

Abstract
Axons of the mammalian CNS lose the ability to regenerate soon after development due to both an inhibitory CNS environment and the loss of cell-intrinsic factors necessary for regeneration. The complex molecular events required for robust regeneration of mature neurons are not fully understood, particularly in vivo. To identify genes affecting axon regeneration in Caenorhabditis elegans, we performed both an RNAi-based screen for defective motor axon regeneration in unc-70/β-spectrin mutants and a candidate gene screen. From these screens, we identified at least 50 conserved genes with growth-promoting or growth-inhibiting functions. Through our analysis of mutants, we shed new light on certain aspects of regeneration, including the role of β-spectrin and membrane dynamics, the antagonistic activity of MAP kinase signaling pathways, and the role of stress in promoting axon regeneration. Many gene candidates had not previously been associated with axon regeneration and implicate new pathways of interest for therapeutic intervention.
Introduction
The capacity of neurons to regenerate rapidly decreases with age due to an inhibitory CNS environment and ineffective activation of the intrinsic regeneration program (Yiu and He, 2006; Liu et al., 2011). In the case of DRG neurons, treatment with a preconditioning lesion in the peripheral axon can overcome the barrier to CNS regeneration (Neumann and Woolf, 1999). In experiments with retinal neurons, embryonic axons regrow into the tectum from older animals with an established inhibitory glial environment (Chen et al., 1995). Conversely, older retinal axons fail to regrow, even into embryonic tectum with its permissive glial environment. These results demonstrate that neurons are capable of regenerating but lose this capacity over time, possibly favoring stabilized connectivity. Therapeutic interventions aimed toward treating axonal injury would ideally target the intrinsic mechanisms responsible for regeneration in preconditioned or embryonic neurons.
Axon severing activates a retrograde signal that is delivered to the cell body from the site of injury (Abe and Cavalli, 2008). Diverse cellular processes are recruited to mount a successful response. The neuron reverts to its developmental program, upregulating transcription factors along with new protein and lipid synthesis to promote cell survival, neurite outgrowth, and guidance (Harel and Strittmatter, 2006). Many individual molecules have been implicated in the intrinsic mechanisms of axon regeneration (Raivich and Makwana, 2007; Sun and He, 2010; Liu et al., 2011; Bradke et al., 2012). A key component of the injury signal is phosphorylated JNK, which activates the AP-1 transcription factor c-Jun. Other transcription factors such as STAT3, CREB, ATF3, CEBP, and SOX11 are induced upon peripheral nerve injury. Axonal transport of mRNA is also an important outcome of the injury response. Hundreds of mRNAs have been identified and localized to axons in studies of regenerating neurons (Willis et al., 2005; Gumy et al., 2010). Activation of the mTOR pathway, which is involved in regulating protein translation and ribosome biogenesis, is emerging as an important indicator of regenerative capacity (Park et al., 2010). Although many pro-regenerative molecules are known, the essential mechanisms needed for successful regeneration of CNS neurons remains elusive.
Caenorhabditis elegans has emerged as an attractive model with which to study axon regeneration (Wu et al., 2007; Gabel et al., 2008; Chen and Chisholm, 2011). The availability of mutants makes it possible to perform candidate screens (Chen et al., 2011). High-throughput screening of drug compounds influencing regeneration has also been performed (Samara et al., 2010). However, unbiased genetic screens for regeneration genes have not been practical in any model system. We discovered that β-spectrin mutation causes axons to break spontaneously due to mechanical stress (Hammarlund et al., 2007). Once broken, the axon responds by forming a growth cone and extending the axon back toward its target. In the β-spectrin mutant, this results in successive rounds of breakage and regeneration. We used this phenotype as the basis for an RNAi screen for genes affecting regeneration and identified >70 candidate genes. Candidates include growth-promoting and growth-inhibiting factors. Several candidate genes have been implicated previously in regeneration and others define new and conserved pathways of interest.
Materials and Methods
C. elegans strains and isolation of Mos1-targeted deletions.
Hermaphrodites were maintained on HB101 E. coli-seeded NGM plates according to standard methods (see Tables 1, 2, 3, and 4 for the alleles used in this study). Generation of the basDf1 (cst-1) deletion was described in detail previously (Frøkjaer-Jensen et al., 2010). sav-1(bas1), F26A3.4(bas2), Y57G11C.33(bas3), and nex-2(bas4) were targeted according to the same method starting with the Mos1 insertion alleles ttTi35282, ttTi6567, ttTi40320, and ttTi35004, respectively.
RNAi screening.
RNAi was performed by feeding (Kamath and Ahringer, 2003). Bleached embryos from MJB1046 [basIs1, oxIs268, unc-70(s1502), eri-1(mg366); lin-15(n744)] gravid hermaphrodites were placed on RNAi bacteria at a concentration of 60–80 embryos per plate. Plates were maintained at 15°C for 12–13 d. L4-stage F1 progeny were then mounted onto slides and scored by counting all GABA commissures contacting the dorsal cord. A minimum of 10 animals were scored for each RNAi clone. For axotomy experiments, early larval EG5375 [oxIs268, eri-1(mg366), lin-15(n744)] hermaphrodites were placed on RNAi bacteria and grown at 20°C. L4-stage F1 progeny were prepared for axotomy and imaged as described in Hammarlund et al. (2009).
Molecular biology and transgenics.
Plasmids were constructed using multisite Gateway cloning (Invitrogen) in which a promoter region was cloned into the [4–1] donor vector, the gene of interest cloned into the [1–2] donor vector, and the unc-54 3′ UTR cloned into the [2–3] donor vector. Promoter-gene combinations used in this study are indicated in Table 3. Kinase-dead jnk-1 constructs were generated by PCR-based site-directed mutagenesis to change Lys148 and Lys149 to Ala (Pdpy-30:jnk-1 KK/AA) and Thr276 and Tyr278 to Ala (Pdpy-30:jnk-1 TY/AA). The activated sek-6 construct (Psek-6:sek-6 DD) was designed to change Ser219 and Thr223 to Asp. fos-1 T304A was generated by subcloning the StuI-NcoI fragment from pDRS53 (Sherwood et al., 2005) into Litmus28 (NEB), followed by PCR-based site-directed mutagenesis to change Thr 304 to Ala. The mutant StuI-NcoI fragment was then used to replace the wild-type fragment in pDRS53. The dominant-negative fos-1 was cloned from fos-1b cDNA and encodes a truncated protein including the first 240 aa of the fos-1b sequence. Transgenic animals were obtained according to standard methods and all constructs were injected at 20–30 ng/μl.
Laser axotomy and time-lapse imaging.
Axotomy and time lapse microscopy was performed as described previously (Hammarlund et al., 2009; Williams et al., 2011). L4 hermaphrodites (unless noted) were subjected to axotomy, recovered 18–24 h, and then prepared for confocal imaging. Regeneration was quantified by scoring the percentage of severed axons that formed a new growth cone and/or grew a distance of 5 μm or more.
Results
An RNAi-based screen for axon regeneration genes
Embryonic neurons lacking the cytoskeletal component β-spectrin develop normally. After hatching, unc-70/β-spectrin mutant neurons undergo spontaneous, movement-induced axotomy followed by regeneration (Hammarlund et al., 2007). Most commissural axons eventually break and regenerate before the animal reaches adulthood. As the animal ages, there is a progressive failure of regeneration with each cycle of axotomy and regeneration so that adults display a severely abnormal nervous system. We exploited this phenotype of unc-70/β-spectrin mutants to screen for genes required for axon regeneration. We reasoned that RNAi knock-down of a gene required for regeneration would result in an increased number of broken axons in the unc-70 mutant. The axons continue to break due to movement, however, regeneration would fail due to RNAi of the candidate gene. This screen provides an unbiased approach to identify novel gene candidates with a function that may not have been associated previously with neuronal regeneration.
We used the OrthoMCL database (www.orthomcl.org) to identify a subset of 5500 C. elegans genes with human orthologs. A majority of these genes were represented in the existing RNAi-feeding libraries (Kamath and Ahringer, 2003). In total, we screened 5076 RNAi clones in an unc-70 mutant sensitized for enhanced RNAi in neurons (Wang et al., 2005). We used a fluorescent marker to visualize the 19 D type motor neurons. The d-type neuron cell bodies lie in the ventral nerve cord and each extends a process anteriorly, which then branches circumferentially and grows to the dorsal nerve cord (Fig. 1A). In unc-70 mutants, the motor neurons have variable and highly age-dependent defects. To assay regeneration, we selected animals at the L4 stage and quantified the number of commissures that contacted the dorsal nerve cord. In wild-type animals, it is generally possible to score 16–17 commissures (two commissures often exit from the left side of the ventral nerve cord and are out of the plane of focus or commissures may fasciculate and be counted as a single commissure; Fig. 1A). unc-70 mutants grown on control RNAi show a range of 8–10 commissures contacting the dorsal cord (average 9.6 ± 1.8, n = 110; Fig. 1B). Branched axons, visible growth cones, path-finding defects, and gaps in the dorsal nerve cord are all characteristics of axons that have broken and regenerated. We scored unc-70 animals fed RNAi clones and selected candidates with the following classification criteria: strong (average commissure ≤4.5), moderate (average commissure number between 4.6 and 5.5), and weak (average commissure number between 5.6 and 6.9) (Fig. 1C).

Axon regeneration genes identified by genetic screening. A–F, GABA neurons visualized with Punc-47:GFP. Animals are arranged with posterior at left, ventral side down. For axotomy, five to six posterior commissures were severed at the midline. Scale bar, 20 μm. A, Wild-type. B, unc-70 mutant fed empty vector control RNAi bacteria. C, unc-70 mutant fed an RNAi clone targeted against a candidate regeneration gene. D, Wild-type axon regeneration after axotomy. E, Axotomy in a mutant with improved regeneration. F, Axotomy in a mutant with blocked regeneration. G–H, Percentage of growth cone formation in candidate genes characterized by axotomy. Mutants showing significantly improved regeneration are in green and mutants showing significantly decreased regeneration are in red. G, Candidate genes identified in the unc-70 RNAi screen. pxn-2 was characterized by Gotenstein et al. (2010) and is not included on this graph. H, Additional candidate genes identified by axotomy. Error bars indicate 95% confidence interval.
Of the clones we selected, 25% (1258 clones) resulted in P0 sterility, larval arrest, or lethality and were not screened further and 1% (53 clones) did not grow in culture. Eighty-five percent of the 3765 clones we screened had no phenotype in this assay. The 488 candidate clones resulting in a weak phenotype in the primary screen were not tested further, whereas strong or moderate candidates were repeated and confirmed in additional RNAi experiments, resulting in 70 candidate regeneration genes (Table 1). RNAi of a small number of candidate genes led to improved regeneration in the unc-70 mutant background. However, in these cases, the phenotype was due to paralysis and suppression of axon breakage (Hammarlund et al., 2007). We did not identify RNAi clones improving regeneration outcomes in unc-70; the reasons for this are described in Results and Figure 2. Several of the gene candidates have been implicated previously in cytoskeletal function or axon outgrowth during development. The majority of candidates identified were signaling molecules or proteins involved in transcription and translation. Others represent cell surface or secreted proteins, molecules involved in cellular metabolism, and conserved proteins with unknown function.
Candidate regeneration genes identified by RNAi screening
To characterize the role of candidate genes independently of unc-70, we performed laser axotomy in a mutant background or in animals fed RNAi clones targeted to specific genes (Table 1). We assayed regeneration in the GABA motor neurons of L4-stage animals. In the wild-type, ∼70% of severed axons initiated a growth cone and regrew beyond the site of axotomy (Fig. 1D). However, growth cone motility and guidance is inefficient at this stage and by 24 h postaxotomy few growth cones (5–10%) successfully reached targets in the dorsal nerve cord. Thirty-one of 70 candidate genes we identified by screening displayed a regeneration phenotype upon axotomy (Fig. 1G). Sixteen of 31 candidates showed improved regeneration in the mutant background and 15 of 31 mutants blocked regeneration (Fig. 1E,F). Mutations improving regeneration result in an increased likelihood of growth cone formation and improved migration to the dorsal cord. Mutations blocking regeneration reduced the likelihood of growth cone formation, but in most cases did not eliminate all growth cones (Fig. 1G). We tested 65 additional mutants based on association with screen candidates and/or based on homology to genes of interest from the literature; of these, 13 show improved growth cone formation and nine had reduced growth cone formation (Fig. 1H; Table 2). The result of our screens described here includes at least 50 new genes implicated in C. elegans axon regeneration.
Candidate regeneration genes tested by axotomy
UNC-70/β-spectrin is required to stabilize the axon during regeneration
In designing and executing the screen, we expected to find genes required to either promote or inhibit regeneration, but instead, we only identified RNAi clones that inhibited regeneration in unc-70 mutants. Despite this, several candidates that showed a strong regeneration block in the unc-70 background showed improved regeneration when assayed by axotomy in the mutant strain alone. To resolve this apparent contradiction, we further characterized unc-70 mutant axons by time-lapse microscopy, which provided a more detailed understanding of the defects caused by loss of β-spectrin.
When an axon is severed, there is a brief influx of calcium before the axonal membrane is repaired (Ziv and Spira, 1995). As a result, one of the earliest responses of the cell involves activation of the calcium-dependent protease calpain. Calpain activation, in turn, leads to degradation of cytoskeletal components and cellular restructuring essential for successful regeneration (Spira et al., 2003; Bradke et al., 2012). Spectrin, which links the plasma membrane to the cytoskeleton, is a major target of calpain. It has also been proposed that removal of spectrin facilitates vesicle fusion to the plasma membrane, which is necessary for growth cone expansion (Spira et al., 2003). Given these observations, we predicted that unc-70 mutants lacking β-spectrin would show early growth cone formation. Indeed, after axotomy, growth cone initiation occurred earlier in unc-70 mutants relative to the wild-type (Fig. 2A). We also observed that, upon axotomy, the proximal axon fragment in unc-70 mutants retracted further. The majority of wild-type axon fragments retracted a short distance between 3 and 5 μm, whereas unc-70 mutants displayed a much wider range between 3 and 20 μm, averaging 11 μm (Fig. 2B).

Axon regeneration defects in unc-70/β-spectrin mutants. A, Timing of first growth cone (GC) appearance in unc-70 mutants compared with wild-type. Each point represents an individual time-lapse experiment. B, Axon retraction distance after axotomy. Each point represents an individual cut axon. C, Quantification of spontaneous growth cone formation in unc-70 mutants per hour per commissure. ***p < 0.001; *p < 0.05, t test. Error bars indicate SEM. D, Schematic drawing showing spontaneous growth cone formation in unc-70 and possible consequences to outgrowth: (a) the formerly intact axon breaks, (b) the new growth cone extends to a nearby, intact axon causing it to break, and (c) the migrating growth cone itself breaks and generates a new growth cone. E, Time-lapse recording shows the unc-70 migrating growth cone (arrowhead) extending across the adjacent commissural axon (arrow) and causing it to break. The intact commissure (asterisk) dynamically extends and retracts branches over the course of the time lapse. At 48 minutes, the growth cone has broken the adjacent axon and the proximal end rapidly retracts (lower arrow), leaving behind fragments that are slowly cleared (top arrow, 48–159 min). At 132–159 minutes, the axon behind the migrating growth cone breaks (arrowhead, 132 min) and the proximal axon fragment retracts back to the branch point (left arrowhead, 159 min), leaving the distal process to degenerate (right arrowhead, 159–225 min). At 291–528 minutes, both stumps regenerate new growth cones. Scale bar, 10 μm.
Spontaneous growth cones do not occur in wild-type animals, but they do regularly appear along both severed and intact axons in unc-70 mutants. It is possible that small, mechanical stresses on the axon that are insufficient to cause a break may be enough to trigger a localized calcium increase and thus growth cone initiation. This is supported by the observation that spontaneous growth cones are significantly less frequent when unc-70 is paired with the muscle myosin mutant unc-54 (Fig. 2C). These double mutants are paralyzed, so the axons experience less mechanical stress.
A spontaneous growth cone appearing in an unc-70 mutant will either collapse and be resolved or it may extend for some distance, contributing to the axon's branched morphology. Growth cone extension in unc-70 can lead to several possible outcomes: (1) the intact axon sprouting a growth cone may itself fully break; (2) the growth cone may extend along a nearby, intact commissure, causing it to break; or (3) the newly regenerated axon behind the growth cone may break (Fig. 2D,E). In each case, the tension created by growth cone migration along its substrate is sufficient to induce new breaks. This observation accounts for the fact that we did not identify candidate genes with improved regeneration in the unc-70 mutant background. Improved regeneration increases the frequency of growth cone formation, which ultimately induces a greater number of breaks in mutants lacking β-spectrin. Over time, this leads to the appearance of more failed regeneration attempts. As an example, we characterized unc-130;unc-70 double mutants by time-lapse microscopy. Axotomy in unc-130 single mutants results in increased growth cone formation relative to the wild-type, whereas unc-130 RNAi in unc-70 led to a strong decrease in the number of intact commissures (Table 1). The increased number of growth cones in unc-130;unc-70 was evident by time-lapse microscopy. We observed a nearly threefold increase in the number of spontaneous growth cones in unc-130;unc-70 compared with unc-70 alone (Fig. 2C). The eventual outcome is that, despite increased growth cone formation, by the end of an imaging period, there were fewer intact commissures in unc-130;unc-70 compared with unc-70 (40/65 commissures intact in unc-70 vs 23/65 commissures in unc-130;unc-70). Together, our data suggest that the spectrin cytoskeleton is a barrier to growth cone formation and must be removed, but is later required to reform and stabilize the axon during outgrowth.
Membrane dynamics during axon regeneration
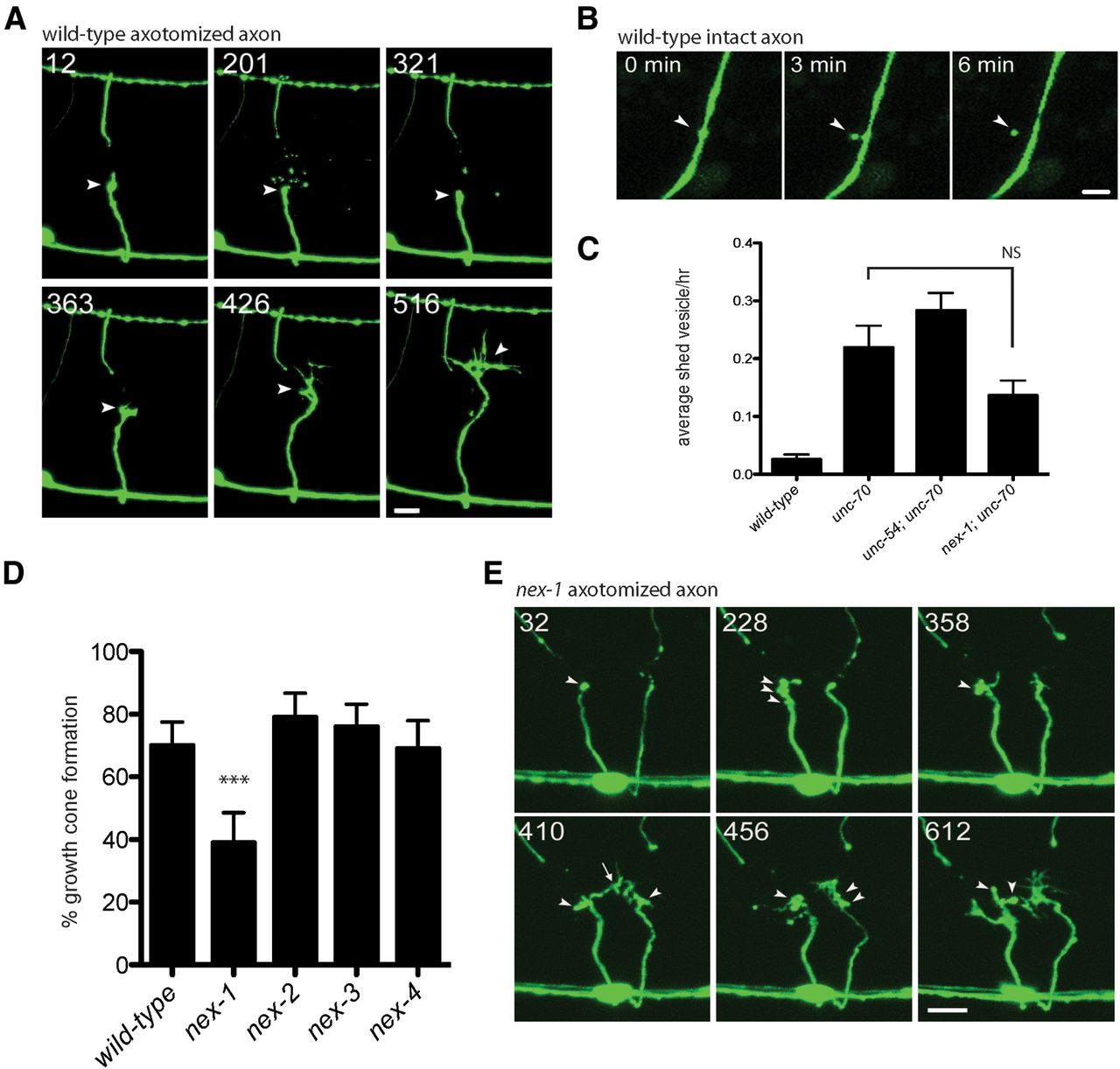
As a result of our time-lapse analysis of unc-70 mutants, we identified a new phenotype associated with axon repair and regeneration. When an axon is severed, the proximal end retracts and the tip becomes enlarged in a structure known as the retraction bulb. This is followed by a quiescent period before growth cone formation and migration begins. During this period, we observed the release of microvesicle particles from the tip of the severed axon (the site of future growth cone formation; Fig. 3A). Microvesicles are also released from intact axons (Fig. 3B). When we compared wild-type and unc-70 axons in time-lapse experiments, we found a tenfold increase in microvesicle shedding in the unc-70 mutant (Fig. 3C). We hypothesized that localized membrane damage and Ca2+ influx activates a membrane repair process that sequesters and expels damaged membrane via microvesicles. The fragility of unc-70 axons would result in a greater amount of membrane damage and shedding of microvesicles. However, the shedding appears to be a property of the membrane itself and not due to mechanical stress, because unc-70; unc-54 double mutants release microvesicles in numbers comparable to unc-70 single mutants (Fig. 3C).

Microvesicle release is an early step in axon regeneration. A, Time-lapse images of an axotomized wild-type axon. The retraction bulb is formed within 12 minutes (arrowhead). At 201 minutes, there is an expulsion of numerous microvesicles, which are then cleared and mostly removed by 321 min. The growth cone forms shortly after at 363 min. Scale bar, 5 μm. B, Microvesicle release from an uninjured wild-type axon. At 0 minutes, a bulge predicts the site of vesicle release. At 3 minutes, the vesicle is connected by a faint neck and, by 6 minutes, the vesicle is separated from the axon. Scale bar, 1 μm. C, Microvesicle shedding per hour per commissure from intact axons is increased in unc-70 mutants. p-values were determined by t test. Error bars indicate SEM. D, nex-1 is required for normal GABA neuron regeneration. ***p < 0.001, Fisher's exact test. Error bars indicate 95% confidence interval. E, Time-lapse images of axotomized nex-1 axons suggest defects in damaged membrane repair. At 228 minutes, membrane blebs are visible and appear to interfere with normal growth cone formation. Scale bar, 5 μm.
Annexins are Ca2+-sensitive phospholipid-binding proteins implicated in membrane repair and therefore are good candidates to mediate this process in regeneration (Draeger et al., 2011). We performed axotomy in the C. elegans annexin mutants nex-1, nex-2, nex-3, and nex-4 and found that nex-1 results in a significant reduction in regeneration compared with wild-type animals (Fig. 3D). Defective regeneration in nex-1 mutants is more apparent in time-lapse images. After axotomy, retraction bulb formation is normal, but excessive blebbing can be seen at the site of damage before the appearance of a growth cone. In some cases, a growth cone may form, but often collapses or fails to progress due to abnormal motility (Fig. 3E). We interpret these blebs as regions of damaged membrane that are not removed from the injury site in the absence of nex-1 activity and think that the damaged membrane interferes with normal growth cone formation. We predicted that microvesicle release would be decreased in nex-1;unc-70 double mutants, but found that vesicle release was unchanged (Fig. 3C). Although nex-1 mutations affect growth cone initiation and may be involved in membrane repair at the site of damage, the trigger for microvesicle release from intact axons remains to be determined.
mtm-1 encodes the C. elegans homolog of human myotubularin (MTM1) that is mutated in X-linked myotubular myopathy (Hnia et al., 2012). MTM1 is one of a large family of myotubularin proteins that function as lipid phosphatases, removing the phosphatidylinositol 3-monophosphate from PI(3)P and PI(3,5)P2; as such, myotubularins play an important role in regulating membrane dynamics and membrane trafficking. We identified mtm-1 as a candidate in the unc-70 screen and confirmed the defect by axotomy in mtm-1(op309) mutants (Fig. 4A; Table 1). Regeneration was moderately decreased; however, it is likely that mtm-1(op309) still retains some function because deletion of the mtm-1 gene causes lethality. In addition, mtm-1 may act redundantly with other myotubularin genes. We found significant regeneration defects in mtm-6 and mtm-9 mutants, although regeneration was not further decreased in mtm-1;mtm-6 double mutants (Fig. 4A). Aberrant growth cones are visible after axotomy in mtm-1(op309) time-lapse experiments. The growth cones often appear to be embryonic-like in nature with fewer filopodial extensions than wild-type (Fig. 4B). However, migration of the mutant growth cone is ineffective, eventually collapsing. This phenotype is similar to observations from Drosophila hemocytes in which filopodial protrusions normally appear in the wild-type, but were absent from mtm mutants (Velichkova et al., 2010). mtm-1 overexpression in GABA neurons led to dramatic defects in axonal morphology and decreased regeneration (Fig. 4A,B; Table 3). Large swellings of the axonal membrane and large vacuolar structures are visible within the cytoplasm. We have not yet investigated the cause of the phenotype, but it is suggestive of defects in membrane trafficking. Endocytosis is an important event in remodeling the axonal membrane during growth cone formation (Bloom and Morgan, 2011). Loss of lipid phosphorylation due to increased mtm-1 expression could alter membrane identity, resulting in increased endosomal structures and failure to deliver vesicles to the plasma membrane, where they are needed for growth cone formation.

Axon regeneration defects in myotubularin mutants by loss-of-function and overexpression. A, mtm-1 and other myotubularin family members are required for normal GABA neuron regeneration. **p < 0.01; ***p < 0.001, NS, Fisher's exact test. Error bars indicate 95% confidence interval. B, mtm-1 loss-of-function and overexpression in GABA neurons causes axonal membrane defects. Arrowheads indicate the highly branched wild-type growth cones seen at L4. The mtm-1 growth cones are often large and unbranched (left growth cone), but many fail (right growth cone). Overexpression of mtm-1 may cause failure to initiate growth cones (lower left). Bottom left, right arrowhead, Successful growth cones often leave behind growth cone remnants. Bottom right, Failed growth cone collapsed around a large vacuole (left arrowhead) and a large growth cone remnant along the axon of a successful commissure (right arrowhead). Asterisk shows a large vacuole in a neuron cell body. Scale bar, 5 μm.
Overexpression phenotype of selected candidate genes assayed by axotomy
Multiple MAP kinase pathways influence axon regeneration
The MAP kinase signal involves a three-tiered kinase cascade consisting of a MAP kinase kinase kinase (MAP3K), a MAP kinase kinase (MAP2K), and a MAP kinase (MAPK). Several candidates identified in the unc-70 screen fall into the category of genes involved in MAPK signaling, including dlk-1, mlk-1, kin-18, and cst-1. This led us to complete a broader screen of MAPK-related genes with emphasis on the stress-activated JNK- and p38-related pathways (Sakaguchi et al., 2004), candidate target genes and candidate coregulators (Table 4). We screened nine MAP3Ks, eight MAP2Ks, and seven MAPKs within the p38 and JNK families of C. elegans. Our previous work described two MAPK modules (DLK-1-MKK-4-PMK-3 and MLK-1-MEK-1-KGB-1) that are both essential to promote axon regeneration (Hammarlund et al., 2009; Nix et al., 2011). Here, we identify two additional MAPKs that function as negative regulators of regeneration.
Regeneration phenotype of MAPK pathway mutants
Loss-of-function mutants in the MAPKs jnk-1 and kgb-2 show improved regeneration (Fig. 5A). Improved regeneration in the jnk-1 mutant was dependent on normal activation of the pmk-3 MAPK pathway, because dlk-1(MAP3K);jnk-1 double mutants, like dlk-1 single mutants, failed to regenerate. However, loss of jnk-1 moderately suppressed the regeneration phenotype of kgb-1 MAPK mutants (Fig. 5A). JNK-1 and KGB-2 overexpression inhibited new growth cone formation (Fig. 5A; Table 3). In particular, the JNK-1 overexpression phenotype was as severe as the loss of dlk-1. One possibility is that JNK-1 overexpression competes with PMK-3 and/or KGB-1 for binding to an activator or substrate, thereby preventing proper activation and thus blocking regeneration. Alternatively, JNK-1 kinase activity itself could inhibit regeneration. To distinguish between these possibilities, we overexpressed either a kinase-dead (TY/AA) or an inactive, nonphosphorylated (KK/AA) form of JNK-1. These constructs eliminate kinase activity without affecting putative protein interactions. Overexpression of either construct failed to inhibit regeneration, indicating that kinase activity is essential to the function of JNK-1 in regeneration (Fig. 5A).

Multiple MAPK pathways antagonistically influence axon regeneration. A, dlk-1 and kgb-1 mutants represent two MAPK modules essential for axon regeneration. MAPK signaling via JNK-1 inhibits axon regeneration and requires functional kinase activity. Improved regeneration by loss of jnk-1 weakly suppresses loss of kgb-1, but is not sufficiently to suppress the loss of dlk-1. B, The MAP2Ks jkk-1 and sek-6 may function upstream of JNK-1 to inhibit axon regeneration. Loss of jkk-1 and sek-6 partially suppresses the effects of JNK-1 overexpression. C, The MAP3K kin-18 may inhibit axon regeneration via JNK-1. D, FOS-1 transcription factor is necessary for regeneration and is a target of KGB-1 MAPK signaling. E, Summary of MAPK signaling components affecting axon regeneration. *p < 0.05; **p < 0.01; ***p < 0.001, NS, Fisher's exact test. Error bars indicate 95% confidence interval.
As part of its characterized function in locomotion and longevity, JNK-1 is phosphorylated by JKK-1 (Villanueva et al., 2001; Oh et al., 2005). jkk-1 mutants did not show a regeneration defect, suggesting that JNK-1 activity may be stimulated by a different MAP2K upon axotomy or that additional MAP2Ks may function redundantly (Fig. 5B). The MAP2K sek-1 activates the p38 MAPK pmk-1 as part of the immune response (Kim et al., 2002). The jkk-1 sek-1 double mutant was indistinguishable from either single mutant, indicating that these MAP2Ks are unlikely to function redundantly in regeneration. One MAP2K, sek-6, did show improved regeneration in the mutant background and may function upstream of jnk-1 or kgb-2. However, neither overexpression of wild-type sek-6 nor the phosphomimetic sek-6(DD) resulted in a regeneration defect (Table 3). The jnk-1 overexpression phenotype is partially suppressed by sek-6 and jkk-1 single mutants and by the sek-6 jkk-1 double mutant, suggesting that sek-6 may function redundantly with jkk-1. Despite this, another, as-yet-unidentified MAP2K could also function, because JNK-1 activation is not completely eliminated by the absence of jkk-1 and sek-6 (Fig. 5B). It is also possible that JNK-1 activity may be artificially elevated from overexpression, even in the absence of the upstream signal.
The MAP3K associated with JNK-1 activation is currently unknown. We identified the MAP3K kin-18 as a candidate in the unc-70 regeneration screen. kin-18 mutants had significantly improved growth cone initiation, although overexpression of kin-18 in GABA neurons did not affect regeneration (Fig. 5C; Table 3). Loss of kin-18 suppressed the jnk-1 overexpression phenotype, which is consistent with a model in which kin-18 acts upstream of JNK-1 (Fig. 5C).
The dual-specificity MAPK phosphatases (MKPs) remove phosphate groups from activated MAPKs and represent an important control point for regulating MAPK activity. We demonstrated that VHP-1 interacts with both PMK-3 and KGB-1 to inhibit kinase activity (Nix et al., 2011). Loss of vhp-1 increases levels of activated PMK-3 and KGB-1, improving regeneration. There are at least eight additional genes in C. elegans with homology to MKPs (Kim et al., 2004). We predicted that loss of one of these might result in blocked regeneration due to increased activity of either JNK-1 or KGB-2. We assayed mutant alleles for each of the eight genes (Table 4). Although none showed reduced regeneration, we found that three alleles, lip-1(zh15), ZK757.2(ok3597), and F28C6.8(ok2013), improved growth cone formation.
A number of scaffolding proteins coordinate the activity of MAPK modules by bringing the kinases into the proximity of each other. unc-16 encodes the homolog of mammalian JIP3, a JNK-interacting protein that links the activity of JNK-1 and JNK kinases (Byrd et al., 2001). unc-16 mutants improve regeneration to the same extent as jnk-1 and kgb-2 MAPK mutants (Table 2). Loss of unc-16 also suppresses the blocked regeneration in jnk-1-overexpressing animals (Fig. 5C). Mutations in another JNK-interacting protein, jip-1, did not show a regeneration phenotype in our assay (Table 4). unc-16 was initially identified by its defect in synaptic vesicle localization (Byrd et al., 2001). UNC-16 interacts with JNK-1 and the microtubule motor protein UNC-116 (kinesin) to deliver cargo vesicles to the synapse. The regeneration phenotypes observed in the jnk-1 loss-of-function or overexpression strains may be explained by its role in vesicle transport.
In summary, we have reported previously the requirement for PMK-3 and KGB-1 MAPK signaling in promoting axon regeneration (Hammarlund et al., 2009; Nix et al., 2011). Here, we show that additional MAPK signaling factors antagonistically influence the outcome of regeneration in the worm (Fig. 5E). The coordinated regulation of positive and negative signals could function to preserve the balance between maintaining a fixed nervous system and allowing for plasticity under certain conditions, including injury. Further experiments will be necessary to confirm the newly defined MAPK components, including the MAP3K KIN-18, the MAP2K SEK-6, and the MAPKs JNK-1 and KGB-2.
KGB-1 MAPK pathway targets FOS-1 during axon regeneration
The downstream target of PMK-3 signaling in regeneration is the MAPK-activated kinase MAK-2, which influences stability of cebp-1, the C. elegans CCAAT/enhancer-binding protein (Yan et al., 2009). The downstream targets of JNK-like MAPKs (KGB-1, KGB-2, and JNK-1) are unknown. Two well known targets of JNK signaling in other species include the transcription factors Jun and Fos. In mammals, axonal activation and retrograde transport of JNK, followed by upregulation of c-jun, is a consistent marker of successful regeneration (Raivich et al., 2004; Raivich and Makwana, 2007). In Drosophila, the axonal injury signal is mediated by the DLK/JNK pathway and activates the transcription factor Fos (but not Jun; Xiong et al., 2010). We examined the C. elegans Jun and Fos homologs for a potential role in regeneration. Growth cone formation was normal in jun-1 mutants, whereas fos-1-null mutants show significantly reduced growth cone formation (Fig. 5D; Table 2). Expression of a dominant-negative form of fos-1 that encodes the DNA-binding and dimerization domains but lacks the transcriptional activation domain also inhibited regeneration when expressed specifically in the GABA neurons (Fig. 5D). Overexpression of a genomic fos-1 transgene rescued the sterility associated with fos-1(km30)-null mutants and rescued the regeneration phenotype back to wild-type levels. These data are consistent with a model in which FOS-1 functions downstream of MAPK signaling. Hattori et al. have recently demonstrated that KGB-1 phosphorylates FOS-1 directly at Thr-304 during heavy-metal stress (Hattori et al., 2013). We investigated whether phosphorylation at Thr-304 also influences axon regeneration by expressing a mutant transgene, fos-1 T304A, in which Thr-304 is mutated to alanine. The mutant rescued fos-1(km30) sterility, but failed to rescue regeneration, suggesting that activated KGB-1 phosphorylates FOS-1 to promote axon regeneration (Fig. 5D).
Stress response pathways are necessary for axon regeneration
The p38 and JNK family of MAP kinases are also known as stress-activated protein kinases (SAPKs). A wide variety of environmental conditions induce a stress signal by stimulating MAP kinase activity. As a result, the cell mounts an adaptive response through changes in gene expression to manage a particular stress. Because both the p38 and JNK pathways are stimulated in response to axotomy, we reasoned that axotomy itself is a form of stress on the cell. Does activating the stress response by other means alter regeneration? To address this question, we characterized axon regeneration in hsf-1(heat-shock factor 1) and hif-1 (hypoxia inducible factor) mutants and under conditions of heat stress and starvation.
A 1 h period of heat stress at 33°C, followed by recovery at 20°C, resulted in significantly improved regeneration (Fig. 6A). Similarly, growth cone formation was improved when axotomized animals were recovered on plates in the absence of food. The upregulation of heat-shock proteins (HSPs) is an important response to cellular stress, protecting the cell from aggregated and/or misfolded proteins. A key regulator of HSP transcription is heat shock factor 1 (Anckar and Sistonen, 2011). The improved regeneration we observed after a short period of heat shock was dependent on HSF-1 activity. Regeneration was blocked in heat-shocked hsf-1 mutants (Fig. 6A). Furthermore, hsf-1 mutants showed severely reduced regeneration even in the absence of heat shock. We observed a small but significant increase in growth cone initiation when hsf-1 mutants were heat shocked. This difference may be explained by the fact that the hsf-1(sy441) allele is not a complete null and some activity could remain in these animals. Alternatively, it suggests that the majority of the heat shock response occurs through HSF-1, but that secondary pathways contributing to regeneration may also be activated by heat shock. Nevertheless, these results indicate that axotomy itself is a form of cellular stress and requires mobilization of the stress response. hif-1 also showed reduced regeneration, but a formal test of the effects of hypoxia on regeneration was not done.

Stress response pathways are necessary for efficient axon regeneration. A, Stress-inducing conditions improve regeneration outcomes and require hsf-1. Regeneration in hif-1 is also reduced, but was not tested under hypoxic conditions. WT, Wild-type; HS, heat shock. B, Increased IRE-1 activity in xbp-1 mutants and by ire-1 overexpression inhibits regeneration. *p < 0.05; **p < 0.01; ***p < 0.001, NS, Fisher's exact test. Error bars indicate 95% confidence interval. C, Example of candidates affecting axon regeneration. Proteins “positively” required for axon regeneration are shown in green; those that “inhibit” axon regeneration are shown in red. Mammalian orthologs are in parentheses where known and different from the C. elegans name.
While HSF-1 and HIF-1 dictate the response to stress in the cytoplasm, a similar mechanism exists in the ER and mitochondria to manage accumulation of misfolded proteins. The so-called unfolded protein response (UPRER) leads to upregulation of ER-specific HSPs and an overall downregulation of protein synthesis as a means to lower the protein load entering the ER (Ron and Walter, 2007). A core component of the UPRER is IRE1, which encodes an ER membrane protein with a cytoplasmic endonuclease and kinase domain. Autophosphorylation activates IRE1, leading to precise cleavage of its substrate: the XBP1 mRNA. Splicing of XBP1 mRNA, in turn, produces a reading-frame shift that results in an active transcription factor. We found that loss of xbp-1 results in severely reduced regeneration, whereas the loss of ire-1 had no effect (Fig. 6B). In addition to its function as a transcriptional activator, XBP-1 provides inhibitory feedback on IRE-1 itself (Richardson et al., 2011). As a result, IRE-1 activity is increased in xbp-1 mutants. Is the xbp-1 mutant phenotype due to loss of xbp-1 transcriptional activity or due to upregulation of ire-1? Axon regeneration in ire-1;xbp-1 double mutants was normal, indicating that reduced growth cone formation in the xbp-1 mutant was a result of ire-1 misexpression (Fig. 6B). Indeed, overexpression of ire-1 in neurons also blocked regeneration (Fig. 6B; Table 3). We speculate that increased IRE-1 endonuclease activity might affect mRNAs present in the cytoplasm, causing inappropriate cleavage of the transcripts necessary for regeneration.
The mitochondrial UPR is communicated to the nucleus by ubl-5, a ubiquitin-like protein, along with two transcription factors, dve-1 and atfs-1 (Kirstein-Miles and Morimoto, 2010). We identified ubl-5 as a gene candidate in the unc-70 regeneration screen (Table 1). In addition, we screened the atfs-1(gk3094) mutant by axotomy and found significantly improved regeneration in the mutant background (Fig. 1H). These results also support a shared role for proteins in both the mitochondrial UPR and axon regeneration.
Discussion
In this study, we completed an unbiased genetic screen for genes affecting axon regeneration. We identified 70 candidate regeneration genes by screening for reduced commissure number in unc-70/β-spectrin mutants fed a total of 5076 RNAi clones. Of the 70 candidates isolated from the screen, 31 showed regeneration defects when assayed by axotomy in a mutant strain. These include 15 positive regulators and 16 negative regulators of regeneration. Sixty-five additional genes were selected based on their association with screen candidates. Twenty-two of the 65 mutants showed regeneration phenotypes after axotomy. The total number of genes screened represents about one-quarter of the C. elegans genome and suggests that continued screening by this method may add to the collection of regeneration genes.
Successful growth cone formation and axonal regeneration requires a multitude of coordinated events within the cell (Raivich and Makwana, 2007; Abe and Cavalli, 2008; Bloom and Morgan, 2011; Bradke et al., 2012). An injury signal sent to the cell body is necessary to mount an appropriate response to damage. The damaged membrane is repaired before growth cone formation can begin. Through a combination of local protein synthesis, protein trafficking, and transcriptional regulation, the cell transitions to a developmental growth phase. A large proportion of candidates fall into the category of genes involved in cell signaling. We and others have shown that p38 and JNK family MAP kinases play a critical role in triggering the injury signal (Yan et al., 2009; Ghosh-Roy et al., 2010; Xiong et al., 2010; Nix et al., 2011; Shin et al., 2012). We also identified several components of Ras/MAP kinase signaling, including let-60/Ras, mpk-1, lin-2, sli-1, and soc-2. The Notch and Wnt signal transduction pathways active during development are also implicated from the candidate genes. C. elegans Notch signaling was recently shown to function as an inhibitor of axon regeneration (El Bejjani and Hammarlund, 2012). Two lipid phosphatases suggest that membrane lipid composition and/or lipid signaling play an important role in regeneration. One of these, mtm-1, encodes the C. elegans homolog of human myotubularin. Mutations in myotubularin genes are associated with several neuromuscular disorders (Hnia et al., 2012). In C. elegans, mtm-1 functions as a negative regulator of cell corpse engulfment during apoptosis (Zou et al., 2009). Its main enzymatic activity is dephosphorylation of PI(3)P and PI(3,5)P2, so mtm-1 could be involved in signaling membrane damage or in cytoskeletal dynamics necessary for growth cone formation.
A number of candidate genes are associated with RNA binding and/or the protein synthetic machinery. Mammalian cells transport molecules involved in protein synthesis in response to injury, either to the site of injury itself or back to the nucleus to further influence gene expression (Michaelevski et al., 2010a). Neurons in particular use the subcellular localization of mRNAs as a mechanism to regulate protein synthesis both spatially and temporally. C. elegans cebp-1 mRNA is localized to synapses and activation of the DLK-1 MAP kinase pathway induces local translation of the CEBP-1 transcription factor (Yan et al., 2009). Both CEBP-1 and DLK-1 are essential for regeneration. One model consistent with our results involves putative RNA-binding proteins (bcc-1, F16A11.2, tdp-1, zbp-1) binding to and/or sequestering mRNAs until they are necessary to promote axon regeneration. Indeed, bcc-1 and F16A11.2 mutants show enhanced regeneration, whereas overexpression of either gene or zbp-1 strongly inhibits regeneration.
Evidence suggests that many transcription factors in addition to CEBP-1 are required to convert the injured cell from a maintenance program to a growth-promoting program (Michaelevski et al., 2010b; Tedeschi, 2011; Ben-Yaakov et al., 2012; Patodia and Raivich, 2012). This is partly accomplished through transcriptional activation or repression of specific target genes. In our screen, we found that the transcription factors FOS-1, HSF-1, and HIF-1 are required for GABA neuron regeneration, whereas UNC-130, CEH-13, and KLF-1 are inhibitors of regeneration. As an independent validation, mammalian KLF family members were shown to be negative regulators of axon regeneration (Moore et al., 2009). We also found that regeneration was inhibited in klf-1-overexpressing strains, indicating that KLF-1 function in regeneration is conserved.
Several gene candidates identified in the screen did not show a regeneration phenotype when assayed by axotomy. One class of candidates showed reproducibly decreased commissure counts after RNAi in the unc-70 background, but no phenotype upon axotomy in the mutant or RNAi-treated strain. This raises the possibility that loss of the spectrin cytoskeleton in unc-70 sensitizes the background to further defects in regeneration. This could be the case for the candidate genes that function in the Hippo kinase pathway (cst-1, sav-1, egl-44). Hippo activity is regulated by Merlin, a FERM-domain-containing protein that binds to several cytoskeletal proteins, including β-spectrin, and may transmit signals from the cell membrane (Scoles, 2008; Halder and Johnson, 2011). Alternatively, genes affecting axon outgrowth and path finding could show defects in the unc-70 background but not by axotomy. Despite this, we recovered relatively few known path-finding mutants in our screen and most candidate mutants showed normal nervous system development. Because the assay primarily quantifies the incidence of growth cones, certain genes identified in the screen will require further analysis using additional criteria for validation.
An important outcome of our screen was the identification of the MAP3Ks DLK-1 and MLK-1. Through our analysis of these genes, we gained an understanding of the essential nature of two MAP kinase signaling modules in promoting axon regeneration (Hammarlund et al., 2009; Nix et al., 2011). This led us to complete a broader characterization of other MAP kinase-related genes. We found that activation of the JNK-1 MAP kinase inhibits GABA neuron regeneration. Loss of jnk-1 resulted in animals with improved regeneration, whereas jnk-1 overexpression completely inhibited regeneration. The inhibitory effect of jnk-1 overexpression was dependent on kinase activity because overexpression of a kinase-dead form of the protein no longer blocked regeneration. In addition to JNK-1, we identified potential upstream activators of MAPK signaling, including the MAP3K kin-18, MAP2K sek-6, and the Jnk-like MAPK kgb-2. The mechanism of JNK-1 activation is currently unclear. JNK signaling in mammals and other species activates a diverse array of cellular targets, including many transcription factors, mitochondrial proteins, microtubule-associated proteins, and cytoskeletal factors (Bogoyevitch and Kobe, 2006). In C. elegans, JNK-1 and UNC-16 function in neurons to regulate vesicle transport (Byrd et al., 2001). More recently, JNK-1 and UNC-16 were shown to function as axonal gatekeepers for specific organelles, including Golgi and endosomes (Edwards et al., 2013). Mutants accumulate membrane-enclosed compartments in axonal regions beyond the axon initial segment and in synaptic regions. Based on this model, jnk-1 and unc-16 mutants may show improved regeneration due the immediate availability of membranous compartments for axonal growth and repair after injury. Conversely, jnk-1 overexpression may inhibit regeneration by preventing the release of organelles from the cell soma and axon initial segment, which would limit the amount of accessible membrane necessary for growth cone formation and regeneration.
MAP kinases respond to extracellular signals, including changing environmental conditions. Therefore, they function as stress-sensors influencing a wide range of cellular processes. We found that axon regeneration improved when animals were exposed to stress-inducing conditions such as heat, starvation, and possibly hypoxia. Other stress-activated proteins are also important for regeneration. The main integrator of the stress response in eukaryotic cells is the transcription factor HSF1 (Anckar and Sistonen, 2011) and, to a lesser extent, HIF-1 (Powell-Coffman, 2010). The improved regeneration we observed in animals exposed to heat stress requires functional HSF-1. Furthermore, loss of hsf-1 prevents regeneration even under nonstressed conditions, as does hif-1. Efficient regeneration requires an injured neuron to manage a profusion of new protein synthesis and recapitulate much of the developmental program. Therefore, the success of regeneration to some extent becomes an issue of protein homeostasis. As an organism ages, it is less able to cope with errors in protein translation and folding and this can in part explain the age-dependent decline in regeneration. Indeed, the DNA-binding activity of HSF1 is diminished over time, although protein levels remain stable (Fawcett et al., 1994). Loss of proteostasis defines an early stage in the C. elegans aging process even before the physical manifestations of aging appear (Ben-Zvi et al., 2009). Overexpression of HSF-1 and DAF-16 can suppress the protein-folding defects of temperature-sensitive mutations and delay the onset of age-related defects.
Overall, the candidate genes identified in this screen (Fig. 6C) are conserved among species and should present several new targets for therapeutic interventions. Genes inhibiting regeneration are of particular interest because these pathways may be easier to eliminate via drug application rather than by upregulating growth-promoting pathways. In addition, combinatorial approaches may prove more effective (Kadoya et al., 2009). Sun et al. demonstrated that CNS neurons in adult mice show improved and sustained regeneration in PTEN- and SOCS3-deleted double mutants compared with single mutants or the wild-type (Sun et al., 2011). An important approach will be to determine whether elimination of particular pathways in C. elegans can also enhance regeneration in older animals or induce regeneration in neurons that do not normally regenerate.
Footnotes
This research was supported by the National Science Foundation, the McKnight Endowment Fund for Neuroscience, the Christopher and Dana Reeve Foundation, and the Amerisure Charitable Foundation (to M.B.) and the Howard Hughes Medical Institute (to E.M.J.). We thank L. Avery, K. Blackwell, C. Haynes, H.R. Horvitz, A. Hsu, Y. Jin, P. Mains, K. Matsumoto, B.J. Meyer, D. Sherwood, and K. Shen for sharing strains and reagents used in this study; the C. elegans Gene Knockout Consortium and the Japanese National Bioresource Project for deletion mutants; the NemaGENETAG consortium for Mos1 transposon strains; the Caenorhabditis Genetics Center for additional strains; and members of the Jorgensen laboratory for helpful discussions and critical reading of this manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Michael Bastiani, Department of Biology, University of Utah, 257 South 1400 East, Salt Lake City, UT 84112. bastiani{at}bioscience.utah.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}