

Abstract
Excitatory synaptic activity governs excitotoxicity and modulates the distribution of NMDA receptors (NMDARs) among synaptic and extrasynaptic sites of central neurons. We investigated whether NMDAR localization was functionally linked to excitotoxicity by perturbing F-actin, a cytoskeletal protein that participates in targeting synaptic NMDARs in dendritic spines. Depolymerizing F-actin did not affect NMDA-evoked whole-cell currents. However, the number of dendritic NMDAR clusters and the NMDAR-mediated component of miniature spontaneous EPSCs were reduced, whereas the number of AMPA receptor clusters and AMPA receptor-mediated component of EPSCs was unchanged. This selective perturbation of synaptically activated NMDARs had no effect on neuronal death or the accumulation of 45Ca2+evoked by applying exogenous NMDA or l-glutamate, which reach both synaptic and extrasynaptic receptors. However, it increased survival and decreased 45Ca2+accumulation in neurons exposed to oxygen glucose deprivation, which causes excitotoxicity by glutamate release at synapses. Thus, synaptically and extrasynaptically activated NMDARs are equally capable of excitotoxicity. However, their relative contributions vary with the location of extracellular excitotoxin accumulation, a factor governed by the mechanism of extracellular neurotransmitter accumulation, not the synaptic activation of NMDARs.
- NMDA receptors
- actin filament
- latrunculin A
- cytochalasin D
- excitotoxicity
- oxygen glucose deprivation
- cortical neurons
Excitotoxic neuronal damage is the consequence of excessive stimulation of postsynaptic receptors byl-glutamate, the major excitatory neurotransmitter in the mammalian CNS (Rothman and Olney, 1986). NMDA receptors (NMDARs), a subtype of ionotropic glutamate receptors, are key participants in excitotoxicity owing to their ubiquitous distribution in central neurons and to their high permeability to Ca2+ ions (Choi, 1988; Tymianski, 1996). NMDAR overactivity is thought to trigger excitotoxicity by permitting the accumulation of intracellular Ca2+ions to levels that exceed the regulatory capacity of the cell (Hartley et al., 1993; Eimerl and Schramm, 1994). However, it has recently become apparent that the quantity of Ca2+ that accumulates in the cell is not the sole determinant of Ca-mediated neuronal damage. Whereas neurons are rapidly damaged when loaded with Ca2+ions through NMDA receptors, a similar Ca2+ load incurred through alternative influx pathways, such as voltage-sensitive Ca2+ channels, is innocuous (Tymianski et al., 1993; Sattler et al., 1998). Consequently, NMDARs must possess as yet uncharacterized properties that permit them to trigger Ca-mediated neuronal damage more effectively than other Ca2+ sources. We have previously hypothesized that such properties may include a unique association of NMDARs with rate-limiting substrates or enzymes that trigger neurotoxicity, or a unique compartmentalization of NMDARs with subcellular sites essential to cell survival (Tymianski et al., 1993;Tymianski, 1996; Sattler et al., 1998).
NMDARs and other glutamate receptor subtypes are clustered in dendritic spines (Craig et al., 1994; Kornau et al., 1995; Rao and Craig, 1997;O'Brien et al., 1998a), which serve as integrative units in synaptic circuitry and participate in synaptic plasticity (for review, see Harris and Kater, 1994; Yuste and Denk, 1995). The accumulation of glutamate receptor clusters in spines is governed by excitatory synaptic activity, and increases when activity is suppressed (Rao and Craig, 1997; O'Brien et al., 1998b). Conversely, excitotoxicity produces a rapid and profound loss of dendritic spines in cultured neurons (Halpain et al., 1998), mimicking the loss in dendritic spine synapses in neurological conditions including epilepsy, schizophrenia, aging, and prion protein-related diseases (Jeffrey et al., 1997; Jiang et al., 1998; Garey et al., 1998). This suggests that receptor localization at synapses might be critical to excitotoxicity, and that dendritic spines constitute the subcellular sites that govern neuronal vulnerability to excitotoxicity. However, NMDARs are also found at extrasynaptic sites (Rosenmund et al., 1995; Clark et al., 1997; Rao and Craig, 1997), raising the possibility that the synaptic and extrasynaptic subsets of NMDA receptors play different physiological and pathological roles in the cell.
The localization of NMDARs to synaptic sites is achieved through interactions between their intracellular domains and cytoskeletal elements (Wyszynski et al., 1997; Allison et al., 1998; Ehlers et al., 1998) and with cytoplasmically located submembrane proteins in the postsynaptic density (Gomperts, 1996; Ponting et al., 1997). F-actin, a cytoskeletal protein that is concentrated in dendritic spines (Matus et al., 1982; Kaech et al., 1997) may be responsible for targeting NMDARs to synaptic sites, because treatment with actin-depolymerizing agents selectively reduces the numbers of synaptic NMDAR clusters without affecting nonsynaptic clusters (Allison et al., 1998). F-actin is bound to NR1 and NR2B subunits via the actin-binding protein α-actinin-2 (Wyszynski et al., 1997). Ca influx through NMDARs causes a depolymerization of F-actin (Bonfoco et al., 1996; Shorte, 1997) and inhibits its interaction with NMDARs through the competitive inhibition of α-actinin binding by Ca–calmodulin (Wyszynski et al. 1997, 1998;Zhang et al., 1998). This induces a Ca-dependent inactivation of NMDA currents (Rosenmund and Westbrook, 1993; Krupp et al., 1999) that may protect neurons against excitotoxicity (Furukawa et al., 1995, 1997;Furukawa and Mattson, 1995).
We used the actin-depolymerizing agents latrunculin-A and cytochalasin-D to disrupt F-actin in cultured cortical and hippocampal neurons. Cytochalasin-D binds to the (+) end of the actin filament, preventing its growth and resulting in an abundance of short actin filaments (Cooper, 1987). Latrunculin A, a compound isolated from the Red Sea sponge Negombata (Spector et al., 1983), affects actin polymerization by the formation of a 1:1 molar complex with G-actin, causing net actin depolymerization (Spector et al., 1989). The cytochalasins have been used to examine the relationship between the actin cytoskeleton and NMDA currents (Rosenmund and Westbrook, 1993), calcium influx (Shorte, 1997), and excitotoxicity (Furukawa et al., 1995). Latrunculin-A was recently used by Allison et al. (1998) to study the role of actin in anchoring NMDARs to synaptic sites. Here we show that depolymerizing dendritic F-actin selectively reduces the activity of synaptically activated NMDARs, allowing us to functionally separate the effects of synaptic and extrasynaptic receptors on excitotoxicity and neuronal Ca2+homeostasis. Our data indicate that NMDARs can trigger excitotoxicity both within and outside of synapses, and that the mechanism of receptor activation, not receptor location, determines excitotoxic consequences.
MATERIALS AND METHODS
Tissue culture
Cortical neuronal cultures. Mixed cortical cell cultures containing both neurons and glia were prepared from embryonic Swiss mice at 15 d of gestation as previously described (Sattler et al., 1997), with minor modifications from Choi (1987). In brief, cerebral cortices from 10–12 embryos were incubated for 10–12 min in 0.05% trypsin in EDTA, dissociated by trituration, and plated on poly-l-ornithine-coated 24 well plates (Corning, Corning, NY) or glass coverslips at a density of 0.43 × 106 cells per well or 0.9 × 106 cells per coverslip. Plating medium consisted of Eagle's minimum essential medium (MEM; Earle's salt) supplemented with 10% heat-inactivated horse serum (ICN Biochemicals, Costa Mesa, CA), and (in mm) 2 glutamine, 25 glucose, and 26 bicarbonate. The cultures were maintained at 37°C in a humidified 5% CO2 atmosphere. After 3–5 d in vitro, growth of non-neuronal cells was halted by a 24–48 hr exposure to 10 μm FDU solution (5 μm uridine and 5 μm(+)-5-fluor-2′-deoxyuridine). This produces cultures in which >85% of the cells were neurons, based on immunohistochemical staining for glial fibrillary-associated protein (exclusive to astrocytes), and for the NMDAR1 subunit (data not shown). The cultures were used for experiments after 12–14 d in vitro. In all experiments, the culture medium also contained 100 μmdl-2-amino-5-phosphonovaleric acid (APV) from day 2 until the cultures were used. This chronic APV treatment causes the number of synaptic NMDAR clusters to increase (Rao and Craig, 1997). Low-density cortical cultures (see Fig. 4) were grown as above, except that they were plated at a density of 0.06 × 106 cells per coverslip and switched to serum-free media at 24 hr [Neurobasal with B27 supplement (Life Technologies, Gaithersburg, MD)]. They were fed every other day with fresh serum-free media and used after 12 d in vitro.
Low-density hippocampal neuronal cultures. These were prepared as previously described (Banker and Cowan, 1977; Goslin and Banker, 1991). In brief, hippocampi were dissected from 18 d mouse embryos and dissociated using trypsin and by trituration through a Pasteur pipette. The neurons were plated on coverslips coated with poly-l-lysine in MEM with 10% horse serum at an approximate density of 3000 cells/cm2. After the neurons had attached to the substrate, they were transferred to a dish containing a glial monolayer and maintained for up to 3 weeks in serum-free MEM with N2 supplements.
Drugs and solutions
The control solution contained (in mm): 121 NaCl, 5 KCl, 20 d-glucose, 10 HEPES acid, 7 HEPES-Na salt, 3 NaHCO3, 1 Na-pyruvate, 1.8 CaCl2, and 0.01 glycine, adjusted to pH 7.4 with NaOH. Oxygen glucose deprivation (OGD) was performed in a glucose-free bicarbonate-buffered solution containing (in mm): 121 NaCl, 5 KCl, 1 Na-pyruvate, 1.8 CaCl2, 25 NaHCO3, and 0.01 glycine, adjusted to pH 7.4 with HCl.
Stock solutions of nimodipine (Miles, Elkhart, IN), 6-cyano-7-nitroquinoxaline (CNQX; Research Biochemicals, Natick, MA), cytochalasin-D, and Latrunculin-A (Molecular Probes, Eugene, OR) were prepared in DMSO and kept at −20°C until used. APV and MK-801 stocks were prepared in distilled water and also stored at −20°C until used.
Stock solutions of NMDA were prepared daily in control solution. Propidium iodide (PI; 1 mg/ml stock; Molecular Probes) was prepared in control solution and dissolved to a final concentration of 50 μg/ml. This concentration of PI produced no observable effects on cell morphology or survival, as demonstrated by the low cell mortality in all control groups. All compounds were always diluted to their final concentrations in the experimental solution. Nimodipine, CNQX, and MK-801 in all experiments were applied at final concentrations of 2, 10, and 10 μm, respectively (Sattler et al., 1998). All solutions were sterile-filtered before use. Unless otherwise noted above, all chemicals were obtained from Sigma (St. Louis, MO).
Determination of cell death
Cell death was determined by serial quantitative measurements of PI fluorescence using a multiwell plate fluorescence scanner (Cytofluor II; PerSeptive Biosytems, Framingham, MA) as described previously (Sattler et al., 1997, 1998). In brief, the culture medium in each tissue culture well was replaced with control solution containing 50 μg/ml PI, and a baseline fluorescence reading was taken. Sequential readings were then taken at appropriate intervals over the 24 hr after the experimental manipulations. The fraction of dead cells in each culture at a given time was calculated as: fraction dead = (Ft −Fo)/FNMDA, where Ft = PI fluorescence at timet, Fo = initial PI fluorescence at time 0, and FNMDA = background subtracted PI fluorescence of identical cultures from the same dissection and plating, 24 hr after a 60 min exposure to 1 mm NMDA at 37°C. Based on manual observations at the time of validation of this technique, this NMDA exposure routinely produced near complete neuronal death in each culture but had no effect on surrounding glia (Bruno et al., 1994; David et al., 1996;Sattler et al., 1997). Adding Triton X-100 (0.1%) to cultures treated in this manner produced an additional 10–15% increase in PI fluorescence caused by permeabilization of non-neuronal cell membranes, consistent with a 10–15% glial component in the cultures.
Experimental protocols
Treatment with actin-perturbing agents. Cultures were treated with varying concentrations of cytochalasin-D or latrunculin-A for 12 hr by applying the drugs from concentrated DMSO stocks into the medium. The actin-depolymerizing agents were not present during subsequent toxicity assays,45Ca2+accumulation measurements, or electrophysiological recordings (below).
Excitotoxicity assay. In most experiments, the cultures were washed one time in control solution, and then subjected to a challenge (usually 60 min) with a range of concentrations of NMDA orl-glutamate. Nimodipine and CNQX, antagonists of voltage-sensitive Ca2+ channels and of AMPA/kainate glutamate receptors, respectively, were present to restrict Ca2+ loading to NMDARs (Sattler et al., 1998). After the insult, the cultures were washed two times and maintained in control solution containing MK-801, an NMDAR antagonist, to ensure that toxicity recorded at later times was triggered by the initial insult rather than by delayed depolarization and/or EAA release. Cell survival was measured as described above at 24 hr. All experiments were performed at 24°C.
OGD. After taking a baseline PI fluorescence reading (see above) the cultures were transferred to an anaerobic chamber containing a 5% CO2, 10% H2, and 85% N2 (<0.2% O2) atmosphere (Goldberg and Choi, 1993). They were washed three times with 500 μl of deoxygenated glucose-free bicarbonate solution in the presence of nimodipine and CNQX as above and maintained anoxic for 2 hr at 37°C. OGD was terminated by washing the cultures with oxygenated glucose-containing (20 mm) bicarbonate solution containing all three antagonists (nimodipine, CNQX, and MK-801). The cultures were maintained for a further 22 hr at 37°C in a humidified 5% CO2 atmosphere. PI fluorescence readings were taken at 24 hr.
Measurements of Ca2+ load
Neuronal Ca2+ loading was determined using measurements of45Ca2+accumulation in the cells as described previously (Sattler et al., 1998). In brief, cultures were washed one time with control solution and then challenged for 60 min with NMDA, l-glutamate or 2 hr OGD. The solution contained nimodipine, CNQX, and45CaCl2 (0.85 μCi/well; 0.5–0.6 mm). Thereafter, the cells were rinsed four times in cold control solution, lysed with 0.2% SDS and counted in a scintillation counter. The counts were normalized to the45Ca2+ counts obtained from sister cultures exposed for 1 hr to 1 mm NMDA in the presence of nimodipine and CNQX at room temperature, which selectively destroyed all neurons in the culture. The result of this normalization is thus the fraction of the maximal45Ca2+ load obtainable in the neurons under study. This normalization method is similar to that used in previous reports (Hartley et al., 1993), was validated for this purpose (Sattler et al., 1998, 1999), and was selected over normalizing the45Ca2+reading to the total protein content in the cultures because the latter measurement reflects both the neuronal and glial compartments.
Immunostaining
F-actin labeling. Cultures were fixed in warm 4% paraformaldehyde and 4% sucrose in PBS for 15 min and were permeabilized with 0.25% Triton X-100 for 5 min. They were blocked at 37°C with 10% bovine serum albumin (BSA) for 30 min and incubated at 37°C with rhodamine phalloidin (Molecular Probes; 1:4000) in 3% BSA for 2 hr.
Glutamate receptors. After treatment with F-actin depolymerizing agents, the cells were fixed first with 4% paraformaldehyde in PBS + 4% sucrose for 20 min at 4°C. Cultures were subsequently fixed in ice cold 100% methanol for 10 min at 4°C. After repeated washing, they were permeabilized with 0.02% Triton X-100 in PBS for 10 min at 4°C and blocked in 10% goat serum in PBS for 45 min at room temperature (RT), followed by incubation with primary antibodies in 10% goat serum in PBS for 3 hr at RT or 37°C. The antibodies were mouse monoclonal anti-NR1 (generous gift from R. L. Huganir, Johns Hopkins University, Baltimore, MD; 1:50 dilution) or rabbit polyclonal anti-GluR1 (Upstate Biotechnology, Lake Placid, NY; 1:3000 dilution). The cultures were then washed and incubated with secondary antibody for 1.5 hr at RT using goat anti-rabbit (Amersham, Arlington Heights, IL) or anti-mouse (Jackson ImmunoResearch, West Grove, PA) IgG conjugated to CyTM3. Immunostaining was visualized with a laser-scanning confocal microscope (Bio-Rad, Hercules, CA; MRC 1000) through a 60× oil immersion lens.
Glutamate receptor cluster counts. All cultures within a given series of experiments were imaged using identical confocal settings established in pilot experiments to cover the widest possible range of gray level intensities. Images were then intensity thresholded at 150 gray levels, and all areas smaller than 2 pixels were removed. The remaining clusters were counted and expressed as clusters per unit of dendrite length. This approach was validated in pilot experiments against manual counts obtained by two independent blinded observers from original (unthresholded) confocal images. Cluster counts in primary and secondary dendrites (arising from the cell soma and from the primary dendrites, respectively) were done separately. Examples of this approach to cluster counting are provided in Figure 4.
Immunoblotting
Cultures were treated with sham (0.1% DMSO) or 1 μm latrunculin-A for 12 hr. Tissue was then harvested and pooled from two cultures per lane. Immunoblotting was performed exactly as described in Jones et al. (1997). The blotted proteins were probed using a rabbit polyclonal anti-NR1 IgG (Upstate Biotechnology, 1:1000 dilution) or rabbit polyclonal anti-GluR1 (Upstate Biotechnology; 1:3000 dilution). Secondary antibodies were donkey antibody to rabbit Ig conjugated to horseradish peroxidase (Amersham).
DiI staining/assessment of cell morphology
Neuronal morphology after treatment with actin-depolymerizing agents was examined by labeling the cultures with the membrane tracer DiI (Park et al., 1996; Faddis et al., 1997; Sattler et al., 1998). Briefly, they were fixed in 4% paraformaldehyde with 0.025% glutaraldehyde in PBS for 30 min at RT. DiI was used from stock solution (0.5 mg/ml absolute ethanol) diluted 1:100 in PBS. Fixed cultures were incubated in DiI suspension for 70 min at RT, washed with PBS, and imaged on an inverted laser-scanning confocal microscope (MRC 1000; Bio-Rad; Nikon lens CF UV-F; 40×; NA, 1.3). This method stains a small (<3%) fraction of neurons in each culture, allowing detailed visualization of membrane and dendritic spine morphology.
Electrophysiology
NMDA currents. Whole-cell patch-clamp recordings in the cultured neurons were performed and analyzed as described in Xiong et al. (1997). During each experiment, a voltage step of −10 mV was applied from the holding potential, and the cell capacitance was calculated by integrating the capacitative transient. The extracellular solution contained (in mm): 140 NaCl, 5.4 KCl, 1.3 CaCl2, 25 HEPES, 33 glucose, 0.01 glycine, and 0.001 tetrodotoxin, pH 7.3–7.4, 320–335 mOsm. A multibarrel perfusion system was used to rapidly exchange NMDA-containing solutions. The pipette solution contained (in mm): 140 CsF, 35 CsOH, 10 HEPES, 11 EGTA, 2 tetraethylammonium chloride, 1 CaCl2, 4 MgATP, pH 7.3 at 300 mOsm.
Miniature EPSCs. Spontaneous miniature EPSCs (mEPSCs) were recorded as described previously (Lu et al., 1999). The extracellular solution contained (in μm): 0.5 tetrodotoxin, 1 strychnine, 10 bicuculline methiodide, and 1 glycine. mEPSCs were filtered at 2 kHz and stored on tape before off-line acquisition and analysis with an event detection program (SCAN, Strathclyde software; courtesy of Dr. J. Dempster). For event detection, the trigger level was set at approximately three times of the baseline noise. False events were eliminated by subsequent inspection of the raw data. In general, >80 events were acquired for averaging.
After establishing the whole-cell configuration and stable baseline recordings, mEPSCs with both AMPA and NMDA components (without APV and Mg2+ added to extracellular solution) were recorded for ∼5 min to acquire sufficient numbers of events. The perfusion solution was then changed to one containing 20 mmAPV plus 2 μm Mg2+ to record AMPA-only mEPSCs. The NMDA-only mEPSCs were obtained by subtracting AMPA-only mEPSCs from the total mEPSCs.
All recordings of NMDA currents and of EPSCs were obtained from cultures taken from at least two platings. An approximately equal number of neurons were used for each experimental group from each plating.
Data analysis
Data were analyzed by ANOVA, with a post hocStudent's t test using the Bonferroni correction for multiple comparisons. All means are presented with their SEs.
RESULTS
Effects of actin-depolymerizing agents on neuronal F-actin and morphology
We studied neuronal actin using rhodamine phalloidin, a fluorescent actin-stabilizing compound used to stain, visualize, and quantify F-actin (Cooper, 1987; Allison et al., 1998; Shorte, 1997). Rhodamine phalloidin labels F-actin in cell bodies and processes, and is highly fluorescent in dendritic spines where F-actin is concentrated. The cultures were treated for 12 hr with the depolymerizing agents, stained, and viewed with a confocal microscope.
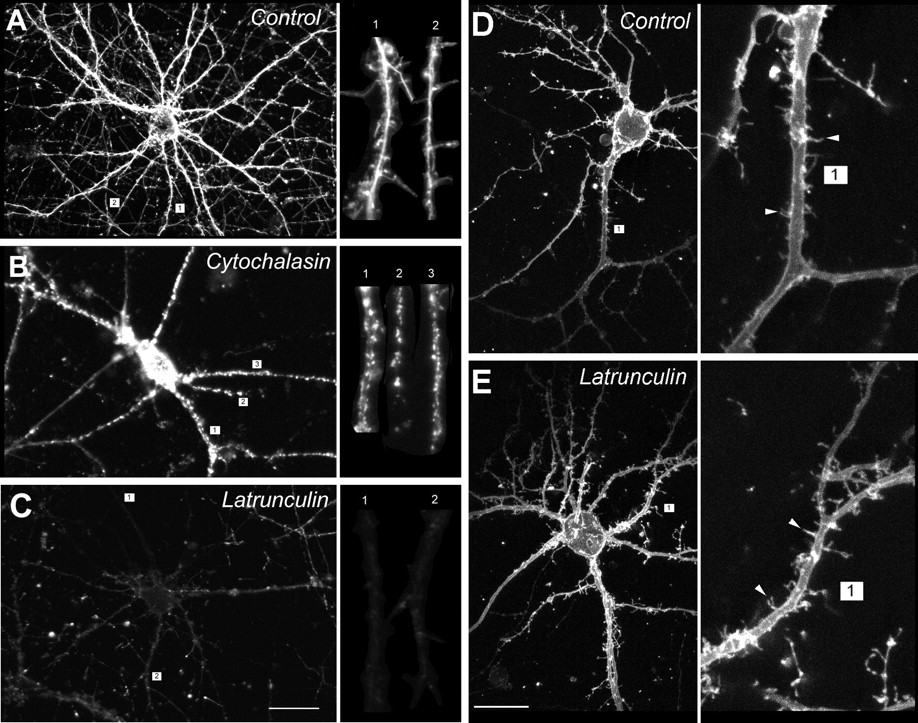
In hippocampal neurons, F-actin staining appeared homogeneous in dendritic shafts and spines of sham-treated cells (Fig.1A). This was changed by treatment with cytochalasin-D (10 μm) to a punctate pattern suggestive of actin breakdown, but not complete destruction (Fig. 1B, insert). Treatment for 12 hr with latrunculin-A completely dissolved F-actin in the cell, including actin in dendritic shafts and spines (Fig. 1C). In cortical neurons, the cell type used in the remaining experiments in this paper, treatment with cytochalasin-D (1–10 μm) caused actin to agglomerate (Fig.2A) without attenuating the overall rhodamine phalloidin fluorescence (Fig.2B). By contrast, latrunculin-A treatment (0.1–5 μm) reduced rhodamine phalloidin staining and fluorescence by 70% (Fig. 2A,B). The lack of a quantitative effect of cytochalasin-D on rhodamine phalloidin fluorescence (Fig. 2B) is consistent with the anticipated action of the cytochalasins which, while causing F-actin to break down into shorter filaments, still bind phalloidin (Cooper, 1987). The more dramatic effect of latrunculin-A suggests a more potent actin depolymerization by this agent.

Effects of depolymerizing agents on actin filaments and morphology of dendrites and spines of cultured hippocampal (A–C) and cortical (D, E) neurons. A–C, The cultures were treated with 10 μm cytochalasin-D or 1 μm latrunculin-A for 12 hr, stained with rhodamine–phalloidin, and imaged with the confocal microscope using identical settings. A, Homogeneous actin staining in control neuron treated with sham (0.1% DMSO) solution. Inset, Homogeneous staining of dendritic shafts and spines. B, Cytochalasin-D induces agglomeration of F-actin throughout the entire neuron.Inset, Maintenance of actin clumps in dendritic spines.C, Latrunculin-A dissolves F-actin throughout the cell.Inset, Uniform loss of rhodamine–phalloidin staining in dendritic shafts and spines. Scale bar: C, 20 μm.Insets of A–C show magnified views of the indicated dendrites (numbers). D, E,Effect of latrunculin-A on the morphology of cortical neurons in high-density cultures as assessed with confocal imaging of DiI staining. D, Control neuron treated with sham (0.1% DMSO) solution. E, Neuron treated with 1 μm latrunculin-A for 12 hr. Insets ofD and E, Higher power views of the numbered dendrites and representative spines (arrowheads). Scale bar: E, 20 μm. Images in A–E are representative of neurons inN > 6 cultures per condition.

Differential effects of depolymerizing agents on actin in cultured cortical neurons. A,Rhodamine–phalloidin fluorescence images of neurons treated with 0.1% DMSO (sham), cytochalasin-D (10 μm), or latrunculin-A (1 μm), obtained using identical confocal excitation, emission, and gain settings. Treatment with cytochalasin-D caused actin to agglomerate into clumps (arrow), whereas latrunculin-A reduced actin staining throughout the cells. Scale bar, 30 μm. B, Quantification of rhodamine–phalloidin fluorescence in cultured cortical neurons treated with actin-depolymerizing agents. The cultures were grown in 24-well plates, treated with the agents at the indicated concentrations for 12 hr, and imaged with the confocal microscope using identical settings for each well. An averaged, background-subtracted fluorescence intensity was derived from 15–20 randomly selected fields taken from six separate cultures per condition (shown as mean ± SE averaged pixel values). One micromolar latrunculin-A induces a 70% decrease in rhodamine–phalloidin fluorescence. Asterisk,Statistically different from sham (t38= 24.6; p < 0.0001)
Surprisingly, the potent actin-depolymerizing effects of latrunculin-A were not accompanied by changes in neuronal morphology as observed with phase-contrast optics over 24 hr (data not shown). However, owing to the high density of neurons in the cortical neuronal cultures, we could not reliably visualize the fate of dendrites and their spines after treatment with actin-depolymerizing agents. Therefore, we visualized the cortical neurons and their arbors in greater detail by staining a small fraction of neurons in each dish (<3%) with the membrane tracer DiI (see Materials and Methods). This revealed that the cell soma, dendrites, and even dendritic spines were preserved in neurons treated with latrunculin-A as compared with controls (Fig.1D,E). Thus, despite the abundance of actin in dendritic spines (Matus et al., 1982), it may not be essential to the preservation of dendritic morphology.
We also examined the effects of concentration and of treatment duration with the actin-depolymerizing agents in both cortical and hippocampal cultures. The effects of cytochalasin-D (concentrations tested: 0.1, 3, 10, and 30 μm) on producing a punctate distribution of actin was maximal at 10 μm. The effects of latrunculin-A (concentrations tested: 0.1, 0.3 1, 3, and 5 μm) on reducing actin staining reached a peak at 1 μm (data not shown). There were no differences between 12 and 24 hr treatment periods for either agent. Also, 24 hr cell survival was unchanged by the drugs (see sections below). For subsequent experiments, the effects of the depolymerizing agents on rhodamine phalloidin staining were used as the main criterion in selecting their concentrations and duration of application.
Disruption of F-actin does not affect NMDA-evoked ionic currents
Previous studies have established an intimate structural and functional relationship between F-actin and NMDARs. F-actin is bound to NMDARs via α-actinin (Wyszynski et al., 1997), and its state of polymerization may play an important role in regulating NMDA channel activity (Rosenmund and Westbrook, 1993). Therefore, to determine the functional consequences of depolymerizing actin in our cortical neurons, we recorded whole-cell NMDA-evoked currents from cultures treated with the depolymerizing compounds. The concentrations selected were based on their effects on F-actin in the imaging studies (Figs.1,2).
Cortical neuronal cultures were treated for 12 hr before recordings with either cytochalasin-D (10 μm), latrunculin-A (1 μm), or DMSO (0.1%). F-actin depolymerization had no effect on passive membrane properties, including input resistance and membrane capacitance [capacitance: DMSO, 51.4 ± 2.5 pF (n = 23); cytochalasin-D, 56.1 ± 3.0 pF (n = 20); latrunculin-A, 48.8 ± 2.4 pF (n = 27); one-way ANOVA; F = 1.98;p = 0.15]. Figure3A shows representative traces of whole-cell recordings of currents elicited by brief applications of 3–300 μm NMDA. Peak NMDA currents were not significantly different in cultures treated with cytochalasin-D or latrunculin-A: DMSO, 1970 ± 142 pA (n = 23); cytochalasin-D, 2231 ± 224 pA (n = 20); latrunculin-A, 2020 ± 126 pA (n = 27) (Fig.3A, one-way ANOVA; F = 0.68;p = 0.51). NMDA concentration–response relationships were also unaffected (Fig. 3B; EC50: DMSO, 23.2 ± 2.5 μm (n = 8); cytochalasin-D, 22.4 ± 2.9 (n = 8); latrunculin-A, 18.1 ± 2.1 (n = 7); one-way ANOVA;F = 1.26; p = 0.30). Also, there were no observable differences in NMDA current density (Fig. 3C;ANOVA; F = 0.23; p = 0.79) and desensitization (Fig. 3D; ANOVA; F = 0.12;p = 0.92). Identical results were obtained with higher concentrations of latrunculin-A (5 μm) in separate studies (data not shown). Thus, in spite of the dramatic alterations produced by the actin-depolymerizing agents on rhodamine–phalloidin staining (Figs. 1, 2), macroscopic currents evoked by adding exogenous NMDA were unchanged.

NMDA-evoked ionic currents are not affected by the actin-perturbing agents. Cultured cortical neurons were treated for 12 hr with latrunculin-A (1 μm) or cytochalasin-D (10 μm) and maintained in solutions containing the agents until recordings were made. A,Representative NMDA-evoked currents obtained with 3–300 μm NMDA in control (sham-treated) cultures and in cultures treated with the depolymerizing agents. B, NMDA concentration–response curves: EC50 for control, 23.2 ± 2.5 μm (n = 8); latrunculin-A, 18.1 ± 2.1 μm (n = 8); cytochalasin-D, 22.4 ± 2.9 μm(n = 7), one-way ANOVA; F = 1.26; p = 0.30. Symbols represent mean ± SE. Error bars are shown where they exceed symbol size.C, NMDA current density measurements elicited with 300 μm NMDA. In picoamperes per picofarad: control, 39.1 ± 2.6 (n = 23); latrunculin-A, 41.8 ± 2.0 (n = 27); cytochalasin-D, 40.9 ± 4.4 (n = 20); one-way ANOVA; F = 0.23; p = 0.79. D, Analysis of NMDA current desensitization. Iss, Steady-state current; Ipeak, peak current. Control, n = 23; latrunculin-A,n = 27; cytochalasin-D, n = 20.Columns in C and Dindicate the mean + SE. Data in B–E were pooled from neurons taken from two separate culture platings.
Depolymerizing F-actin in dendritic spines targets synaptic NMDARs selectively
Next, we used both imaging and electrophysiological approaches to study the effects of perturbing F-actin on the distribution and function of NMDARs and of AMPA receptors (AMPARs). We used latrunculin-A (1–5 μm), because this compound had the most pronounced effects on actin in the cells (Figs. 1, 2). First, cortical neurons grown at low density (Materials and Methods) were treated with latrunculin-A for 12 hr and were then stained for the NMDAR subunit NR1 or for the AMPAR subunit GluR1. Both sham and latrunculin-treated cortical neurons exhibited punctate NR1 and GluR1 immunostaining that indicates receptor clusters, as reported by others (Fig. 4A–H; Kornau et al., 1995; Halpain et al., 1998). The staining was quantified by counting individual clusters per unit dendrite length as shown in Figure 4, B and D for NMDARs and Figure 4,F and H, for GluR1 (see Materials and Methods). Depolymerizing F-actin with latrunculin-A reduced the total number of NMDAR clusters in primary dendrites from 14.3 ± 0.6 to 8.8 ± 0.5 (t68 = 6.23; p= 0.0001) and in secondary dendrites from 12.7 ± 0.9 to 6.6 ± 0.4 (t54 = 6.55; p= 0.0001) clusters per 10 μm of dendrite length (Fig.5A). The reduction in the numbers of NMDAR clusters likely reflected a change in receptor aggregation rather than a loss of NMDAR protein, because the level of NR1 was unchanged on Western blot analysis (Fig. 5C). Our results are consistent with the experiments of Allison et al. (1998) in hippocampal neurons, in which depolymerizing F-actin with latrunculin-A reduced the total number of dendritic NMDAR clusters by a similar degree. Using colocalization studies with the presynaptic marker synaptophysin, these authors showed that this decrease was entirely attributable to a selective loss of synaptic NMDAR clusters, because clusters that did not colocalize with synaptophysin were unaffected.

Effect of disrupting F-actin on the distribution of NMDAR and AMPAR clusters in dendrites of cultured cortical neurons. The cells were treated with 1 μmlatrunculin-A for 12 hr and immunostained for the NMDAR subunit NR1 (A–D) or the AMPAR subunit GluR1 (E–H). A, NR1 immunostaining of a neuron treated with sham (0.1% DMSO) solution illustrating the punctate appearance of NMDAR clusters. B, Higher power view of a representative cortical primary dendrite immunostained for NR1. B1, Original (unprocessed) view. B2,Same dendrite after image processing for cluster counts (see Materials and Methods). C, NR1 immunostaining of a neuron treated with latrunculin-A. D, Representative NR1 immunostained dendrite from latrunculin-A-treated neuron, unprocessed (D1) and image-processed (D2) as inB. Note reduction in the number of clusters per unit dendrite length. E, GluR1 immunostaining of a neuron treated with sham (0.1% DMSO) solution illustrating the punctate appearance of GluR1 clusters. F, Representative GluR1-immunostained dendrite from control neuron, unprocessed (F1), and image processed (F2) as inB. G, GluR1 immunostaining of a neuron treated with latrunculin-A. H, Representative GluR1-immunostained dendrite from latrunculin-A-treated neuron, unprocessed (H1) and image-processed (H2) as in B. Scale bars, 10 μm. Images in A, C, E, and H are representative of six separate experiments per condition.

Dissolving F-actin selectively reduces the number of dendritic NMDAR clusters and the NMDA component of spontaneous mEPSCs, but leaves AMPAR staining and mEPSCs unchanged.A, Effect of 1 μm latrunculin-A versus sham (0.1% DMSO) on the numbers of NMDAR clusters per 10 μm of primary (1ry; n = 35 dendrites per group) and secondary (2ry; n = 28 dendrites per group) dendrites in cultured cortical neurons.Asterisk, Different from sham, Student'st test; p = 0.0001.B, Effect of 1 μm latrunculin-A versus sham (0.1% DMSO) on the numbers of AMPAR clusters per 10 μm of 1ry (39–42 dendrites per group) and 2ry (31–36 dendrites per group) dendrites in cultured cortical neurons. C, Immunoblot of GluR1 and NR1 protein expression in cultures treated with sham (0.1% DMSO) or 1 μm latrunculin-A (Lat-A) for 12 hr. Representative of three experiments. D, E, Cultured cortical neurons were treated with 5 μm latrunculin-A for 12 hr. mEPSCs were recorded for 5 min in the whole-cell configuration.Nsham = 13 neurons,Nlatrunculin = 11 neurons pooled from two platings. D, Summary data showing the effect of latrunculin on the area (picoamperes × milliseconds) of different components of mEPSCs. The area (A) of mEPSCs was integrated over 50 msec.A-total-sham, 137.0 ± 12.0;A-total-latrunculin, 88.3 ± 7.2 (p = 0.002);A-AMPA-sham, 60.8 ± 5.8;A-AMPA-latrunculin, 61.0 ± 3.5 (p = 0.974);A-NMDA-sham, 76.0 ± 12.0;A-NMDA-latrunculin, 27.3 ± 6.6 (p = 0.003). Asterisks,Statistical difference as compared with sham (Student'st test). E, Representative averaged traces showing mEPSCs recorded from cultured mice cortical neurons without (left) and with latrunculin-A treatment (right). Both AMPA and NMDA-containing mEPSCs (1) were recorded without APV and Mg2+. AMPA-only mEPSCs (2) were recorded with 20 μm APV and 2 mmMg2+ in the perfusion solution. NMDA-only mEPSCs (3) were obtained by subtracting trace 2 from trace 1.
In contrast with the effect of depolymerizing F-actin on NMDAR cluster counts, treatment with latrunculin-A (1–5 μm) failed to similarly affect the distribution of AMPARs in both primary and secondary dendrites of cortical neurons (Figs. 4E–H,5B; data for 1 μm latrunculin-A, similar results with 5 μm not shown). GluR1 levels were likewise unaffected on Western blot analysis (Fig.5C).
The above findings support the use of latrunculin-A to perturb NMDA receptor clusters, but do not establish a functional significance for this effect. Because the actin-depolymerizing agents had no effect on macroscopic whole-cell NMDA currents (Fig. 3), latrunculin treatment might indicate that although NMDAR localization might be rearranged, function is grossly unaffected. It is difficult, based on imaging experiments, to determine whether the NMDAR clusters migrate away from dendritic spines, whether they dissociate, are degraded, or internalized. Regardless, if the number of NMDAR clusters is reduced, then their activity might be affected. Because many clusters are localized at synapses (Allison et al., 1998), we next examined mEPSCs in our cells because these currents are mediated by synaptic receptors.
Recordings were made in cortical neuronal cultures pretreated with latrunculin-A (5 μm) for 12 hr. Using the whole-cell configuration, spontaneous mEPSCs were recorded for ∼5 min to acquire sufficient numbers of events. Representative averaged traces from these recordings are shown in Figure 5E for sham and latrunculin-treated cultures. Miniature EPSCs were first recorded without APV and Mg2+ in the extracellular solution (Fig. 5E, trace 1). The AMPA receptor-mediated component of the mEPSCs was then recorded after switching the neurons to an extracellular solution containing 20 μm APV and 2 mmMg2+ (Fig. 5E, trace 2). Subtracting the fast AMPA receptor-mediated component from the total mEPSC revealed the slower NMDAR-mediated component of spontaneous mEPSCs (Fig. 5E, trace 3). Integration of the area (A) of the different mEPSC-components revealed that latrunculin-A-treated neurons exhibited a significantly reduced total mEPSC (Fig.5D). Consistent with the immunohistochemical data (Fig.5A,B), this was attributable entirely to a selective reduction of the NMDAR-mediated component, because the AMPA receptor-mediated component was unaffected (Fig. 5D). [In picoamperes × milliseconds:A-total-sham, 137.0 ± 12.0;A-total-latrunculin, 88.3 ± 7.2 (p = 0.002);A-AMPA-sham, 60.8 ± 5.8;A-AMPA-latrunculin, 61.0 ± 3.5 (p = 0.974);A-NMDA-sham, 76.0 ± 12.0;A-NMDA-latrunculin, 27.3 ± 6.6 (p = 0.003), ANOVA]. The effect of disrupting F-actin with latrunculin-A was postsynaptic, because it only affected the NMDA component of the response to released neurotransmitter. These data support the use of latrunculin-A to selectively perturb the synaptic activation of NMDARs.
Depolymerizing F-actin does not affect NMDAR-mediated Ca2+ loading or neurotoxicity produced by exogenously applied NMDAR agonists
We next examined the effects of depolymerizing actin on NMDAR-mediated excitotoxicity using two established in vitromodels. First, by applying exogenous NMDA orl-glutamate to the cultures (Choi et al., 1988;Sattler et al., 1998). Then, by exposing the cultures to oxygen glucose deprivation (Goldberg et al., 1987; Abdel-Hamid and Tymianski, 1997).
Applying NMDA or l-glutamate directly to the bath should produce a uniform concentration of the agonist in the extracellular medium and affect all NMDA receptors, irrespective of their distribution. The agonists were always applied in the presence of CNQX (10 μm) and nimodipine (2 μm), antagonists of non-NMDARs and voltage-gated Ca2+channels, to isolate both Ca2+ entry and neurotoxicity to NMDARs (Sattler et al., 1998, 1999). The cortical neuronal cultures were treated with either latrunculin-A (1–5 μm) or cytochalasin-D (0.1–30 μm) for 12 hr. The cells were then exposed to NMDA (0, 30, or 100 μm) or to l-glutamate (10–1000 μm) for 60 min. They were then washed and observed for a further 23 hr in control solution containing CNQX, nimodipine, and MK-801 (10 μm). Cell survival was monitored by measuring propidium iodide fluorescence as an index of cell death (Materials and Methods; Sattler et al., 1997; Tymianski et al., 1998). Sister cultures were identically treated and used immediately after the insult for determinations of NMDAR-mediated45Ca2+accumulation (Sattler et al., 1998, 1999).
Figure 6, A and B,summarizes the effects of latrunculin-A (1–5 μm; Fig. 6A) and cytochalasin-D (0.1–30 μm; Fig.6B) on toxicity and Ca2+loading incurred after the NMDA application. Even the highest concentrations of depolymerizing agents were well tolerated by the cells under control conditions (0 μm NMDA groups in Fig. 6A1,B1). However, despite the profound effects of these compounds on the polymerization state of F-actin (Figs. 1, 2), neither one affected NMDA excitotoxicity produced by a range of concentrations (Fig. 6A1,B1). The accumulation of45Ca2+ in the cells throughout the NMDA application was also unaffected (Fig.6A2,B2). Similarly, treating the neurons with latrunculin-A (1 μm) had no effect on excitotoxicity (Fig. 6C1) or45Ca2+accumulation (Fig. 6C2) produced by applyingl-glutamate, the endogenous neurotransmitter. These results are consistent with our electrophysiological observations that indicated a lack of effect of both depolymerizing agents on macroscopic ionic currents mediated by exogenous NMDA (Fig. 3) and indicate that NMDAR activation can trigger excitotoxicity, irrespective of receptor localization.

Disrupting F-actin has no impact on excitotixicity or neuronal Ca2+ loading evoked by exogenous NMDA or l-glutamate. Cultured cortical neurons were pretreated with the indicated concentrations of latrunculin-A (0–5 μm) or cytochalasin-D (0–30 μm) before undergoing exposure to 0, 30, or 100 μm NMDA (A, B) or 10–1000 μml-glutamate (C) for 60 min (in 2 μm nimodipine and 10 μm CNQX; see Materials and Methods). The cultures were then maintained for a further 23 hr to measure neuronal survival (A1–C1) or used for45Ca2+ accumulation measurements (A2–C2). A, Effects of treatment with latrunculin-A on NMDAR-mediated excitotoxicity (A1) and45Ca2+ loading (A2).B, Effects of treatment with cytochalasin-D on NMDAR-mediated excitotoxicity (B1) and45Ca2+ loading (B2).C, Effect of treatment with 1 μmlatrunculin-A on glutamate-mediated excitotoxicity (C1) and 45Ca2+ accumulation (C2). Symbols in A1 andB1 represent the mean survival (± SE) of 4–64 cultures per experimental condition, obtained from at least two (usually 4–6) different platings. The lines indicate the least-squares linear regression curves obtained for each NMDA concentration.Columns in A2 and B2represent the mean (± SE) 45Ca2+accumulation averaged from 16–36 cultures obtained from 4–6 different culture platings. Columns in C1 and C2were obtained from 16 cultures per condition pooled from two separate platings. Error bars are shown where they exceed symbol size.
Depolymerizing F-actin reduces NMDAR-mediated Ca2+ loading and neurotoxicity evoked by OGD
Oxygen glucose deprivation releases glutamate at synapses and causes neurotoxicity that is mediated primarily by NMDA receptors and Ca2+-dependent mechanisms (Rothman, 1983,1984; Goldberg et al., 1987; Goldberg and Choi, 1993). In cultured cortical neurons, OGD causes vesicular glutamate release, because both glutamate accumulation and OGD toxicity are blocked by pretreatment with tetanus toxin (Monyer et al., 1992), which prevents synaptic vesicle exocytosis (Bergey et al., 1987; Ahnert-Hilger and Bigalke, 1995). OGD also causes nonvesicular transmitter release via reverse operation of glutamate transporters (Attwell et al., 1993; Szatkowski and Attwell, 1994), which are enriched in neurons at presynaptic and postsynaptic sites (Rothstein et al., 1994). Thus, OGD may cause glutamate to accumulate at synaptic sites preferentially. Therefore, unlike excitotoxicity produced by exogenous NMDA orl-glutamate, which target both synaptic and extrasynaptic receptors (Fig. 6), OGD is anticipated to injure neurons by activating synaptic NMDARs perferentially. Because disrupting actin with latrunculin-A selectively perturbs the localization and function of synaptically activated NMDARs (Figs. 4, 5), we asked whether neurons treated with this agent would exhibit altered vulnerability to OGD.
We exposed cultured cortical neurons to combined oxygen glucose deprivation in the presence of CNQX (10 μm) and nimodipine (2 μm) to block non-NMDARs and Ca2+ channels. After 2 hr, the cells were washed and kept for further 22 hr in oxygenated glucose-containing bicarbonate solution containing CNQX, nimodipine, and MK-801. Sister cultures were equally exposed to OGD and used for45Ca2+accumulation assays to measure Ca2+loading through NMDARs.
Cultures that had been pretreated with latrunculin-A for 12 hr were significantly less vulnerable to OGD-induced NMDAR-mediated neurotoxicity than controls (Fig.7A; n = 6 cultures from two different dissections;t10 = 4.18; p = 0.002). This protective effect was exactly paralleled by reduced45Ca2+accumulation in the latrunculin-treated neurons (Fig. 7B;n = 6 cultures from two different dissections;t10 = 4.07; p = 0.002). Thus, neurons in which the function of synaptically activated NMDARs was selectively perturbed by depolymerizing actin (Figs. 4,5) also exhibited reduced vulnerability to excitotoxic insults that are preferentially mediated by synaptic NMDARs.

Depolymerization of F-actin attenuated NMDR-mediated cell death and neuronal Ca2+ loading when excitotoxicity was evoked by OGD. Cultures were pretreated with 1 μm latrunculin-A for 12 hr, after which they were exposed to 2 hr of OGD in the presence of non-NMDAR and Ca2+channel antagonists (CNQX and nimodipine, respectively) to isolate Ca influx to NMDARs (Materials and Methods). The cells were observed for a further 22 hr to measure cell death, whereas sister cultures were used to measure 45Ca2+ accumulation after the 2 hr OGD exposure. A, Cell death was significantly reduced in the latrunculin-A-treated cultures as compared to sham cultures (t10 = 4.18;p = 0.002). Each bar represents the mean ± SE of six cultures obtained from two different dissections.B, 45Ca2+ accumulation was significantly reduced in the latrunculin-A-treated cultures (t10 = 4.07; p = 0.002). Each bar represents the mean (± SE) of six cultures obtained from two different platings.
DISCUSSION
Treating cortical and hippocampal neurons with cytochalasin-D and latrunculin-A disrupted neuronal F-actin to different degrees (Figs.1,2), with no apparent effect on macroscopic NMDA-evoked whole-cell currents (Fig. 3) or NMDAR-mediated45Ca2+accumulation (Fig. 6A2,B2,C2). Latrunculin-A, the agent that disrupted actin in dendritic spines most effectively (Fig.1C), caused a reduction in the total number of NMDAR clusters in the dendrites (Fig. 4,5) and selectively reduced the activity of synaptically activated NMDARs (Fig. 5). Perturbing F-actin with latrunculin did not affect excitotoxicity evoked by exogenous NMDAR agonists (Fig. 6), but reduced excitotoxicity caused by OGD, an insult that preferentially releases excitatory neurotransmitter at synapses (Fig. 7).
Our data indicate that signaling mechanisms regulating NMDAR-mediated excitotoxicity are not governed by the synaptic localization of NMDARs. Because conditions that preferentially attenuated the function of synaptically activated NMDARs had no effect on the toxicity of exogenous NMDA or l-glutamate (Fig. 6), extrasynaptic NMDARs must still be linked to the second messenger pathways that trigger neuronal damage. We have previously hypothesized that this may be attributable to the physical association of NMDARs with distinct macromolecular complexes that initiate and/or propagate neurotoxic signaling (Tymianski et al., 1993; Sattler et al., 1998). Recent data on the molecular organization of neuronal synapses support this idea and suggest candidate molecules that may be involved. Glutamate receptors interact, via their intracellular domains, with submembrane proteins in the postsynaptic density (PSD; Gomperts, 1996; Ponting et al., 1997). Several mammalian homologous families of PSD proteins known as membrane-associated guanylate kinases have been identified and shown to interact with high specificity with distinct classes of glutamate receptors. For example, PSD-95/synapse-associated protein 90 (SAP90) (Cho et al., 1992; Kistner et al., 1993), chapsyn-110/PSD-93 (Brenman et al., 1996a; Kim and Sheng, 1996), and SAP102 (Muller et al., 1996), are submembrane proteins that interact preferentially with NMDARs. The analogous but distinct proteins GRIP (Dong et al., 1997) and Homer (Brakeman et al., 1997) interact with AMPA and with metabotropic glutamate receptors, respectively.
PSD-95 is specifically bound to NMDARs via the second of its three PDZ domains (Kornau et al., 1995; Kim and Sheng, 1996; Niethammer et al., 1996), forming multimers that are thought to lead to NMDAR clustering. PSD-95 also interacts with other intracellular signaling molecules, including neuronal nitric oxide synthase (nNOS; Brenman et al., 1996a,b; Stricker et al., 1997). This enzyme catalyzes the production of nitric oxide, a neurotoxic signaling molecule (Dawson et al., 1991,1993; Brorson et al., 1997). We have recently demonstrated that reducing the expression of PSD-95 in cultured cortical neurons reduces the interaction of NMDAR-mediated Ca2+influx with nNOS and excitotoxicity (Sattler et al., 1999). Thus, the complex that includes NMDARs, PSD-95, and its associated molecules is one that likely mediates neurotoxic NMDAR signaling. The actin cytoskeleton does not seem to be involved, because previous reports have examined the association of PSD-95 with synaptic NMDARs under conditions that perturb actin. Allison et al. (1998) demonstrated that destabilizing actin with latrunculin-A reduced the numbers of synaptic NMDA receptors, but that they remained associated with PSD-95. Halpain et al. (1998) further showed that the association of NMDARs and PSD-95 remained unperturbed by excitotoxic insults with NMDA, which causes F-actin depolymerization (Shorte, 1997). Taken together, these data suggest a model in which F-actin plays a structural role in targeting NMDARs and their associated signaling complexes to synaptic sites (Fig.8A). The optimal positioning of such complexes may maximize the efficiency of activating NMDARs and their associated signal transduction pathways. Depolymerizing F-actin perturbs the efficiency of postsynaptic receptor activation and reduces the probability of activating neurotoxic signaling molecules (Fig. 8B). However, this occurs without disrupting the association of NMDARs with the molecules responsible for initiating these neurotoxic signaling cascades.

Schematic depiction of one mechanism that could account for the distinct contributions of synaptic and extrasynaptic NMDA receptors to OGD-mediated excitotoxicity. A, NMDARs are tethered to the synapse by interactions with the F-actin cytoskeleton via α-actinin. During an excitotoxic insult, presynaptically released glutamate binds to the NMDAR and induces Ca2+ influx. Ca2+ ions entering the postsynaptic cell through NMDAR channels then trigger neurotoxicity by interacting with a macromolecular complex that is linked to the NMDAR. B, Depolymerization of the F-actin cytoskeleton reduces the number of synaptic NMDAR clusters. Glutamate is released into the synaptic cleft, but activates a smaller number of NMDARs, resulting in decreased Ca2+ influx and a decreased activation of NMDAR-associated neurotoxic signaling molecules. This results in decreased neuronal cell death.
Despite the above-proposed model (Fig. 8), it remains difficult to determine the precise mechanism by which treatment with latrunculin-A reduced the activation of synaptic NMDARs. Consistent with our results (Figs. 4,5), Allison et al. (1998) showed in hippocampal neurons that this compound reduced the numbers of NMDAR clusters in dendrites. However, they also showed a similar reduction by latrunculin of the numbers of AMPA receptor clusters using immunostaining for the GluR1 subunit. Because depolymerizing actin in spines selectively attenuated the NMDA component of spontaneous mEPSCs while leaving the AMPA component intact (Fig. 5D,E), the loss of receptor cluster immunostaining may not translate directly to an effect on receptor function. Rather, the effect may occur at a level of organization that is not easily detectable by conventional optical means. For example, AMPA receptors are readily solubilized from adult rat hippocampal tissue and cultured neurons using Triton X-100 extraction (Wenthold et al., 1996; Allison et al., 1998), whereas NMDA receptors and other core components of the PSD such as PSD-95 are relatively detergent-insoluble (Cohen et al., 1977; Kennedy, 1997). This may indicate that AMPA receptors are less tightly anchored to structures in the PSD and that their submicroscopic localization may be less affected than that of NMDARs after treatment with actin-perturbing agents. Studies using immunogold histochemistry suggest that AMPARs are preferentially localized at the periphery, whereas NMDARs are found at the highest concentration in the middle of the synaptic apposition (Bernard et al., 1997; Kharazia and Weinberg, 1997). Thus, the impact of destabilizing the cytoskeleton may have different implications for the function of these two receptor classes.
In this paper, the neuronal cultures were treated with actin-depolymerizing agents for 12–24 hr to visibly perturb the F-actin cytoskeleton before experiments (Fig. 1,2). Recently, using rat hippocampal slices, Kim and Lisman (1999) examined the effects of shorter applications (several minutes) of similar depolymerizing agents on synaptic transmission and long-term potentiation. They showed that bath-applying latrunculin B produced an early (within minutes) attenuation of field EPSPs in the CA1 region and a reduction of both AMPA and NMDA receptor-mediated components of the response. This effect was felt to be in part presynaptic, because paired-pulse facilitation was increased and mEPSC frequency was reduced during latrunculin B application. Furthermore, by dialyzing the agents into postsynaptic neurons via patch pipettes, they showed no effect of latrunculin-B on NMDAR-mediated currents. However, the actin-stabilizing compound phalloidin reduced the AMPAR-mediated component of EPSCs. None of these experiments undertook a direct visualization of the actin cytoskeleton to determine to whether the effects observed were related to perturbed F-actin content or to other direct or indirect drug actions. However, these data warn that the depolymerizing agents might have diverse effects on synaptic mechanisms within minutes of drug application. As the timing, duration, and specificity of the diverse effects of actin-depolymerizing agents is poorly understood, it was helpful in our present study to obtain a separate confirmation of the effects of these compounds by visualizing the actin cytoskeleton directly (Figs. 1,2) and by visualizing the effects of the compounds on NMDA and AMPA receptors by immunohistochemistry (Fig. 4).
It has been reported that cytochalasins can protect cultured hippocampal neurons against glutamate and amyloid β-peptide toxicity by stabilizing neuronal calcium homeostasis (Furukawa and Mattson, 1995; Furukawa et al., 1995). The authors achieved this by pretreating the cells with cytochalasin-D for 1 hr at concentrations of 1–100 nm. These findings are consistent with a proposed action of cytochalasins in promoting NMDA channel rundown by mimicking the effects of Ca2+-mediated actin depolymerization (Rosenmund and Westbrook, 1993). In the present study, we found no effect of cytochalasin-D on NMDA-evoked ionic currents (Fig. 3), NMDA excitotoxicity (Fig. 6B1), or NMDA-evoked45Ca2+accumulation (Fig. 6B2). However, our cortical neurons were treated with this depolymerizing agent for a longer duration (12 hr), and generally at higher concentrations (up to 30 μm). In the present experiments, our aim was to achieve a maximal depolymerizing effect on actin based on imaging with rhodamine–phalloidin (Figs. 1,2). Thus, we cannot exclude the possibility that brief exposure to low concentrations of cytochalasins might affect receptor activity, Ca2+homeostasis, or survivability by mechanisms that were not addressed in our experiments.
The finding that extrasynaptic NMDARs are fully capable of triggering excitotoxic neuronal damage may imply different mechanisms in different human neurological diseases. Excitotoxic neuronal damage is thought to play a role in traumatic brain and spinal cord injuries (Faden et al., 1989; Tecoma et al., 1989), because CNS trauma may produce a rapid disruption of cellular membranes, causing a diffuse rise in extracellular glutamate levels (Brown et al., 1998). By its nature, traumatic injury may result in excitotoxic neuronal damage via both synaptic and nonsynaptic receptors. Conversely, transient focal cerebral ischemia, transient global ischemia, and epilepsy are disorders of synaptic overactivity that occur in the absence of overt early neuronal damage (Kirino, 1982; Petito et al., 1987; During and Spencer, 1993). Under these pathological circumstances that do not cause the loss of neuronal membrane integrity, the glutamate receptors that trigger excitotoxicity would likely be located in synaptic sites.
Our data reveal a distinct functional role of the actin cytoskeleton. This protein appears to target NMDARs with their associated scaffolding, clustering, and signaling macromolecules to synaptic sites, but not to affect the function of these complexes. Recent evidence indicates that this targeting is a dynamic process, because the accumulation of both AMPA and NMDA receptors at synapses is highly responsive to the stage of neuronal development and their level of endogenous excitatory activity (Rao and Craig, 1997; O'Brien et al., 1998b; Liao et al., 1999). For example, it is possible to regulate the numbers of synaptic glutamate receptor clusters in cultured neurons by pharmacologically inhibiting or increasing excitatory synaptic activity (Rao and Craig, 1997; O'Brien et al., 1998b). Here we have shown, for the first time, that the converse is also true: That it is possible to influence excitatory synaptic activity and its pathological consequences (excitotoxicity) by regulating the numbers of synaptic receptors.
Footnotes
This work was supported by grants from the Medical Research Council (MRC) to J.F.M. and M.T., and National Institutes of Health Grant NS 39060 to M.T. R.S. is a student of the Ontario Heart and Stroke Foundation. W.Y.L is a fellow of the Ontario Heart and Stroke Foundation. Z.X. is an MRC Centennial Fellow. M.T. is an MRC Clinician-Scientist. We thank E. Czerwinska for technical assistance, Drs. Owen T. Jones and Michael Salter for their critical review of this manuscript, Drs. Mark P. Goldberg (Washington University) and John E. Lisman (Brandeis University) for thoughtful discussions, and Drs. Richard Huganir and Dezhi Lao (Johns Hopkins) for technical advice and antibodies.
Correspondence should be addressed to Dr. Michael Tymianski, Lab 11-416, MC-PAV, Toronto Western Hospital, 399 Bathurst Street, Toronto, Ontario M5T 2S8, Canada. E-mail: mike_t{at}playfair.utoronto.ca.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}