for 4 weeks with icv IL-1ra, but also after a brief and direct incubation of IL-1ra on EAE slices. Although the latter effect was, as expected, not complete, conversely IL-1β incubation on normal slices could mimic EAE alterations, strongly supporting a direct role of Il-1β on glutamate uptake.
Furthermore, consistent with such electrophysiological observations, we demonstrated a direct effect of IL-1β on GLAST/EAAT1 expression. In particular, in the IL-1ra in vivo experiments, we observed normal expression levels of GLAST/EAAT1 likely due to either an attenuation of the inflammatory reaction or to a direct effect of the IL-1β signaling inhibition on BG. Such evidence was corroborated by complementary in vitro experiments in which a brief (10 min) incubation of IL-1β on normal slices was able to induce a rapid and dramatic reduction of GLAST/EAAT1 expression, thus mimicking the alterations observed in EAE cerebellum.
In vitro evidence suggests that the mRNA half-life of GLAST/EAAT1 and of GLT-1, both abundant proteins critical for limiting excitotoxicity, is quite long (24 h; Zelenaia and Robinson, 2000). However, it is becoming clear that in pathological conditions such as ischemia, traumatic brain injury, and EAE, a faster decrease in protein expression or mismatch with the mRNA level respond to further regulatory processes that depend on posttranslational disturbances or on injury/inflammatory-dependent regulation factors (Torp et al., 1995; Rao et al., 1998, 2001; Mitosek-Szewczyk et al., 2008). Accordingly, rapid GluT regulation may occur in response to many factors, including protein kinase C levels, glutamate release, and levels of growth factors and cytokines (Duan et al., 1999; Munir et al., 2000; Sims et al., 2000; Pitt et al., 2003; González et al., 2005). Here, such a rapid effect of IL-1β on protein expression, although it is somewhat speculative at this time, could be dependent either on fast inhibition of the synthesis or degradation of the protein localized in perisynaptic processes (Grosche et al., 2002). Synaptic plasticity events are indeed controlled by local and fast mechanisms of protein regulation (Steward and Schuman, 2003; Villareal et al., 2007) and likely involve BG proximal processes surrounding PC excitatory synapses (Palay and Chan-Palay, 1974) and containing GLAST/EAAT1 mRNA (Schmitt et al., 1997). Which pathway could be activated by IL-1β signaling and be involved in such fast regulation of GLAST/EAAT1 is not known. Activation of the NF-κB transcription factor (Mercurio and Manning, 1999) occurs by phosphorylation, multi-ubiquitination, and degradation of IκBα, a regulatory protein of NF-κB (Ghosh et al., 1998); nascent IκBα begins to degrade 5 min after IL-1β treatment and disappears completely after 15 min (Uehara et al., 1999).
As mentioned above, in addition to investigating the acute effect of IL-1β, we wanted to verify whether a prolonged and preventive effect of IL-1ra was sufficient to ensure adequate synaptic homeostasis in EAE cerebellum. In previous work, we indeed observed an impairment of cerebellar GABAergic transmission that was likely dependent on IL-1β signaling and was correlated with synaptopathy and degeneration of the main cerebellar interneurons impinging on PCs (Mandolesi et al., 2012). A proper balance between excitatory and inhibitory signaling is crucial for a correct cerebellar function. In particular, the duration of the excitatory and inhibitory action on the PCs is critical to ensuring not only the dynamic aspect of the cerebellar control, but also learning and memory processes (Scelfo et al., 2008). Although past efforts have already shown that peripheral or central administration of IL-1ra had a therapeutic effect on EAE (Jacobs et al., 1991; Martin and Near, 1995; Badovinac et al., 1998; Wiemann et al., 1998; Pollak et al., 2003; Furlan et al., 2007), no electrophysiological studies had been conducted so far. Here, we observed the effectiveness of the icv treatment on both glutamatergic and GABAergic transmission in correlation with a modest inflammatory reaction. Such observations strengthen the idea that IL-1β, by acting directly on the neuronal/glial system, can induce important alterations, potentially causing detrimental excitotoxic damage.
There have been reports that IL-1β is elevated in the brain of MS patients (Baranzini et al., 2000) and is localized in the lesion sites (Woodroofe and Cuzner, 1993; Brosnan et al., 1995). In the EAE model, it seems to be a prominent microglia product (Bauer et al., 1993). Here, we characterized IL-1β expression in various inflammatory cells (including astroglia) localized in the WM lesions and in the gray matter of the EAE cerebellum. We observed that this cytokine was produced by largely segregated subsets of activated microglia/macrophages. Evidence of a differential expression of IL-1β and TNF-α in different subsets of innate immune cells has been reported after ischemic stroke in mice (Clausen et al., 2008). Surprisingly, in the ML, where microglia/macrophage activity may provide the insidious and persistent inflammation that mediates the progressive synaptopathy and loss of CNS tissue in MS and EAE, IL-1β was not detectable; on the contrary, the main source seems to be CD3+ infiltrating lymphocytes. Accordingly, we observed that lymphocytes isolated from EAE spleens and releasing high levels of IL-1β could replicate the electrophysiological changing observed in EAE.
In conclusion, the cytokine-dependent synaptic alterations observed in EAE cerebellum and also occurring in other brain regions such as striatum (Centonze et al., 2009; Rossi et al., 2011, 2012) and hippocampus (Nisticò et al., 2013) seem to represent clear hallmarks of the EAE model, playing a crucial role in EAE pathology and likely in MS. Remarkably, recent evidence in MS patients shows that, during an acute MS attack, inflammation increases brain IL-1β signaling, which enhances in turn glutamate-mediated synaptic excitability and neurotoxicity (Rossi et al., 2012). These data provide valuable therapeutic information because they elucidate the molecular mechanisms involved in enhanced neuronal excitability that characterizes the EAE brain and represents a further cause of excitotoxic damage in MS.
Footnotes
This work was supported by the Italian National Ministero della Salute (D.C.), Fondazione Italiana Sclerosi Multipla (D.F.), and the European Community AXREGEN: Axonal Regeneration, Plasticity, and Stem Cells (Grant #21–4003 to P.S.). We thank Vladimiro Batocchi and Massimo Tolu for excellent technical assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Diego Centonze, Clinica Neurologica, Dipartimento di Medicina dei Sistemi, Università Tor Vergata, Via Montpellier 1, 00133 Rome, Italy. centonze{at}uniroma2.it





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}