

Abstract
At most excitatory central synapses, glutamate is released from presynaptic terminals and binds to postsynaptic AMPA receptors, initiating a series of conformational changes that result in ion channel opening. Efficient transmission at these synapses requires that glutamate binding to AMPA receptors results in rapid and near-synchronous opening of postsynaptic receptor channels. In addition, if the information encoded in the frequency of action potential discharge is to be transmitted faithfully, glutamate must dissociate from the receptor quickly, enabling the synapse to discriminate presynaptic action potentials that are spaced closely in time.
The current view is that the efficacy of agonists is directly related to the extent to which ligand binding results in closure of the binding domain. For glutamate to dissociate from the receptor, however, the binding domain must open. Previously, we showed that mutations in glutamate receptor subunit 2 that should destabilize the closed conformation not only sped deactivation but also altered the relative efficacy of glutamate and quisqualate. Here we present x-ray crystallographic and single-channel data that support the conclusions that binding domain closing necessarily precedes channel opening and that the kinetics of conformational changes at the level of the binding domain importantly influence ion channel gating. Our findings suggest that the stability of the closed-cleft conformation has been tuned during evolution so that glutamate dissociates from the receptor as rapidly as possible but remains an efficacious agonist.
Introduction
AMPA receptors are glutamate-gated ion channels and mediate most of the fast excitatory neurotransmission in the CNS (Dingledine et al., 1999). X-ray crystallographic studies on the isolated ligand binding domain (LBD) of the glutamate receptor subunit 2 (GluR2) AMPA receptor subunit (GluR2–S1S2) show that glutamate and other agonists bind within a deep cleft between two globular domains (1 and 2) (Armstrong et al., 1998; Armstrong and Gouaux, 2000). In agonist-bound structures, domain 2 rotates around helix I ∼20° relative to its position in apo structures (Armstrong and Gouaux, 2000; Hogner et al., 2002; Jin et al., 2002), closing the binding cleft and trapping glutamate and other agonists in the binding pocket.
Comparison of binding core structures for different agonists demonstrated that there is also a strong positive correlation between the extent to which domain 2 moves and agonist efficacy, with full agonists all producing similar amounts of LBD closure and partial agonists resulting in less closure of the binding cleft (Armstrong et al., 1998; Armstrong and Gouaux, 2000; Hogner et al., 2002; Jin et al., 2002, 2003). Although there is no direct evidence that LBD closure necessarily precedes channel gating, these and other results strongly suggest that closing of the LBD is the initial large-scale conformational change that triggers the subsequent gating rearrangements that result in channel opening and desensitization (Sun et al., 2002; Robert et al., 2005; Valentine and Palmer, 2005; Armstrong et al., 2006; Mayer, 2006; Ahmed et al., 2007; Hansen et al., 2007). The available evidence therefore indicates that, for glutamate to be an effective and fast neurotransmitter, the kinetics of LBD closing and opening must be such that two apparently opposing requirements are met: the LBD must be closed for effective gating but open for glutamate to dissociate.
In agonist-bound structures of GluR2–S1S2, the closed-cleft conformation is stabilized by a network of direct and water-mediated interdomain hydrogen bonds (Armstrong and Gouaux, 2000). Disruption of one such cross-cleft interaction (between threonine 686 and glutamate 402) significantly increases EC50 values for glutamate and quisqualate (Quis) and speeds deactivation (Robert et al., 2005), results consistent with subsequent work on kainate and AMPA receptors and the idea that the rate at which the LBD opens is a major determinant of agonist affinity (Weston et al., 2006). Serine and alanine mutations at T686 also selectively reduced the efficacy of glutamate and slowed the rate of receptor desensitization, raising the possibility that subsequent gating transitions were also altered, perhaps because the LBD closed differently (Robert et al., 2005).
Here we determined the impact of the T686A mutation on the extent of LBD closure directly by introducing the mutation in GluR2–S1S2 and crystallizing the mutant protein with glutamate and quisqualate. We also compared the properties of single-channel currents through wild-type (wt) and T686A mutant channels evoked by each agonist. The results support the conclusion that destabilizing the closed-cleft conformation of the LBD selectively reduces the open probability of glutamate-bound channels and delays their activation.
Materials and Methods
Mutagenesis.
The T686S and T686A mutations were introduced into GluR2–S1S2 (kindly provided by Eric Gouaux, Vollum Institute, Portland, OR) and full-length GluR2 flip encoding a glutamine at the Q/R editing site (kindly provided by Mark Mayer, National Institutes of Health, Bethesda, MD) with reverse transcription-PCR. All constructs were verified by sequencing.
Patch-clamp recording and single-channel analysis.
tsA201 cells were maintained and transfected as described previously (Robert et al., 2005). Recordings from outside-out patches held at −100 mV were performed 12–24 h after transfection at room temperature with an EPC 9 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). The external solution contained the following (in mm): 150 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES, pH 7.4. The internal solution contained the following (in mm): 135 CsF, 33 CsOH, 2 MgCl2, 1 CaCl2, 11 EGTA, and 10 HEPES, pH 7.4. Patch pipettes were pulled from thick-walled borosilicate glass capillaries (GC-150F; Harvard Apparatus, Edenbridge, UK), coated with Sylgard resin (Dow Corning, Corning, NY) and fire polished. When filled with internal solution, the recording pipettes had open-tip resistances of 10–15 MΩ. Glutamate and quisqualate (Tocris Bioscience, Avonmouth, UK) were added to the external solution and applied with theta pipettes mounted on a piezoelectric bimorph (Morgan Electro Ceramics, Windsor, UK). The rate of solution exchange time estimated from open-tip potentials was 100–200 μs. Solution exchange times were also estimated in some experiments by applying glutamate in normal external solution containing 100 μm cyclothiazide (CTZ) to evoke a steady-state inward current and then stepping the patch into glutamate (+CTZ) in a solution containing 25 mm NaCl and 125 mm tetraethylammonium (TEA)-Cl. The 20–80% fall time of the current during substitution of the impermeant cation TEA was 300–350 μs.
Glutamate-evoked currents were analog low-pass filtered at 10 kHz, sampled at 25 kHz, and written directly to the hard drive of two computers. One computer stored the records in PULSE format (the software used to run the EPC 9), and the other stored the data in QuB (http://www.qub.buffalo.edu) format (the software used for single-channel analysis). Most single-channel analysis was done on currents recorded during 5–10 min continuous applications of glutamate or quisqualate. Before these applications were made, we collected responses to 100–500 concentration jumps (220 ms applications). These applications evoked rapidly decaying currents and, in conjunction with the single-channel currents, allowed us to estimate the number of active channels in the patch. All patches analyzed gave maximum peak currents of <25 pA at −100 mV, and most patches appeared to contain 5–10 active channels.
Single-channel data recorded during continuous agonist applications were subjected to digital Gaussian low-pass filtering at 3.5 or 4 kHz (−3 dB), which resulted in closed-channel root mean square noise values (300–350 fA) that were less than half the smallest difference in current amplitudes between adjacent conductance levels. The closed-channel current was manually set to zero, and the entire filtered dataset from a given patch was idealized with the segmental k means (SKM) algorithm of QuB to identify single-channel transitions and estimate conductance levels and event durations (Qin et al., 1996; Qin, 2004). Because our data appeared to contain many rapid transitions between several closely spaced open levels, we empirically tested different sampling rates and resolutions. Sampling at 25 kHz and a time resolution of two sample intervals (80 μs, 0.9 filter rise times) consistently gave good results. At lower sampling rates or more conservative time resolutions, many large events were missed, and, at less conservative time resolutions, the idealization found many transitions with amplitudes that were smaller that twice the SD of the closed-channel noise.
The kinetic model used for the SKM idealization of the results contained two closed states and one open state for each of the four open levels found in each of the records. The states were connected so that all types of transitions between the five different conductance levels were allowed. To test whether the relatively high sampling rates we used resulted in serious overestimation of the number of direct transitions between adjacent conductance levels, we also analyzed portions of the records with the maximum idealized point likelihood rate estimation (MIP) subroutine of QuB. The conductance levels and the frequency of transitions between them that were obtained with the SKM and MIP routines were consistently similar.
After visually inspecting the idealized record and removing dubious events, open and shut times were exported to Channelab (Synaptosoft, Decatur, GA), and histograms (10–12 bins per decade) of the dwell times were displayed and fitted with log-likelihood log-binned subroutines. Distributions were fitted with one to four exponential components. The idealized record was also used to calculate the proportion of the different possible types of transitions between conductance levels. Statistical comparisons of sample means were made with two-tailed Student's t tests or one-way ANOVAs as indicated.
Protein purification and crystallization.
The GluR2–S1S2J ligand binding core (Armstrong and Gouaux, 2000) has a hexahistidine tag at the N terminus, and the T686 mutants were expressed in Origami (DE3) cells at 20°C overnight. Proteins were initially purified using a nickel affinity column, and the His tag was cleaved with trypsin and removed using a cation exchange column. A final purification step was done with a size exclusion column (Superdex S200; GE Healthcare, Little Chalfont, UK). The protein was dialyzed (40,000-fold dilution) for 48 h at 4°C to remove the 1 mm glutamate present during the purification steps. The protein was concentrated to 10 mg/ml and mixed with 5 mm l-glutamate (or 1.7 mm quisqualate). The mixture was incubated for 15 min, added to crystallization buffer at a 1:1 ratio, and crystallized by the hanging drop vapor diffusion method at 20°C. Initial crystals were obtained from the Crystal Screen (Hampton Research, Aliso Viejo, CA) and optimized in 0.1 m Tris-HCl, pH 8.5, 0.15–0.2 m lithium sulfate, and 18–21% PEG4000.
Structure determination.
The diffraction data for T686S:Glu were collected at BNL X29. All other data were collected using an R-Axis IV detector at Cu Kα energy under cryo-stream at 100°K. Crystals were transferred to 15–20% glycerol in mother liquor before flash cooling in the cryo-stream. The data were processed using either HKL2000 or MOSFLM.
Wild-type GluR2–S1S2J crystal structures [Protein Data Bank (PDB) codes 1FTO and 1FTJ] were used as templates for finding molecular replacement solutions using PHASER (McCoy et al., 2005). Rigid body refinement, energy minimization, simulated annealing, and individual B-factor refinement were performed with the program CNS, followed by the maximum-likelihood-based Refmac5. The refined protein models were submitted to the Model Bias Removal server to avoid over-refinement (Rupp et al., 2002). The molecular coordinates of bound glutamate or quisqualate as well as water were added, and the dataset was subjected to cycles of CNS and Refmac5 refinement. For manual model fitting, Xfit in the XtalView crystallographic package was used. Refined models were superposed on published GluR2–S1S2 wild-type structures for comparison using Swiss PDB viewer and scrutinized using the crystallographic visualizing program SPOCK (http://quorum.tamu.edu). Non-covalent bonds were found using HPlus (McDonald and Thornton, 1994) and visualized with PyMol. Local intermolecular interactions around the bound ligands were illustrated with Ligplot (Wallace et al., 1995), and the backbone geometry of the structures was evaluated using PROCHECK (Laskowski et al., 1993). Percentages of residues in most favored regions, additional allowed regions, and generously allowed regions in Ramachandran plots were 91.2, 8.4, 0.4 for the T686A:Quis structure, 90.7, 9.3, 0.0 for the T686A:Glu structure, and 92.9, 6.9, 0.1 for the T686S:Glu structure. Residues with poor side-chain electron density were changed to alanine but retained their original names in the structure files. In one of four protomers in the asymmetric unit of the T686S:Glu structure, the electron density was poorly defined from S640 to Y700, which resulted in R factors higher than expected given the resolution of the data. Coordinates of the T686A:Quis, T686A:Glu, and T686S:Glu mutant structures are deposited in the Protein Data Bank, and their PDB codes are 3B6T, 3B6Q, and 3B6W, respectively.
Results
T686A alters the relative efficacy of glutamate and quisqualate
Many studies have shown that glutamate and quisqualate are both full AMPA receptor agonists, and both agonists bind similarly to the isolated GluR2 LBD (Jin et al., 2002). When glutamate- and quisqualate-evoked currents were compared after reducing desensitization of GluR2 channels with CTZ, mutation of threonine 686 to serine or alanine altered the relative efficacy of the two agonists, reducing the maximal glutamate-evoked currents to ∼75 and 45%, respectively, of those seen with quisqualate (Robert et al., 2005). To verify that these results still held when desensitization was intact, we first compared peak currents evoked by rapid application of the two agonists in patches containing wild-type GluR2 channels (GluR2-wt) or GluR2 channels carrying the T686A mutation (both GluR2 constructs were flip versions and had a glutamine at the Q/R editing site). As expected, saturating concentrations of glutamate or quisqualate produced similar peak currents in patches expressing GluR2-wt channels (Fig. 1a). In contrast, glutamate gave maximal peak currents for T686A channels that were only ∼20% of those evoked in the same patches by quisqualate (Fig. 1d). Compared with glutamate-evoked currents through wild-type channels or currents evoked by quisqualate, the rate of desensitization was also substantially slower for the T686A mutants and steady-state desensitization was reduced (Fig. 1d), results consistent with our previous observations (Robert et al., 2005).

The T686A mutation selectively reduces the efficacy of glutamate. a, d, Mean inward currents from 100–150 applications of near-saturating concentrations of glutamate (red) or quisqualate (black) to patches containing GluR2-wt (a) and T686A (d) channels. The holding potential was −100 mV, and each application was 220 ms in duration. Gray trace in the inset in d shows the glutamate-evoked current for the T686A mutants scaled to the peak of the quisqualate response. b, c, e, f, Examples of unitary currents seen during individual applications of glutamate and quisqualate. The data were sampled at 25 kHz and digitally low-pass filtered at 3.5 kHz. The peak currents at the onset of the applications are off-scale. g, Diagram of AMPA receptor gating. Transitions between states 0 and 1 represent agonist binding to and dissociation from the open-cleft conformation of the LBD of one subunit in a receptor dimer. Transitions between states 1 and 2 represent opening and closing of the LBD of this subunit. State 2, in which the LBD and the channel pore are closed, is unstable, and the instability is relieved by either channel opening (state 3) or rearrangement of the dimer interface and desensitization (state 4) (Sun et al., 2002). Rearrangement of the dimer interface dissipates the work done on the transmembrane helices by LBD closure and results in a long-lived closed state in which the LBD of one subunit in the dimer is stabilized in the closed-cleft conformation (Robert and Howe, 2003; Armstrong et al., 2006). h, Hypothetical plot of the occupancy of state 2 (relative to state 1) for an individual subunit. The T686A mutation increases the rate constant for LBD opening, CO, which decreases the ratio CC/CO and the occupancy of state 2 more for the low-affinity agonist glutamate (in which the value of CO is larger for GluR2-wt channels). The symbols show the values predicted from the values of CC and CO estimated with Monte Carlo simulations of ensemble currents for wild-type GluR2 and T686A channels in our previous work (Robert et al., 2005). The arrows illustrate the effect of the mutation-associated shifts in CO values for glutamate and quisqualate.
In patches containing a small number of GluR2-wt or T686A channels, currents through individual channels could be distinguished late in the 220 ms applications of glutamate (Fig. 1b,e) or quisqualate (Fig. 1c,f), most of which likely represent channel openings during occasional escapes from desensitization. The only obvious difference in the unitary currents seen with wild-type and mutant receptors was that they were more frequent with glutamate for the T686A channels, as expected from the larger steady-state currents seen with glutamate in patches that gave large-ensemble currents, as well as the effect of the T686A mutation on recovery from desensitization (Robert et al., 2005).
Figure 1g depicts the sequence of events thought to result in activation of AMPA receptor channels. After glutamate binds to the open-cleft conformation of the LBD of an individual subunit (states 0 and 1), the cleft closes, resulting in an intermediate state (state 2) in which both the LBD and ion channel pore are closed. The inherent instability of this state is relieved by either pore rearrangements that open the channel or subunit rearrangements along the dimer interface that result in desensitization (Sun et al., 2002; Robert et al., 2005; Armstrong et al., 2006). In this model, the relative occupancies of the bound closed-channel states in which the LBD is open or closed (states 1 and 2) depend on the ratio of the rate constants CC and CO (Fig. 1h), and the proportion of channels in state 2 (of those in states 1 or 2) will be close to 1 provided that CC is substantially greater than CO. Kinetic simulations we performed previously indicated that the value of CO is ∼15-fold greater for glutamate than quisqualate and that the T686A mutation increased the value of CO for both agonists by 15- to 25-fold (Robert et al., 2005). The arrows in Figure 1h illustrate how the size of these apparent differences could selectively reduce the fractional occupancy of state 2 for a low-affinity agonist such as glutamate.
In the kinetic scheme depicted in Figure 1g, once values of CO are similar to the values of the rate constants that determine channel opening and desensitization (β and δ), additional increases in CO will delay subsequent gating transitions because some channels will cycle multiple times between states 1 and 2 before they open or desensitize. These considerations, and the results of our previous kinetic modeling (Robert et al., 2005; Zhang et al., 2006), provide a possible explanation for the selective decrease in peak currents and the slower desensitization decays seen with glutamate for the T686A mutants. It is equally possible, however, that glutamate is a partial agonist at T686A channels because the mutation alters the nature or extent of LBD closure, and this differs for glutamate and quisqualate (Jin et al., 2003), especially because ligand–protein interactions for the two agonists, although similar, are not identical (Armstrong and Gouaux, 2000; Jin et al., 2002). To determine the effects of the T686A mutation on the conformation of the LBD, we introduced the T686A mutation into a construct encoding the ligand binding core of GluR2 (GluR2–S1S2J) and cocrystallized the mutant protein with glutamate and quisqualate.
The T686A mutation does not reduce the extent of binding cleft closure
Crystal structures of GluR2–S1S2 carrying the T686A mutation in complex with either glutamate or quisqualate were determined by molecular replacement using published wild-type GluR2–S1S2 structures as templates (Armstrong and Gouaux, 2000; Jin et al., 2002). The final glutamate- and quisqualate-bound structures were refined at 2.0 and 2.1 Å resolution, respectively (Table 1). Bound glutamate and quisqualate were clearly identified from their electron density in the T686A structures (Fig. 2). Unlike previous results for wild-type GluR2–S1S2, the asymmetric unit in both the glutamate- and quisqualate-bound T686A crystals contained only one protomer. However, the vast majority of the inter-protomer contacts in these crystals occurred along the twofold crystallographic axis seen in previous structures that contained an LBD dimer within the asymmetric unit. Although we cannot exclude minor effects of differences in crystal packing, there is no reason to think such potential effects significantly influenced the results.
Crystallographic data collection and refinement statistics

Stereo views of the electron density of glutamate and quisqualate bound to the LBDs of T686A S1S2J. Difference Fourier electron density maps (Fo-Fc) were generated without the coordinates of the ligands in their corresponding molecular models to confirm the presence of the ligands in the experimental diffraction data. a, b, Glutamate (a) and quisqualate (b) bound in the ligand-binding pocket are represented by both mesh electron density and stick model. The electron density of both the ligands was contoured at 2.5 σ.
To compare the degree of cleft closure in the wild-type and mutant structures, the domain 1 α carbons (residues 396–498 and 730–770) were superposed (Fig. 3a,b). For both glutamate and quisqualate, comparison of the positions of the α carbon atoms of G630 (one of two amino acids that replace the channel pore and link the S1 and S2 segments of the LBD in GluR2–S1S2J) indicated that domain 2 of the T686A mutant moved at least as close to domain 1 as it did in the corresponding wild-type structures (Table 2), although the rotation axis along which domain 2 moved was slightly different. The degree of cleft closure in the two T686A structures was similar for the two agonists. Although several partial AMPA receptor agonists cause incomplete closure of the LBD (Armstrong et al., 1998, 2003; Jin et al., 2003), the T686A structures argue against the possibility that the T686A mutation reduces the relative efficacy of glutamate because it selectively reduces the extent to which the LBD closes when glutamate is bound.

The T686A mutation disrupts cross-cleft hydrogen bonds but does not reduce LBD closure. a, b, Ribbon diagrams of the glutamate-bound (a) and quisqualate-bound (b) wild-type and T686A GluR2–S1S2J structures. Glutamate and quisqualate are shown in stick representation within the binding cleft (boxed) between domain 1 (top) and domain 2 (bottom). The wild-type (blue) and T686A (red) structures were superposed using the domain1 residues. Domain 2 has moved 2–3° farther in the T686A structures, closing the binding cleft more completely and displacing the bound ligands toward domain 1. c, d, Enlarged views of the boxed regions in a and b. The interdomain hydrogen bond present in the wild-type protein between E402 and T686 and the hydrogen bond mediated by a water molecule (W2) are shown as blue dashed lines. Both hydrogen bonds and W2 are absent in the T686A structures. The intradomain hydrogen bond between E402 and Y450 is shown in blue and red for the wild-type and mutant structures, respectively. The domains to which individual residues belong are indicated in parentheses.
Comparison of the extent of LBD closure in wild-type and T686 mutant protomers
We also obtained diffraction data for GluR2–S1S2 carrying the serine mutation at T686 in complex with glutamate or quisqualate. We were unable to obtain a satisfactory refinement of the dataset for quisqualate, but the glutamate-bound structure was refined at 1.7 Å (Table 1) and the LBD was closed to an extent similar to the corresponding structure for the T686A mutant (Table 2). In total, comparison of the different structures shows that the T686 mutations have minimal effects on the extent to which the LBD closes.
Changes in hydrogen bond interactions within the binding cleft
Detailed comparison of the GluR2-wt and T686A structures demonstrated that elimination of the direct cross-cleft hydrogen bond between the hydroxyl group of T686 and the ε oxygen of E402 resulted in reorientation of the χ2 torsion angle of the E402 side chain in the agonist-bound T686A structures, as well as reduction of the angle defined by Cα, Cβ, and Cγ atoms of the same residue. This geometric change moved the carboxyl group of E402 toward domain 1 (by 0.56 Å), without altering the distance of the hydrogen bond made with the Y450 hydroxyl group (Fig. 3c,d). In the glutamate-bound T686A structure, the side-chain of M708 is also oriented slightly differently, eliminating a hydrogen bond present in one wild-type protomer between the hydroxyl of T686 and the δ sulfur of M708 (Fig. 3c). In wild-type structures, T686 makes a water-mediated hydrogen bond with both ligands (Fig. 3c,d). The water molecule (W2) mediating this interaction is missing in the T686A structures, resulting in the loss of this bond as well as hydrogen bonds between the ligand and the domain 2 residue Y702 (see below). The loss of these direct- and water-mediated cross-cleft hydrogen bonds would be expected to destabilize the closed-cleft conformation of the LBD.
Effect of the T686 mutations on interactions at the mouth of the binding cleft and the hinge region
In wild-type GluR2–S1S2 structures, the peptide backbone between residues D651 and S652 assumes two different conformations. In all AMPA-bound structures and four of five quisqualate-bound protomers, the trans peptide bond between these residues is “flipped” 180° relative to its configuration in the apo structure or LBD structures in which the bound ligands are competitive antagonists or partial agonists (Armstrong et al., 1998; Armstrong and Gouaux, 2000; Jin et al., 2002), resulting in the formation of additional interdomain hydrogen bonds across the mouth of the binding cleft between the backbone atoms of these residues and the domain 1 residues Y450 and G451. The additional interactions are missing in two of three glutamate-bound protomers in which the peptide bond is “unflipped,” which may contribute to the lower potency of this agonist (Armstrong and Gouaux, 2000; Jin et al., 2002).
In both T686A structures, the trans peptide bond between D651 and S652 is in the flipped conformation and the carbonyl oxygens of these residues make interdomain hydrogen bonds like those seen in flipped wild-type conformers for both agonists. Similarly, in the glutamate T686S structure, the D651–S652 bond is flipped in both protomers that contain electron density for bound glutamate (A and B). The results indicate that changes in interactions across the mouth of the binding cleft are unlikely to contribute substantially to the effect of the mutations on channel properties.
The rotation of domain 2 evident in agonist-bound GluR2–S1S2 structures occurs around an axis that projects through helix I (Armstrong et al., 1998; Armstrong and Gouaux, 2000; Hogner et al., 2002), and the anti-parallel β strands that form the back of the binding cleft act as a hinge for the movement of domain 2. Comparison of wild-type apo and agonist-bound structures shows that the pivot point of the hinge occurs at the level of residues L498 and S729 in the two β strands. Two differences were noted in this region when agonist-bound structures were compared for wild-type GluR2–S1S2 and the T686A mutant. First, although the pivot point of hinge movement was unaltered, the kink in the two β strands was slightly increased in the mutants relative to wild-type protomers. Second, when Y405 and Y732 from domain 1 and T707 from domain 2 were aligned along the axis of hinge motion, the hydrogen bond distance between the residues from the two domains was increased in the T686A structures. The weakened interdomain hydrogen bonds appear to result because the LBD of GluR2–S1S2 and the T686A mutant closed along slightly different axes of rotation. Compounds that interact with the side chains of residues at the back of the hinge region have been suggested to slow deactivation of AMPA receptors by stabilizing the closed-cleft conformation of the LBD (Jin et al., 2005). The weakened interdomain hydrogen bonding seen in this region in the T686A mutant may contribute to destabilizing the closed-cleft conformation, thereby increasing the rate at which the binding cleft opens. This would be expected to hasten the dissociation of glutamate and speed deactivation, as was observed (Robert et al., 2005).
Changes in the solvent structure within the binding cleft
Although in general the ligand–protein interactions seen in the wild-type GluR2–S1S2 structures were very similar in the corresponding T686A structures, there were differences in the positions of water molecules in the T686A binding cleft that likely reflect its slightly more closed conformation relative to wild-type GluR2. Figure 4, a and b, shows detailed views of the binding cleft in the glutamate- and quisqualate-bound T686A mutants. The gray spheres show the position of waters in previously published wild-type structures, and the red spheres are waters in the T686A structures. In wild-type structures (Armstrong and Gouaux, 2000; Jin et al., 2002), water molecules W1 and W2 interact directly with glutamate or quisqualate, participating in a network of water-mediated hydrogen bonds between the ligands and the domain 2 residues L650, T686, and Y702 (Fig. 4a,b). In the T686A structures, the hydrogen bonds mediated by W1 are maintained, although the greater degree of cleft closure displaces W1 toward domain 1. Unlike the T686 hydroxyl group, the A686 methyl group cannot hydrogen bond, and W2 is missing in both mutant structures. However, in the quisqualate-bound structure, the mutant has gained a new water molecule, W3, that hydrogen bonds between the 4-nitrogen of quisqualate and the carboxyl oxygen of E402 (Fig. 4b). There is also a hydrogen bond between W3 and the hydroxyl group of Y450, making a tight hydrogen-bonding network between these domain 1 residues and the ligand. The additional interactions mediated by W3 in the quisqualate-bound structure may tether quisqualate to domain 1 once the cleft opens and slow its dissociation (Abele et al., 2000).

The T686A mutation alters water-mediated hydrogen bonds within the binding cleft. The T686A structures with glutamate (a) or quisqualate (b) are shown as ribbon diagrams (top). Domains 1 and 2 are colored green and blue, respectively. Water molecules from the wild-type structure (gray) and those from the T686A mutant structure (red) are represented as spheres. The regions around the water molecules are shown in detail in the zoomed views looking down the longitudinal axis of the binding cleft (middle). Glutamate and quisqualate bound to the T686A mutant are shown with the domain 1 and 2 residues colored green and blue. The corresponding wild-type residues are gray. The arrows indicate the apparent displacement of water molecules in the T686A mutant. The water molecule (W2) that is present in both wild-type structures and absent in the mutant structures, and the new water molecule (W3) in the T686A quisqualate structure, are circled in magenta and cyan, respectively. The zoomed views of the ligand binding cleft are rotated by 90° in the bottom row.
Some displacement of waters W6–W9 was also evident, which in both wild-type and mutant structures are just outside the mouth of the binding cleft (Fig. 4a,b). In both wild-type and mutant structures, these waters participate in cross-cleft hydrogen bonds between residues in domains 1 and 2. The minor displacements seen in the T686A structures may reflect the slightly different axis along which the cleft closes in the mutants.
The structural changes in the T686 mutants are consistent with destabilization of the closed-cleft conformation of the LBD
Although previous studies have shown that the extent to which the LBD closes is a major determinant of agonist efficacy (Armstrong et al., 1998, 2003; Armstrong and Gouaux, 2000; Jin et al., 2003), our structural results on the LBD of the T686A mutant suggest strongly that the mutation does not reduce the efficacy of glutamate by decreasing the extent to which the binding cleft closes. If anything, the LBD is slightly more closed in the agonist-bound T686A and T686S structures. Other structural differences between the wild-type and mutant structures are minor and likely reflect the slightly greater extent of cleft closure and the somewhat different axes along which the LBD closes in the mutants. Although we cannot exclude that these minor differences result in conformational changes in regions outside the LBD in the full-length receptor, nor have we measured LBD kinetics directly, the loss of interdomain interactions seen in the T686 mutants would be expected to destabilize the closed-cleft conformation of the LBD and is consistent with our previous proposal that the stability of this conformation influences channel gating (Robert et al., 2005).
Analysis of single-channel currents
To understand better the effect of the T686A mutation on gating, we compared single-channel currents through GluR2-wt and T686A channels. Because AMPA receptors desensitize strongly, we could routinely discriminate unitary currents in patches that contained a small number of channels (Fig. 1) and analyze their steady-state characteristics during long continuous applications of glutamate or quisqualate. The small size of the peak currents recorded in the patches analyzed (5–25 pA at −100 mV) and the low open probability under steady-state conditions make it likely that the vast majority of the events analyzed were currents through individual channels. In Figure 5, we show typical examples of such currents at the 4 kHz low-pass filtering imposed on the data before they were analyzed (digital Gaussian filter, −3 dB). The gray traces show the results of the idealization of the record with the SKM algorithm of QuB software (for details, see Materials and Methods).

Both GluR2-wt and T686A channels open to small, closely spaced, conductance sublevels. a–d, Inward single-channel currents (downward) through GluR2-wt channels (a, b) and T686A mutant channels (c, d). The currents were recorded at −100 mV during continuous applications of near-saturating concentrations of glutamate (a, c) or quisqualate (b, d). The top two traces in each panel show 2 s of continuous record. The bottom trace in each panel shows the openings marked by an asterisk in the traces above on an expanded timescale. The time and current calibrations in d apply to all panels. The data were sampled at 25 kHz and digitally low-pass filtered at 4 kHz. For each dataset, the entire record (2–3 min of continuous recording) was idealized with the SKM algorithm of QuB. The gray lines superposed on the expanded traces are the fits from this global idealization. The currents shown consist of multiple transitions between the three smallest open levels detected in each patch. The idealized record was used to calculate the proportion of time spent at each conductance level, as well as mean open and shut times (Fig. 6). The mean results of this analysis are given in Table 3.
As found previously for both recombinant and native AMPA receptors in patches containing only one channel (Rosenmund et al., 1998; Smith and Howe, 2000), both wild-type and mutant channels displayed multiple open levels. Three commonly visited open levels were detected in all patches analyzed, and channel activations typically contained many transitions between adjacent open levels (Fig. 5). Although infrequent and not evident in the traces in Figure 5, each record also contained openings to a larger conductance level of ∼36 pS (Table 3). The three smallest levels seen for both GluR2-wt and T686A channels are similar to conductance levels reported previously for homomeric recombinant AMPA receptor channels (Swanson et al., 1997; Derkach et al., 1999; Banke et al., 2000; Jin et al., 2003). Although rare, the 36 pS events seen with either glutamate or quisqualate in all patches analyzed are similar to large conductance openings reported previously for both recombinant and native AMPA receptors (Smith et al., 2000; Tomita et al., 2005).
Single-channel properties for wild-type and T686A GluR2 channels
The relative proportions of open time spent at each conductance level are given in Table 3. For both wild-type and T686A channels, most openings were to the two smallest open levels, although all our results were obtained at agonist concentrations at which the channels should often be fully occupied. Similar results were obtained before for GluR4 channels (Tomita et al., 2005), and results for GluR2-Q channels and native channels in hippocampal neurons are also consistent with a predominance of openings to small conductance states at high glutamate concentrations (Mansour et al., 2001; Gebhardt and Cull-Candy, 2006).
In previous studies in which large conductance openings were common at high receptor occupancy, channel desensitization was greatly reduced (Rosenmund et al., 1998; Smith and Howe, 2000). In our experiments, desensitization was intact and the openings we observed presumably occur during short-lived escapes from the desensitized states that predominate during prolonged applications of agonist. Therefore, one possible explanation for the rarity of large conductance openings is that channels return from desensitization preferentially to smaller conductance levels. If this were true, openings to larger open levels would be expected to be more frequent at the beginning of applications of high concentrations of agonist than we observed under steady-state conditions (Tang et al., 1989).
To investigate this possibility, we examined currents evoked at the beginning of rapid agonist applications (10 or 50 mm glutamate and 1 or 5 mm quisqualate) in patches that appeared to contain one to three channels. The high open probability at the beginning of the applications and the presence of multiple channels in most patches prohibited a quantitative analysis, but examination of hundreds of records in each patch gave no evidence that the channels preferentially open to large conductance levels at the onset of the applications. We cannot exclude the possibility that large conductance openings do preferentially occur early in the applications but are brief and therefore incompletely resolved. However, when desensitization is intact, both our steady-state and rapid application results suggest that openings to the two smallest levels predominate even at agonist concentrations at which the receptors are fully occupied.
T686A decreases the proportion of large conductance openings
If the LBD of an individual subunit must be closed for it to gate, then the unitary conductance of AMPA-type channels may depend not on the number of subunits occupied but rather on the number of subunits with closed binding domains. If the kinetics of LBD closing and opening are rapid, then even receptors with four agonist molecules bound may rarely have four binding domains closed simultaneously, and destabilizing the closed conformation of the LBD should further decrease the proportion of openings to large conductance levels (because reducing the time each LBD is closed will reduce the probability that multiple binding domains are closed simultaneously). As can be seen in Table 3, the T686A mutants do indeed open to larger conductance levels less often with both agonists, and the difference from wild-type channels is greater for the low-affinity agonist glutamate. We also determined the relative proportion of transitions between the different conductance levels (including the closed level) for both agonists and each type of channel (supplemental Table 1, available at www.jneurosci.org as supplemental material). The proportion of transitions between the two smallest levels and the closed level was increased in the T686A mutants, and the frequency of transitions between the three largest conductance levels was reduced. Although glutamate and quisqualate gave similar results for GluR2-wt channels, the increase in closed-state transitions was greater for glutamate than for quisqualate. The results of this analysis support the prediction that decreasing the stability of the closed-cleft conformation reduces the probability that individual subunits contribute to activation gating, decreasing the likelihood that the channels gate to large conductance states.
T686A changes closed-time distributions
Although the T686A mutation altered the substate prevalence of GluR2 channels, the differences found between glutamate and quisqualate are not large enough to account for the differences in the size of the peak currents seen with the two agonists for the T686A mutant channels (Fig. 1d). We therefore also analyzed open and shut times for wild-type and mutant channels. Although statistically significant differences in mean apparent open times were observed for glutamate (Fig. 6a), the most consistent and substantial changes were seen for closed times. With each agonist, closed-time distributions for GluR2-wt channels contained four distinguishable components (Fig. 6b). The briefest two components, which were similar for glutamate and quisqualate, are likely to reflect the gating kinetics of individual channels and arise from shuttings with mean durations of 300–400 μs and ∼3 ms, respectively (Table 3). For both agonists, shuttings with durations <1 ms were common for wild-type channels but much less frequent for T686A mutant channels. The time constants of the two briefest shut-time components were approximately fivefold larger for glutamate. The T686A mutation also increased the duration of the briefest shuttings seen with quisqualate, but the effect of the mutation was significantly less than observed for glutamate (Table 3).

Comparison of open and shut times for GluR2-wt and T686A mutant channels. a, Weighted mean ± SEM open times for the three smallest conductance levels for GluR2-wt and T686A channels activated by glutamate or quisqualate (n = 4–6 patches). For each patch, log-binned histograms of open times for each conductance level were fitted with exponential functions. Two exponential components were evident in most distributions of mean open times for each of the three smallest conductance levels, although brief open times predominated (>90% of total area). When two components were evident, the biexponential fits to the results were used to calculate weighted mean open times. By two-tailed Student's t test, *p < 0.5 and **p < 0.01. b, Examples of shut-time distributions for GluR2-wt and T686A channels activated by glutamate or quisqualate and fitted with four exponential components using log-binned fitting procedures (dashed lines). The y-axis is arithmetic to facilitate comparison of the relative areas of the different components.
It is tempting to conclude from the results in Table 3 that the T686A mutation caused an increase in the mean duration of both types of brief shutting. However, interpretation of the results is complicated by the effect of the mutation to speed recovery from desensitization (Robert et al., 2005). Under steady-state conditions, channel openings occur when individual channels escape from desensitized states. Wild-type receptors spend most of their time desensitized, and it is unlikely that two channels recover at nearly the same time in patches containing only a few receptors. Brief shuttings in wild-type records are therefore likely to arise from activation gating transitions that reflect the behavior of the same receptor molecule. The similarity of the two brief shut-time components seen with glutamate and quisqualate, agonists that display significantly different rates of recovery (Zhang et al., 2006), also makes it unlikely that recovery contributes substantially to these events. For the T686A mutant, however, the channels recover from desensitization faster, increasing the likelihood that brief shuttings reflect recovery, especially when multiple channels are present. Such shuttings probably contribute substantially to the second component discriminated in shut-time distributions for the T686A mutant, especially with glutamate, and the apparent slowing of this component may not reflect changes in activation gating. An equally plausible conclusion from the shut-time results in Table 3 is that the mutation greatly reduced the brief component of shuttings and had minimal effects on the duration of shuttings that contribute to the second component seen for wild-type receptors. Deciding between these two possibilities requires some indication of the gating rearrangements that give rise to the brief shuttings seen for wild-type channels.
One striking feature of the single-channel currents we recorded was that channel activations consisted primarily of many direct transitions between adjacent open levels (Fig. 5). In total, transitions between open and closed levels represented only approximately one-third of the events detected with either agonist for wild-type channels; brief shuttings within channel activations were relatively rare and occurred mostly from the smallest conductance levels (supplemental Table 1, available at www.jneurosci.org as supplemental material). These findings are in stark contrast to NMDA-type channels in which open–closed transitions predominate and closings occur primarily from the largest open level (Howe et al., 1991; Stern et al., 1994). If the conductance of AMPA-type channels is directly correlated with the number of subunits with closed LBDs, then the brief closings detected in our records may reflect the opening and closing of the LBD of a single subunit (in a channel in which the LBDs of the other subunits are open), and the duration of the brief shuttings that occur between small conductance openings may be influenced by the kinetics of LBD opening and closing. To understand better the effect of the T686A mutation on activation gating, we recorded single-channel currents evoked by rapid applications of glutamate and quisqualate.
The T686A mutation delays channel activation
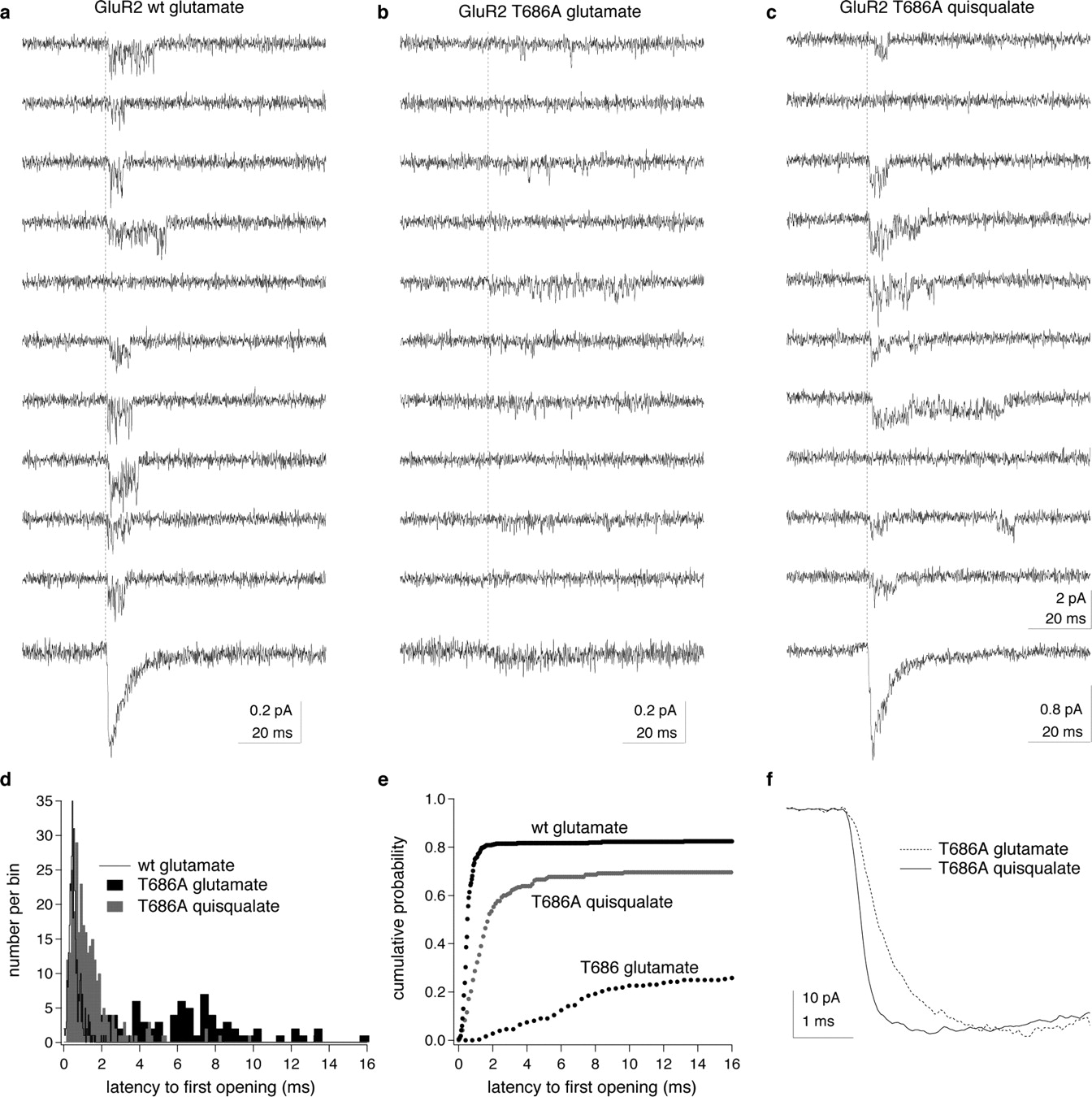
The simple model of AMPA-receptor gating in Figure 1 suggests that, once the rate constant CO is larger than β, individual subunits will cycle multiple times between states 1 and 2 before they revisit state 3. Such cycling would tend to increase the duration of shuttings from small conductance levels and delay opening of the channels when they are rapidly exposed to a high concentration of agonist. Inspection of individual concentration jumps in patches containing at most three active channels strongly indicated that the T686A mutation delayed channel activation. To quantify these observations, we measured latencies to first opening in our two best patches; one contained a single GluR2-wt channel and the other had two T686A channels.
Figure 7 shows example records from the two patches and the results of our analysis. In the patch containing the single wild-type channel (Fig. 7a), the vast majority of the rapid jumps into 10 mm glutamate resulted in a series of channel openings that began within 1 ms of exposure to the agonist. Similar results were obtained with 5 mm quisqualate in the patch containing two T686A mutant channels (Fig. 7c). In contrast, channel openings occurred significantly later in the applications when 50 mm glutamate was applied to the T686A patch, and multiple brief openings were often separated by sojourns at the closed level that had durations >1 ms (Fig. 7b). The proportion of records that appeared to contain no openings also increased. The probability that a channel opened at least once in response to agonist was 0.84 for glutamate and the GluR2-wt channel, whereas the corresponding values for quisqualate and glutamate in the patch containing the T686A mutant channels were 0.73 and 0.66, respectively.

The T686A mutation delays channel opening. a–c, Examples of unitary currents evoked by rapid applications of 10 or 50 mm glutamate (a, b) or 5 mm quisqualate (c). The vertical dotted lines show the onset of the applications, and the bottom traces are the mean waveforms obtained from averaging all the individual records obtained with the respective agonists in each patch. The patch in a contained one active channel. The records in b and c are from the same patch. The quisqualate data were collected first, and two channels were active. The relative sizes of the average currents evoked by the two agonists suggest that only one of these channels was active during the subsequent application of glutamate (bottom traces in each column). d, Latency-to-first-opening distributions from the data illustrated in a–c. For the patch in a, 68 of 440 trials did not contain detectable openings. For the patch in b and c, 56 of 167 trials with glutamate and 23 of 313 trials with quisqualate contained no detectable openings. The probability that an individual channel opened at least once in response to agonist was therefore 0.84 for glutamate and GluR2-wt channels, and 0.66 and 0.73 for T686A channels with glutamate and quisqualate, respectively (Aldrich et al., 1983). e, Same results as in d displayed as cumulative probability density functions. The T686A mutation delays channel activation for both agonists but more so for glutamate than for quisqualate. For glutamate and T686A, 36 trials contained openings in which the latency-to-first-opening was >16 ms. f, Activation phase of inward currents evoked by 50 mm glutamate and 5 mm quisqualate in a patch containing multiple T686A channels (same patch as in Fig. 1d–f). Note the slower rise of the current with glutamate.
For glutamate and the T686A mutant, the delay in channel activation that is evident in the first-latency distributions shown in Figure 7, d and e, desynchronizes channel openings and substantially reduces the peak inward ensemble current. It is this delay, not longer openings, that slows the decay of the ensemble glutamate-evoked current relative to that seen for wild-type channels or quisqualate (Fig. 1d). The agonist-selective delay in activation also selectively slows the rise time of macroscopic currents evoked by glutamate (Fig. 1f). On average, the 10–90% rise times of macroscopic currents evoked with 50 mm glutamate (984 ± 234 μs) were twofold slower than the corresponding rise times for currents evoked in the same T686A patches by 5 mm quisqualate (500 ± 120 μs; n = 5 patches). The results are consistent with the conclusion that the T686A mutation selectively reduces the efficacy of the low-affinity agonist glutamate and slows both activation and entry of glutamate-bound channels into long-lived desensitized states by substantially reducing the probability that the channels occupy closed-cleft states from which gating proceeds.
Discussion
The T686A mutation does not alter the extent to which the binding domain closes
Previous work has shown clearly that the extent to which the LBD closes is a major determinant of agonist efficacy and that partial agonists cause less closure of the binding cleft than full agonists (Armstrong et al., 1998; Armstrong and Gouaux, 2000; Jin et al., 2003). Although the T686A mutation reduces the relative efficacy of glutamate, our crystallographic results strongly indicate that the T686A mutation does not reduce the extent to which the LBD closes during agonist binding. All glutamate- and quisqualate-bound T686 mutant protomers are closed to an extent similar to that observed previously for wild-type GluR2–S1S2, and comparison of the wild-type and T686 mutant structures indicate that both the global conformation of the LBD and ligand–protein interactions are minimally affected by the mutation. The similarity of the glutamate- and quisqualate-bound T686A structures make it highly unlikely that glutamate is a partial agonist at T686A channels because the mutation selectively reduces the extent to which the LBD closes for this agonist.
We did observe partially closed conformations for protomers C and D in the T686S structure (Table 2), which are somewhat reminiscent of previous work on a binding cleft mutation (L650T) that rendered AMPA a partial agonist (Armstrong et al., 2003). In this study, one of seven AMPA-bound protomers showed intermediate domain closure, which led Armstrong and colleagues to conclude that the LBD can assume multiple agonist-bound conformations and that the L650T mutation decreased the efficacy of AMPA by increasing the equilibrium occupancy of AMPA-bound conformations in which the LBD is only partially closed. Our results differ from the previous L650T results, however, because there was no detectable electron density for bound ligand in the partially closed protomers (C and D), although the electron density for side chains in the binding pocket that normally interact with glutamate was well defined in these protomers. It is possible that the partially closed conformation of protomers C and D is a crystallographic artifact or that protomer D contains bound glutamate that is too disordered to give rise to detectable electron density, but we favor the interpretation that promoters C and D in the T686S structure represent authentic apo conformations.
As expected, the T686A mutation results in the loss of direct cross-cleft hydrogen bonds with E402, as well as hydrogen bonding to both glutamate and quisqualate that is mediated by the water molecule (W2) missing in the T686A structures. The loss of these hydrogen bonds would be predicted to reduce the stability of the closed-cleft conformation of the LBD. Most other changes are consistent with the somewhat greater cleft closure seen when compared with the wild-type counterparts (which may result because the loss of the T686–E402 interactions results in reorientation of the side chain of E402 toward domain 1), as well as the slightly different axes along which the LBD closes. The changes are relatively minor and their effects are difficult to quantify, but overall the T686A structures reveal a loss of interdomain interactions. The structural data support the hypothesis that the primary effect of the mutation is to increase the rate at which the LBD opens.
Binding domain closure and ion channel gating
The effect of the T686A mutation on single-channel currents provides the most direct evidence to date that closure of the LBD necessarily precedes channel gating. The delayed activation of the channels seen as increased latencies to first opening in the rapid application experiments, as well as the shift in substate prevalence away from large conductance events under steady-state conditions, are strong indications that the kinetics of conformational changes at the level of the LBD directly impact AMPA receptor gating. The changes in single-channel behavior we observed are consistent with the predictions illustrated in Figure 1, g and h, in which increases in the rate constant for LBD opening (CO) not only decrease the apparent affinity of agonist binding and hasten deactivation (Robert et al., 2005) but also reduce the occupancy of states from which gating occurs.
Although increases in the value of CO are predicted to delay gating, they should not decrease the likelihood that an individual channel opens in response to a rapid and sustained application of agonist. One explanation for the increased proportion of “failures” seen for T686A channels (Fig. 7) is that openings are missed more often for the mutant channels, because both large conductance openings are less common and sequential openings do not occur in rapid succession. Alternatively, the T686A mutation may produce some alteration of the rate constants for channel opening or entry into desensitization (smaller β/δ ratio).
Incorporation of the LBD closing and opening steps into a kinetic model for the complete tetrameric receptor provides a possible explanation for the unusual character of bursts of openings quantified here and the rarity of openings to the largest conductance levels even at saturating agonist concentrations (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). For GluR2-wt and T686A channels, bursts of openings consist primarily of step-like transitions between adjacent conductance levels (supplemental Table 1, available at www.jneurosci.org as supplemental material). One interpretation of these findings is that the transitions occur when the LBDs of individual subunits open and close. When the receptors are suddenly exposed to a high concentration of agonist, the binding domains of some subunits are likely to close before all subunits are occupied and many channels may desensitize before full occupancy is reached (supplemental Results and Discussion, available at www.jneurosci.org as supplemental material).
In summary, our results strongly support the view that closure of the LBD is a necessary prerequisite for channel gating and support our previous proposal that the stability of the closed-cleft conformation influences agonist efficacy. Recent work on glycine (Burzomato et al., 2004; Plested et al., 2007) and nicotinic acetylcholine receptors (Purohit et al., 2007), as well as NMDA-type glutamate receptors (Popescu et al., 2004; Erreger et al., 2005; Maier et al., 2007), also underscores the importance of pregating conformational changes for channel function. The ability of synapses to follow high-frequency firing requires a transmitter that produces rapid and effective receptor activation and also dissociates quickly. The large decrease in peak open probability associated with the relatively modest decreases in agonist potency that result from the T686A mutation (Robert et al., 2005) suggest that the rates at which the LBD opens and closes in response to glutamate binding are nearly optimal to meet the dual requirements that glutamate be a strong agonist and yet dissociate quickly from the receptor.
Footnotes
-
This work was supported by National Institutes of Health Grants NS047712 and NS057725. We thank Aiwei Sui for excellent technical assistance and Eric Gouaux, Neali Armstrong, and Elizabeth Valentine for helpful advice on expression and purification of GluR2S1S2J. W.Z. performed the electrophysiology experiments and analyzed the single-channel results. Y.C. crystallized the proteins, collected the diffraction data, and solved the T686S and T686A structures. E.L. and J.R.H. directed the crystallographic and patch-clamp studies, respectively. All authors contributed to the interpretation of the results, preparation of the figures, and writing the manuscript.
- Correspondence should be addressed to James R. Howe, Department of Pharmacology, Yale University School of Medicine, SHM B-251, 333 Cedar Street, New Haven, CT 06520-8066. james.howe{at}yale.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}