

Abstract
Axonal degeneration is an active process that has been associated with neurodegenerative conditions triggered by mechanical, metabolic, infectious, toxic, hereditary and inflammatory stimuli. This degenerative process can cause permanent loss of function, so it represents a focus for neuroprotective strategies. Several signaling pathways are implicated in axonal degeneration, but identification of an integrative mechanism for this self-destructive process has remained elusive. Here, we show that rapid axonal degeneration triggered by distinct mechanical and toxic insults is dependent on the activation of the mitochondrial permeability transition pore (mPTP). Both pharmacological and genetic targeting of cyclophilin D, a functional component of the mPTP, protects severed axons and vincristine-treated neurons from axonal degeneration in ex vivo and in vitro mouse and rat model systems. These effects were observed in axons from both the peripheral and central nervous system. Our results suggest that the mPTP is a key effector of axonal degeneration, upon which several independent signaling pathways converge. Since axonal and synapse degeneration are increasingly considered early pathological events in neurodegeneration, our work identifies a potential target for therapeutic intervention in a wide variety of conditions that lead to loss of axons and subsequent functional impairment.
Introduction
Axonal degeneration is implicated in neurodegeneration for both the PNS and CNS (Coleman, 2005). Degeneration of denucleated distal injured nerve fibers has been extensively studied since the seminal observation of Waller (1850). The discovery of the Wlds mutant mouse, which exhibits delayed Wallerian degeneration after nerve injury (Lunn et al., 1989; Perry et al., 1990), suggested that axons are endowed with a genetically controlled self-destruction program. Axonal degeneration and synapse loss have been proposed as early events in several human neurodegenerative conditions (Coleman and Perry, 2002; Saxena and Caroni, 2007), and expression of Wlds protein in some animal models of neurodegeneration can delay symptoms and signs of neuronal loss (Ferri et al., 2003; Sajadi et al., 2004; Mi et al., 2005). These observations suggest that Wallerian degeneration and axonal damage triggered by toxins and genetic defects share molecular mechanisms that converge onto a common execution program for axonal degeneration.
In the Wlds mouse, expression of a chimeric protein consisting of a fragment of the polyubiquitination factor UFD2a/UBE4b and the full-length of the nicotinamide mononucleotide adenylyltransferase-1 (NMNAT1) delays axonal degeneration (Mack et al., 2001). Overexpression of the NMNAT1 moiety is partially responsible for the phenotype in Wlds, probably accompanied by retargeting of this moiety to axons (Sasaki et al., 2009; Babetto et al., 2010). There are three vertebrate NMNATs with different subcellular localization, NMNAT1 concentrates in the nucleus, NMNAT2 localizes to the Golgi complex and NMNAT3 localizes to mitochondria (Berger et al., 2005). Overexpression of NMNAT3, also delays Wallerian degeneration (Yahata et al., 2009). Continuous transport of endogenous NMNAT2 along the axon appears to provide a survival factor for intact axons (Gilley and Coleman, 2010).
Although the site of NMNAT's protective action has been narrowed down to the axonal compartment (Gilley and Coleman, 2010), a unifying mechanism to integrate the different insults leading to axonal degeneration into a single downstream mediator is currently lacking. Mitochondria have been suggested to play an active role in axonal degeneration (Yahata et al., 2009), but the canonical apoptotic pathways have been ruled out (Deckwerth and Johnson, 1994; Sagot et al., 1995; Whitmore et al., 2003). Mitochondrial permeability transition has recently been implicated in several neurodegenerative conditions (Forte et al., 2007; Du et al., 2008; Martin et al., 2009). This takes place after mitochondrial Ca2+ overloading, especially when accompanied by oxidative stress, elevated free PO4, and adenine nucleotide depletion. This leads to the opening of the nonspecific mitochondrial permeability transition pore (mPTP), followed by loss of mitochondrial membrane potential (ΔΨm), ATP depletion, increased generation of reactive oxygen species (ROS), release of Ca2+, and mitochondrial swelling (Bernardi et al., 2006). Although the molecular identity of the mPTP remains uncertain, different components have been proposed including the voltage-dependent anion channel (VDAC), the adenine nucleotide translocator (ANT), the mitochondrial phosphate carrier, and the cyclosporin A (CsA) target cyclophilin D (CypD) (Halestrap, 2009).
Genetic ablation studies have only confirmed a role for CypD in mPTP-mediated cell death (Baines et al., 2005; Nakagawa et al., 2005; Schinzel et al., 2005). Notably, deletion of the murine CypD gene reduces axonal degeneration in an autoimmune encephalomyelitis model (Forte et al., 2007), and treatment of animals with CsA protects against Wallerian degeneration (Sunio and Bittner, 1997; Taskinen and Röyttä, 2000). CsA can inhibit the mPTP through its interaction with CypD, but it also has immunosuppressant actions through calcineurin inhibition (Liu et al., 1991). The axonal protective effects of CsA were presumed to reflect immunosuppression rather than any intrinsic effects on the axon.
Here, we show that axonal degeneration triggered by mechanical and toxic stimuli requires mPTP activation. By perturbing activities of known mediators of axonal degeneration, we show that mPTP is a central executioner of axonal degeneration, acting as a convergence point for several degenerative signals. These data indicate that mitochondrial integrity, and in particular the mPTP, may provide a therapeutic target for preventing axonal degeneration in a wide variety of conditions affecting the nervous system.
Materials and Methods
Animals.
Mice of the C57BL/Wlds and wild-type (WT) (C57BL/6J) strain were originally obtained from Harlan. Adult mice (∼25 g) were used. Male Sprague Dawley rats (∼150 g) were purchased from Harlan Biosciences. Experiments with animals followed protocols approved by the Institutional Animal Care and Use Committees and complied with National Institutes of Health guidelines.
Explant cultures.
To study axonal degeneration ex vivo, sciatic and optic nerve segments of ∼10 and 4 mm, respectively, were dissected from adult WT and Wlds mice and cultured in 24 well dishes containing 400 μl of Neurobasal medium (Invitrogen), 2% B27 (Invitrogen), 0.3% l-glutamine, and 1% streptomycin/penicillin. Explants were cultured at 37°C and 5% CO2 for up to 9 d, with daily changes of media supplemented with drug or corresponding vehicle control. Drug concentration used in nerve explants or in vivo by perineural injections are at least one order of magnitude higher than in cultured neurons (Court et al., 2009), probably due to the presence of the hematoneural barrier of the nerve (epineurium, perineurium, and collagen and extracellular matrix of the endoneurium); the presence of myelinating Schwann cells also contributes to lower the effective drug concentration in the nerve explants vs cultured neurons. The following drugs were used: cyclosporin A (LC Laboratories, #C6000), MG132 (N-benzoyloxycarbonyl (Z)-Leu-Leu-leucinal) (Sigma-Aldrich, #C2211), c-Jun terminal kinase inhibitor II, SP600125 (Calbiochem, #4201119), EGTA (Sigma-Aldrich, #E3889), BAPTA-AM [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)] (Invitrogen, #B6769), 4,49-diisothiocyanatostilbene-2,29-disulfonic acid (DIDS) (Sigma-Aldrich, #D3514), Ruthenium Red (R.Red) (Calbiochem, #557450) atractyloside (ATR) (Sigma-Aldrich, #A6882), and FK506 (tacrolimus) (LC Laboratories, #F4900).
Biochemistry.
For cytoskeleton protein analysis, nerve explants were homogenized using a plastic Dounce homogenizer at room temperature (RT) in 1% Triton X-100, 50 mm Tris-HCl, pH 6.8, 2 mm EDTA, 1% PMSF, and 1% Protease Inhibitor Cocktail (PIC) Sigma-Aldrich, #P8340); samples were heated for 5 min at 100°C and centrifuged at 13,000 rpm for 10 min at RT. To analyze cytoskeletal proteins, the insoluble fraction was resuspended in 5% SDS, 10 mm Tris-HCl, pH 7.4, 2% β-mercaptoethanol, 1 mm NaF, 1 mm Na3VO4, and 1% PIC, and boiled/centrifuged as described above. The supernatant from this second lysis step was used as the cytoskeletal fraction. For analysis of soluble proteins, nerve explants were homogenized using a plastic Dounce homogenizer at RT in 95 mm NaCl, 25 mm Tris-HCl, pH 7.4, 10 mm EDTA, pH 8.0, 1% SDS, 1 mm NaF, 1 mm Na3VO4, and 1% PIC; lysates were heated for 5 min at 100°C and then centrifuged at 13,000 rpm for 10 min at RT. The supernatant was used as the cytosolic (soluble) proteins. Western blot (WB) was performed using SDS-PAGE and polyvinylidene difluoride (PVDF) membranes (Pierce) by standard techniques. Band analysis was performed using ImageJ software (http://rsbweb.nih.gov/ij/). Band intensity was normalized to histone H3, a housekeeping protein from Schwann cells, which represents an axonal-independent loading control for the nerve lysates.
Immunofluorescence and electron microscopy.
For immunofluorescence (IF) analysis, nerve explants were fixed by immersion in 4% paraformaldehyde in 0.1 m PBS (1× PBS, pH 7.4) for 1 h, followed by three 10 min washes in 1× PBS, sucrose gradient (5%, 10%, 20% in 1× PBS), and then embedded in OCT (Sakura Finetek). Cryostat sections from the middle of the explants were cut transversely at 10 μm thickness and mounted on Superfrost Plus slides (Thermo Fisher Scientific). Sections were washed in 1× PBS for 10 min and then blocked/permeabilized in 0.1% Triton X-100, 2% fish skin gelatin (Sigma-Aldrich) in 1× PBS for 1 h at RT. Sections were incubated in primary antibodies in blocking/permeabilizing solution overnight at 4°C, washed in 1× PBS 3 × 10 min, and incubated in secondary antibodies for 2 h at RT. Sections were washed 3 × 10 min in 1× PBS and mounted in Vectashield (Vector Laboratories). Number of axons per area of nerve tissue was assessed in confocal images of neurofilament-immunostained explant sections (matched for laser power, photomultiplier tube gain/offset, and postprocessing) using the particle analysis macro of ImageJ.
For EM analyses, nerves were fixed overnight by immersion in 2.5% glutaraldehyde, 0.01% picric acid, 0.1 m cacodylate buffer, pH 7.4. Nerves were rinsed in the same buffer, immersed in 1% OsO4 for 1 h followed by in block incubation with 2% uranyl acetate for 2 h. Nerves were dehydrated with a graded series of ethanol, propylene oxide and infiltrated with Epon (Ted Pella Inc.). Ultrathin sections from the middle of the explants were contrasted with 1% uranyl acetate and lead citrate. Grids were examined with a Philips Tecnai 12 electron microscope operated at 80 kV. Negative films were developed and scanned.
Primary/secondary antibodies.
The following antibodies were used for IF and/or WB analysis: rabbit anti-histone H3 (Millipore, #06-755) at 1:1000 for WB; rabbit anti-neurofilament heavy chain (Sigma-Aldrich, #N4142) at 1:5000 for WB or 1:1000 for IF; mouse anti-neurofilament light chain (Sigma-Aldrich, #N5139) at 1:5000 for WB; chicken anti-neurofilament medium chain (Millipore Bioscience Research Reagents, #AB5753) at 1:2000 for IF; rabbit anti-SAPK/JNK (Cell Signaling Technology, #09252) at 1:1000 for WB; rabbit anti-Phospho-SAPK/JNK (Cell Signaling Technology, #09251) at 1:1000 for WB; goat anti-rabbit HRP (Bio-Rad, #166-2408) at 1:5000 for WB; goat anti-mouse HRP (Bio-Rad, #172-1011) at 1:5000 for WB; donkey anti-rabbit TRITC (tetramethylrhodamine isothiocyanate) (Jackson ImmunoResearch Laboratories, #711-025-152) at 1:300 for IF; and, goat anti-chicken Alexa Fluor 647 (Invitrogen, #A-21449) at 1:300 for IF.
Brain mitochondria purification.
Brains from WT and Wlds mice were dissected. The cortex was weighed and resuspended in 4°C brain mitochondrial isolation solution (BMIS) (100 mm Tris-Cl, pH 7.4, 10 mm EDTA, 12% Percoll) at 10% wet weight/volume. The tissue was manually homogenized at 4°C with a loose glass pestle. The homogenate was subject to a discontinuous Percoll gradient centrifugation as described previously (Sims and Anderson, 2008). The mitochondrial fraction was diluted fourfold in BMIS and centrifuged for 10 min at 16,700 × g. The pellet was resuspended in 500 ml of 10 mg/ml BSA and then brought to 3 ml of BMIS. The resuspended solution was centrifuged for 10 min at 6900 × g and the supernatant was discarded. Finally, the pellet was gently resuspended in 300 μl of BMIS protein concentration was determined using the biuret method (Gornall et al., 1949).
Tetramethylrhodamine methyl ester measurements of mitochondrial membrane potential.
Mitochondria were diluted to 2 mg/ml protein in a buffer containing 200 mm HEPES, 20 mm Tris, 100 mm KCl, 10 mm NaCl, 5 mm Na2 succinate, 2 μg/ml oligomycin, and 2 μm rotenone. After dilution, mitochondria were incubated with 5 μm CsA or vehicle solution. Tetramethylrhodamine methyl ester (TMRM) was added to a final concentration of 1 μm and incubated for 20 min at 37°C protected from light. After incubation, Ca+2 was added to 5–100 μm final concentrations; controls with or without vehicle solution were included. TMRM is taken up electrophoretically by mitochondria in response to the negative mitochondrial membrane potential and its fluorescence is quenched. As mitochondria depolarizes, TMRM is released from mitochondria and fluorescence increases (Blattner et al., 2001). TMRM fluorescence was determined every 43 s in a fluorescence plate reader with excitation at 544 nm and emission at 590 nm. Change in fluorescence levels after different treatments were normalized to the basal fluorescence and the mean value after reaching the plateau was calculated.
Dorsal root ganglion cultures.
L3–L6 dorsal root ganglia (DRGs) from adult Sprague Dawley rats were isolated in ice-cold Hibernate A medium (BrainBits) and then dissociated with type XI collagenase for 25 min at 37°C (Twiss et al., 2000). Single-cell suspension was achieved by trituration using a fire polished Pasteur pipette followed by washing with DMEM/F-12. For gene silencing, dissociated DRGs were electroporated with cyclophilin D shRNA plus AcGFP (Aequorea coerulescens green fluorescent protein) expression plasmids using the Amaxa Nucleofector (Lonza). For this, cells were resuspended in nucleofector solution with 5 μg of each plasmid DNA immediately after DMEM/F-12 wash step. After electroporation, cells were resuspended in DMEM/F-12 medium supplemented with 10% fetal bovine serum (Hyclone) and 1× N2 (Sigma) and then onto poly-l-lysine (Sigma)- and laminin (Invitrogen)-coated substrates (Willis et al., 2005); medium was replaced after 4 h. For live cell imaging and vincristine treatments, dissociated cells were plated onto coated glass bottomed dishes. For isolation of axons, neurons were plated in tissue culture inserts with polyethylene tetraphthalate (PET) membrane track etched with 8 μm diameter pores (Falcon).
For live cell imaging, a Leica TCS-SP2 confocal fitted onto an inverted microscope with environmental chamber set at 37°C, 5% CO2 was used. For time lapse imaging of axonal degeneration, a glass capillary micropipette fitted to a motorized manipulator was used for axotomy. Complete transection was verified both by differential interference contrast(DIC) and compression of axonal AcGFP signals upon retraction of the severed ends. Posttransection, axons were imaged every minutes over 1 h. Pre- and posttransection DIC and the fluorescence were acquired with 488 nm laser, 4% power and confocal pinhole set to 3 arbitrary units.
To evaluate axonal degeneration in intact neurons, transfected dorsal root ganglion (DRG) cultures were treated with vincrinstine (Sigma) or vehicle control. For this, all neurons extending axons over at least 25% of field of view under 20× objective were imaged at 0–10 h after vincristine treatment. The degenerating axons showed loss of green fluorescent protein signal in processes and/or the partial disintegration of process with clear discontinuity between proximal and distal segments. In the late stages of degeneration, which was only seen at the higher (1 μm) doses of vincristine, the cells tended to detach. Since we could not directly quantify these neurons in the final stage of degeneration, we considered a neuron to be an axon-bearing cell only if a process could be identified as belonging to the original process that was imaged at earlier time point. The unidentified neurons were then calculated by subtracting axon-bearing neurons from total neurons that were imaged at 0 h vincristine treatment and quantified as neurons with degenerated neurons.
To quantify degeneration in axons isolated from cell bodies and Schwann cells, control and shRNA-transfected DRGs were cultured 3 d on PET membranes. Preinjury images of all neurons extending axons over at least 25% of field of view under 40× objective were taken from 4 PET membrane inserts per shRNA plasmid before transection. Neuronal cell bodies and Schwann cells were removed by physically scraping the top membrane surface using a cotton-tipped applicator. The inserts with the severed axons were incubated in standard medium for 4 h at 37°C, 5% CO2. Axonal processes were then imaged by epifluorescence. We considered an axon to have survived from Wallerian degeneration if the remaining portion of process isolated from cell bodies could be identified as belonging to the original process that was imaged before transection.
shRNA and quantitative reverse transcriptase PCR analyses.
shRNA constructs were generated by The Broad Institute (Boston, MA) and correspond to pool 6 (CypD6, PpidD6) and pool 9 (CypD9, PpidD9) against the CypD gene. shRNA against the luciferase gene was used as control. Mouse embryonic fibroblast (MEF) cells with reduced levels of CypD were generated by using methods previously described (Hetz et al., 2009). RNA from MEF cells was extracted using TRIzol and were reverse-transcribed using iScript cDNA synthesis kit (Bio-Rad). Reverse-transcribed RNA samples were then amplified by quantitative PCR (qPCR) using ABI Prism 7700 system (Applied Biosystems) with SybrGreen Master Mix (Qiagen). Transfected DRG cultures were similarly analyzed except RNA was extracted using the RNAqueous kit (Ambion) and a Prism 7900HT system (Applied Biosystems) was used. Cyclophilin D primers (sense, TGCAGGCCCCAATACAAATG; antisense, CCACACCTAGTCCTTTTATTACT) and histone H4 primers (sense, CGACAACATCCAGGGCATTACCAA; antisense, TCTCCTCGTAGATGAGACCCGAGA) were used for qPCR. All samples were assayed in quadruplicate from three separate experiments. The relative levels of CypD mRNA were normalized to mouse histone H4 mRNA internal control by the comparative threshold (Ct) method and RNA levels were then expressed relative to control shRNA-treated cells. To verify knockdown of endogenous CypD protein from cultured DRGs with shRNAs, cells were lysed in 300 mm sucrose, 1 mm EDTA, 5 mm HEPES, 1% Nonidet P-40, 0.25% sodium deoxycholate, pH 7.4, 1% PMSF, and 1% Protease Inhibitor Cocktail. Proteins were normalized by Bradford assay; 20 μg was used for SDS-PAGE and transferred to PVDF membranes. CypD protein was detected using mouse anti-CypD monoclonal antibody (1:1000; MitoSciences, #MSA04) and loading quantity was confirmed using rabbit anti-GAPDH monoclonal antibody (1:1000; Cell Signaling Technology). Blots were developed with HRP-conjugated anti-mouse or anti-rabbit antibodies and ECLADVANCE reagent (GE Healthcare Life Sciences).
Results
Inhibition of mPTP delays axonal degeneration
With its reproducibility and relative rapidity, Wallerian degeneration (WD) triggered by mechanical disconnection of the axon from its cell body has provided a useful experimental tool to study axonal degeneration (Tsao et al., 1999; Beirowski et al., 2004). To explore the mechanisms involved in WD, we used an ex vivo model of injury-induced axonal degeneration (Tsao et al., 1999) to directly test degenerative mechanisms in the absence of any effects of the neuronal cell body and the in vivo effects of immune system. For this, segments of severed mouse sciatic nerve were incubated in vitro, and then the degree of axonal degeneration was quantified by both morphological and biochemical analyses. Axonal levels of neuronal intermediate filament proteins, neurofilament medium (NF-M) and high (NF-H), were analyzed by immunofluorescence and Western blot as a direct readout of axonal degeneration. By immunofluorescence, the percentage of neurofilament-positive axons of WT nerve explants decreased to ∼10% of control after 3 d in culture (Fig. 1 B; supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Similar values were obtained by analyzing intact neurofilament protein levels as a surrogate of decay by Western blot (Fig. 1 D; supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). Consistent with previous ex vivo studies of WD (Beirowski et al., 2005), nerve explants from Wlds mice showed a delayed degeneration for several days (supplemental Fig. S1B,D, available at www.jneurosci.org as supplemental material). To further validate the explant system for testing local mechanisms of WD, we tested the effect of pharmacological agents that have been shown to delays WD either in vivo or in culture models. Consistent with previous reports (George et al., 1995; Zhai et al., 2003; Miller et al., 2009), proteasome inhibition (MG132), c-Jun N-terminal kinase (JNK) inhibition (SP600125) or chelation of extracellular calcium with EGTA delayed axonal degeneration in nerve explants over 3 d ex vivo compared with control nerve explants (supplemental Fig. S2A–D, available at www.jneurosci.org as supplemental material).

Pharmacological inhibition of mPTP delays axonal degeneration. A , Transverse sections of nerve explants stained for NF-H (red) and NF-M (green) isoforms. The NF signals decrease considerably after 3 days (3d) in vehicle solution (Veh) and are almost completely lost by 6 days (6d). The CsA (20 μm)-treated explants show preservation of axonal proteins over 3 and 6 d in culture. Scale bar, 20 μm. B , Quantification of NF-H-positive axons in explant cross sections as shown in A , expressed as axons per 100 μm2. Statistically significant protection is seen after CsA treatment at both 3 and 6 d (n = 3 per each group; # p < 0.05 by Student's t test compared with 0 d; *p < 0.05 by Student's t test compared with 3 d vehicle; error bars indicate SEM). C , Nerve explants after different incubation times and conditions were analyzed by Western blot. Treatment with CsA delays the decrease of NF-H at 3 and 6 d compared with untreated nerves. The same effect is seen for neurofilament light (NF-L) at 3 d, but less overall protection was seen for NF-L in the 6 d incubation samples. histone H3 (His-H3) was used as a loading control. D , Densitometry of NF-H normalized to His-H3 and expressed as percentage of NF-H at day 0. Significant protection at 20 μm CsA for neurofilament decay is seen at 3 d and also at 6 d (n = 3 per each group; # p < 0.05 by Student's t test compared with 0 d; *p < 0.05 by Student's t test compared with 3 d vehicle; error bars indicate SEM). E , Transverse sections of nerve explants stained for NF-H (red) and NF-M (green). Incubation with the mPTP blockers DIDS (250 μm), R.Red (50 μm), and BAPTA-AM (100 μm) for 3 d delays axonal degeneration. Scale bar, 20 μm. F , Quantification of axons positive for NF-H in explant cross sections, expressed as axons per 100 μm2 (n = 3 per group; # p < 0.05 by Student's t test compared with 0 d; *p < 0.05 by Student's t test compared with 3 d vehicle; error bars indicate SEM). G , By Western blot analyses for NF-H, each compound appeared to delay the decay of intact NF-H in the nerve explants over 3 d incubation. H , Densitometry of NF-H normalized to His-H3 and expressed as percentage of NF-H at day 0. Significant protection of NF decay is seen at 3 d (n = 3 per group; # p < 0.05 by Student's t test compared with 0 d; *p < 0.05 by Student's t test compared with 3 d vehicle; error bars indicate SEM).
To assess the potential involvement of the mPTP in axonal degeneration, we exposed severed nerve segments to CsA, which binds to the mPTP regulatory protein CypD (Halestrap and Davidson, 1990). Incubation of WT nerve explants with CsA significantly protected axons from degeneration over 3 and 6 d of culture (Fig. 1 A–D). Since CsA can also inhibit calcineurin, we tested the compound FK506 that inhibits calcineurin but has shown no binding to CypD (Friberg et al., 1998). FK506 did not prevent or appreciably delay axonal degeneration in nerve explants over 3 d in culture (supplemental Fig. S2E–H, available at www.jneurosci.org as supplemental material). We compared the rate of degeneration of WT and Wlds axons with or without CsA treatment. The time course of axonal degeneration for CsA-treated WT axons was comparable to nontreated Wlds axons over 9 d in culture (Fig. 2). However, the axonal degeneration of Wlds axons was not further delayed by CsA treatment (Fig. 2), suggesting that protection from axonal degeneration in Wlds axons might be related to delayed activation of the mPTP after injury (see Fig. 5).

Rate of axonal degeneration in WT and Wlds explants and its modulation by CsA. A–C , WT ( A ) and Wlds ( B ) mouse sciatic nerves were incubated for 3, 6 and 9 d in vehicle (Veh) or CsA (20 μm). Immunostained sections were analyzed quantitatively ( C ). Nerve cross sections were immunostained for NF-H (red). Top row, control nerves, and below, nerves cultured for the indicated times in vehicle solution (Veh, left) or CsA (right). Scale bar, 20 μm. CsA significantly delays axonal degeneration of WT axons ( A ). C , Axonal density (axons per 100 μm of cross sectional area). The reduction of axonal density with time in WT nerves is impaired by CsA to a level comparable to nontreated Wlds explants. CsA do not further protect Wlds axons from injury-induced axonal degeneration up to 6 d, but significant protection is found at 9 d (n = 3 per each group; *p < 0.05 by Student's t test compared with CsA treatment in WT; error bars indicate SEM).
VDAC activation and increased cytoplasmic calcium have been implicated in activation of mPTP (Halestrap, 2009). Thus, we tested the possible contribution of VDAC and/or calcium increases to axonal degeneration in explant cultures. Incubating the nerve explants with DIDS, a blocker of the VDAC, significantly reduced axonal degeneration (Fig. 1 E–H). Treating the nerve explants with the intracellular calcium chelator BAPTA-AM similarly delayed axonal degeneration (Fig. 1 E–H). Finally, inhibiting mitochondrial calcium uptake by targeting the mitochondrial calcium uniporter using R.Red also protected the axons from degeneration (Fig. 1 E–H). Although both DIDS and R.Red might inhibit endoplasmic reticulum VDAC and calcium uniporters, respectively (Shoshan-Barmatz et al., 2004), the collective effects of CsA, DIDS and R.Red on degeneration of severed axons suggest a central role for mPTP in Wallerian degeneration.
Axon degeneration and mitochondrial swelling is delayed by CsA
One of the ultrastructural signatures of mPTP opening is mitochondrial swelling, produced by increase in permeability of the mitochondrion's inner membrane (Hunter et al., 1976). Mitochondrial swelling was reported in early ultrastructural analyses of Wallerian degeneration (Vial, 1958; Webster, 1962), and more recently in neurodegenerative diseases where axonal degeneration is seen (Ferreirinha et al., 2004; Sasaki et al., 2004; Martin et al., 2009). Thus, we asked whether CsA could prevent swelling of axonal mitochondria in severed nerves. Nerve explants were processed for electron microscopy (EM) and mitochondrial diameters were quantified. Nerve explants cultured for 3 d in vehicle solution displayed a mixed population of degenerating axons, where most of them were completely degenerated with collapsed myelin sheaths, whereas a population of axons presented better structural preservation with axoplasm at varying degrees of degeneration (Fig. 3 B), consistent with the immunofluorescence results showing a small proportion of spared axons (Fig. 1 A). Mitochondrial diameters were very constant in transverse sections of intact axons due to their characteristic longitudinal orientation in the nerve fibers (Fig. 3 A). Morphometric analyses of 3 d WT explants showed that mitochondria located in visibly intact axons displayed more than a twofold increase in diameter when compared axonal mitochondria from the control nerves (i.e., time 0) (Fig. 3 A,B,E). These mitochondrial changes are comparable to those seen in distal degenerating axons 3 d after in vivo nerve transection (Fig. 3 F–K). In addition, the axonal mitochondria in our ex vivo model system also presented several ultrastructural features that have been associated with mPTP-related swelling, including cristae reorganization and rupture of the outer mitochondrial membranes (Fig. 3 B,H–K). Other common ultrastructural features observed in both in vivo and ex vivo degenerating axons were clustering of mitochondria near the Schmidt-Lanterman incisures that traverse the myelin sheaths and frequent condensation of an electron-dense material in the axoplasm adjacent to swollen mitochondria (Fig. 3 F–I) (data not shown). Notably, these features of swollen mitochondria were not seen in nerve explants from the Wlds mice after similar incubation periods (see Fig. 5 A,B, below).

Axonal degeneration and mitochondrial swelling are delayed by CsA. Representative electron micrographs of control ( A , B ), CsA-treated ( C ), and JNK inhibitor-treated ( D ) wild-type sciatic nerves from explants cultured for 3 d ( A–D ). For comparison, representative images from distal nerve at 3 d in vivo axotomy are shown ( F–K ). A–D , Nerves were processed immediately for EM or after 3 d incubation in vehicle, CsA (20 μm), or the JNK inhibitor SP600125 (60 μm). Top row, WT nerves at low magnification; bottom row, corresponding mitochondria at high magnification. A , At 0 d the top shows that axons are rounded, Schmidt-Lanterman incisures are visible, and the tissue is well organized. Examples of the morphology of mitochondria from day 0 axons are shown at high magnification in the bottom. B , After incubation for 3 d in vehicle solution, the tissue is disorganized, axonal degeneration is extensive, and myelin sheaths are collapsed. In the high-magnification images at the bottom, mitochondria of axons with conserved axoplasm are clearly increased in diameter. C , After 3 d exposure to CsA, the nerve tissue is less altered compared with A , and more axons show a preserved axoplasm; the high-magnification images show mitochondria comparable those in A . D , After exposure to SP600125, the overall picture is similar to that after CsA, including the appearance of mitochondria. Scale bars: top, 10 μm; bottom, 300 nm. E , Average diameter of mitochondria in WT axons measured in EM transverse sections of axons with preserved axoplasm is shown with error bars for SEM. At 3 d of incubation, the diameter of mitochondria is 225% of the control value. This swelling is prevented substantially by CsA, the JNK inhibitor SP600125, DIDS, R.Red, and BAPTA-AM (n ≥ 45 mitochondria/nerve over 3 separate experiments; # p < 0.05 by Student's t test compared with 0 d; *p < 0.05 by Student's t test compared with 3 d vehicle). F–K , Axons undergoing degeneration in vivo display swollen mitochondria comparable to those in explants. Clusters of mitochondria are a characteristic feature of degenerating axons in explants (data not shown) and in vivo ( F , G ). Other abnormalities include remodeled mitochondrial cristae ( H–K ) and rupture of the outer mitochondrial membrane (arrow in K , which represents a higher magnification of a region in J ). Accumulation of electron-dense material is usually associated with swollen mitochondria ( F–I , arrowhead in I ); the nature of this dense and disorganized material is not clear, but an interesting possibility is that it represents aggregated cytoskeletal proteins in the process of degeneration. Scale bars: ( F , G ), 1 μm; ( H , J ), 200 nm; ( I ), 300 nm. L , Mitochondrial swelling precedes axonal degeneration. Wild-type sciatic nerve explants were incubated in vehicle solution for the indicated durations. Nerve explants were fixed and processed for electron microscopy. The left y-axis shows mean mitochondrial diameter in axons (black circles, solid black line) vs Schwann cells of cross-sectioned sciatic nerves (black triangles, dashed black line), and the right y-axis shows axon degeneration expressed as percentage of degenerated axons (right y-axis, red triangles, solid red line). Axonal mitochondrial diameters but not those in the Schwann cells show a rapid increase after 6 h. In contrast, axonal degeneration is not apparent until after 24 h (n = 3 per each time point, between 30 and 100 mitochondria measured per n; error bars indicate SEM).
When wild-type nerve explants were incubated with CsA for 3 d, there was clear evidence of axonal protection at the EM level (Fig. 3 C). The CsA-treated explants showed statistically smaller diameter mitochondria in their axons compared with the nontreated nerve explants of 3 d in culture (Fig. 3 E). Similarly, explants treated with the JNK inhibitor, DIDS, R.Red or BAPTA-AM also showed significantly smaller diameter mitochondria than control explants (Fig. 3 D,E; supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). Thus, these agents are also able to delays the ultrastructural signature of mPTP activation suggesting that JNK, VDAC activation, and loss of mitochondrial homeostasis precede the mitochondrial swelling.
Comparing the degree of mitochondrial swelling to the number of visibly degenerated axons over time suggests that the mitochondrial changes precede the loss of axon integrity by several hours. Minimal swelling was seen at 6 h and a nearly twofold increase in average mitochondrial diameter was seen at 12 h in culture; significant axon degeneration was not appreciable until beyond 24 h in culture (Fig. 3 L). Importantly, mitochondrial swelling was detected only in axons and not in their associated Schwann cells (Fig. 3 L), suggesting that swelling represents an intrinsic neuronal response triggered by axotomy. Indeed, the mitochondrial swelling of axons as early as 12 h in culture was prevented by incubation of explants with CsA (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material), demonstrating that CsA can also prevent or delay rather than reverse the early mitochondrial changes associated with nerve injury.
Inhibition of mPTP delays axonal degeneration in the CNS
To determine whether axons from the CNS also show evidence of mPTP activation after axotomy, we analyzed explants of mammalian optic nerves using the same approach described above. Explants from WT mouse optic nerves showed a dramatic reduction in NF-positive axons after 4 d in culture, and incubation with CsA similarly protected these CNS axons from degeneration (Fig. 4 A,B). At the EM level, optic nerves displayed widespread axonal degeneration with obvious mitochondrial swelling after 4 d incubation in vehicle solution; the mitochondria also showed cristae reorganization and rupture of their outer membranes (Fig. 4 C–E), similar to changes found in sciatic nerve explants. Importantly, both the axonal degeneration and mitochondrial changes in the optic nerve explants were also clearly delayed by incubation with CsA (Fig. 4 C–E). Together, these results suggest that a common mPTP-dependent mechanism of degeneration takes place in the axons of both the PNS and CNS.
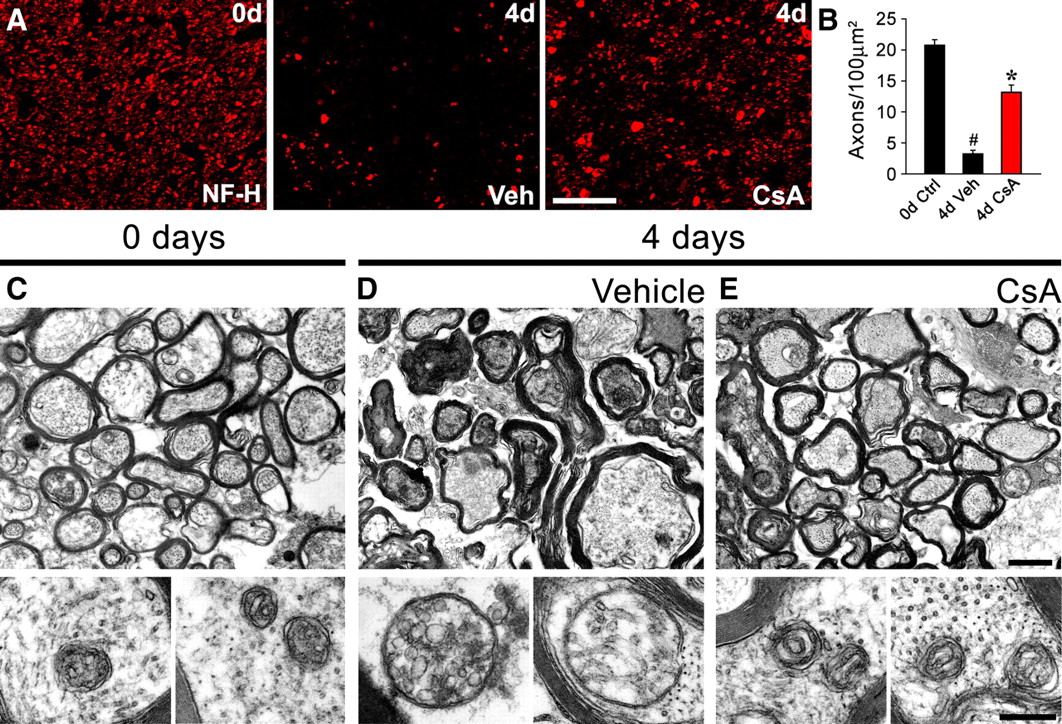
Axonal degeneration in optic nerves is delayed by CsA. Optic nerve (ON) explants from WT mice were incubated in the presence of vehicle or CsA (20 μm) for 4 d and analyzed by immunofluorescence and EM. A , Transverse sections of nerve explants stained for NF-H. After incubation of ON explants for 4 d in vehicle solution, the NF signal drops considerably. Incubation with CsA preserves the NF signals in these ON samples. Scale bar, 20 μm. B , Quantification of NF-H-positive axons in explant cross sections as shown in A , expressed as axons per 100 μm2. Statistically significant protection is seen after the CsA treatment (n = 3 per group; # p < 0.05 by Student's t test compared with 0 d; *p < 0.05 by Student's t test compared with 3 d vehicle; error bars indicates SEM). C–E , Top shows WT nerves at low magnification, and bottom shows corresponding mitochondria at high magnification. D , After incubation for 4 d in vehicle solution, the tissue is disorganized, axonal degeneration is extensive, and myelin sheaths are collapsed. The mitochondria in preserved axons are swollen (high magnification). E , Incubation with CsA protects from injury-induced axonal degeneration and mitochondrial swelling is prevented. Scale bars: top, 10 μm; bottom, 300 nm.
Activation of mitochondrial permeability transition triggers degeneration of Wlds axons
Axonal degeneration is dramatically delayed in axotomized Wlds nerves in vivo and ex vivo (Lunn et al., 1989; Beirowski et al., 2004) (supplemental Fig. S1B,D, available at www.jneurosci.org as supplemental material), but the mechanism of protection of axons remains unknown. Therefore, we tested whether exogenous activation of mPTP would accelerate Wallerian degeneration in nerve explants from Wlds mice. By EM, Wlds sciatic nerve explants cultured for 3 d in vehicle solution showed intact axons with axonal mitochondrial diameters comparable to those of control Wlds nerves (i.e., time 0) and CsA-treated WT nerves (Fig. 5 A,B). Treating Wlds nerve explants for 3 d with the mPTP activator ATR, which stimulates mPTP by inducing a conformational change in ANT (Halestrap et al., 1997), triggered characteristic morphological signs of axonal degeneration (Fig. 5 C–E; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Importantly, mitochondrial diameters in axons of ATR-treated Wlds nerve explants were significantly larger when compared with those of the nontreated Wlds nerve explants (Fig. 5 B,C). These results suggest that the protective mechanism of the Wlds protein may lie upstream of the mPTP.

Activation of mPTP triggers degeneration of Wlds nerves. Sciatic nerve explants from Wlds mice were incubated in vehicle solution or with the mPTP activator ATR (100 μm) for 3 d and analyzed by EM and immunofluorescence. A–C , Top shows WT nerves at low magnification, and bottom show corresponding mitochondria at high magnification. B , Axons from Wlds mice do not show signs of degeneration after incubation for 3 d in vehicle solution. Mitochondria have sizes comparable to those of axonal mitochondria in explants from day 0 ( A ). C , After incubation for 3 d with ATR, axonal degeneration is extensive. Extensive mitochondrial swelling is seen in ATR-incubated axons ( C , bottom). Scale bars: top, 10 μm; bottom, 300 nm. D , Transverse sections of nerve explants stained for NF-H (red) and NF-M (green) isoforms. In the nerve explants incubated with ATR for 3 d, the number of NF-positive axonal profiles is visibly decreased compared with the vehicle-treated nerve explants. Scale bar, 20 μm. E , Quantification of axons positive for NF-H in explant cross sections as shown in D , expressed as axons per 100 μm2. A statistically significant decrease in axonal density is seen after ATR treatment for 3 d (n = 3 per group; *p < 0.05 by Student's t test compared with 3 d vehicle; error bars indicate SEM). Lower ATR doses were also able to trigger degeneration of Wlds nerves as shown in supplemental Figure S5, available at www.jneurosci.org as supplemental material.
The rate of axonal degeneration in the ATR-treated Wlds explants was comparable to the phenotypes observed in nontreated WT explants (Fig. 6 A–C). In addition, axonal degeneration was not accelerated by ATR treatment in the WT nerve explants (Fig. 6 A,C), suggesting the pathway cannot be further activated in WT axons.

Axonal degeneration in WT and Wlds explants after mPTP activation. WT ( A ) and Wlds ( B ) mouse sciatic nerves were incubated for 1, 2, and 3 d in vehicle (Veh) or with the mPTP activator ATR (100 μm). Nerve cross sections immunostained for NF-H (red) are shown [top row, control nerves; bottom row, nerves cultured for the indicated times in vehicle solution (Veh, left) or ATR (right)]. Scale bar, 20 μm. Incubation of Wlds explants with ATR decreases the number of NF-positive axonal profiles compared with the vehicle-treated Wlds nerve explants ( B , C ). Note that the rate of WT axonal degeneration is not modified by ATR treatment ( A , C ) (n = 3 per group; *p < 0.05 by Student's t test compared with any other condition; error bars indicate SEM). D , Calcium-dependent loss of mitochondrial membrane potential (ΔΨm) is similar in purified WT and Wlds brain mitochondria. Purified WT and Wlds brain mitochondria were loaded with the membrane potential-sensitive dye TMRM. This dye accumulates inside mitochondria leading to quenching of its fluorescence. After mitochondrial depolarization, TMRM is released, leading to increase in the measured fluorescence. Thus, increase in TMRM fluorescence reflects mitochondrial depolarization. Mitochondria were incubated with 5, 50, and 100 μm calcium or vehicle. Preincubation with 5 μm CsA was used to demonstrate that the loss of ΔΨm after calcium addition is mPTP dependent. Changes in fluorescence were normalized to basal levels (precalcium or vehicle addition, scale from 0 to 1), and the values shown represent the mean fluorescence of the first 15 measurements (each separated by 43 s) after treatment with calcium or vehicle. As expected, calcium leads to a dose-dependent loss of ΔΨm (increase in TMRM fluorescence) that is inhibited by pretreatment with CsA. WT and Wlds brain mitochondria show no differences in their calcium-dependent loss of ΔΨm (n = 6 for each treatment; 3 animals per strain).
Comparable mPTP activation in isolated WT and Wlds mitochondria
To determine whether the delayed mPTP activation in Wlds is intrinsic to Wlds mitochondria, we isolated mitochondria from Wlds and WT brains and exposed them to increasing [Ca2+], which activates mPTP (Hansson et al., 2003). These purified brain mitochondria showed no appreciable differences in loss of ΔΨm with increasing [Ca2+] (Fig. 6 D). Moreover, WT and Wlds mitochondria were equally protected from loss of ΔΨm by preincubation with CsA (Fig. 6 D). These data indicate that the Wlds protein expression does not change the intrinsic response of the mitochondria to Ca2+ overloading.
As pharmacological inhibition of JNK activation strongly delays axonal degeneration and mitochondrial swelling (supplemental Fig. S2, available at www.jneurosci.org as supplemental material; Fig. 3), we studied activation of this pathway in WT and Wlds nerve explants. JNK phosphorylation increases at 6 and 12 h postinjury in WT, but not in Wlds explants (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). JNK activation in WT explants is inhibited by SP600125 (supplemental Fig. S5, available at www.jneurosci.org as supplemental material), showing the efficacy of the JNK inhibitor in this experimental system. Thus, JNK activation is an early event after nerve injury that must lies downstream of the Wlds protein's protective effects that prevent mPTP activation.
Depleting the mPTP protein CypD from neurons protects them from axonal degeneration
The data outlined above indicate that axonal degeneration in nerve explants can be significantly delayed by CsA, which is known to block opening of the mPTP by binding to CypD. Although the FK506 analyses seemed to rule out calcineurin as a mediator of axonal degeneration, other activities of CsA could contribute to axonal protection beyond its binding to CypD or calcineurin. Direct immunosuppression by CsA is likely not a consideration in the explant system where any effects of inflammation are limited to cells resident to the nerve at the time of explanting. To more directly test the contribution of CypD to axonal degeneration, we used an shRNA strategy to deplete CypD from cultured primary neurons. Two independent shRNA constructs for CypD, D6 and D9, caused a significant reduction of CypD mRNA in both MEF cells and adult rat DRG cultures (supplemental Fig. S6A, available at www.jneurosci.org as supplemental material). There was some variability in the responses to the individual shRNA preparations comparing the MEF and DRG cultures, which could relate to lower transfection efficiency of the primary DRG preparations. However, both shRNAs caused a clear and consistent depletion of CypD mRNA, and in the DRG cultures, there was a corresponding decrease in CypD protein (supplemental Fig. S6B, available at www.jneurosci.org as supplemental material).
Using CypD-depleted DRG cultures, we tested the time course of axotomy- and chemical-induced axonal degeneration. Cultures of adult rat DRG neurons showed axonal degeneration after transection of axons (Fig. 7 A). Similarly, treatment with the chemotherapeutic agent vincristine induced axonal degeneration in the same cell culture model (Fig. 7 B). By live cell imaging, we observed that cultures with reduced levels of CypD showed delayed axonal degeneration after severing of their processes with a micropipette (Fig. 7 A). Considering it took a few hours to image a single axon with this time lapse imaging approach, we evaluated axonal degeneration in intact cultures treated with vincrinstine to quantify the protective effects of CypD depletion. At 50 nm vincristine, axonal degeneration was seen by 10 h in DRGs transfected with control shRNAs, but neurons transfected with the two CypD shRNA constructs showed significantly fewer degenerating axons (supplemental Fig. S6C–E, available at www.jneurosci.org as supplemental material). Importantly, these CypD-targeted cultures were also protected from axonal degeneration at higher doses of vincrinstine that caused an accelerated axonal degeneration (Fig. 7 B).

Knocking down CypD from neurons protects them from axonal degeneration. Three different approaches were used to determine whether depletion of CypD from dissociated adult DRG cultures using shRNA would protect these neurons from axonal degeneration. For this, the DRGs were transfected with CypD shRNAs (D6 and D9) vs a control nontargeting shRNA plus a vector encoding AcGFP to aid in visualizing processes. Efficacy of depletion is shown in supplemental Figure S7, available at www.jneurosci.org as supplemental material. A , Representative images of control and D9 CypD shRNAs (n = 5) before and after transection with a glass micropipette are shown in the top and bottom image sequences, respectively; the inset in the 1 min time point panel shows DIC image of the transected axon indicating the growth cone (arrowhead) and transection site (arrow). The control shRNA-transfected neurons consistently showed rapid degeneration of the distal axon after transection, but the CypD shRNA-transfected neuron showed preservation of the axon over the same time period. Similar findings were seen with D6 CypD shRNA. Scale bar, 50 μm. B , Transfected DRG cultures as in A were treated with 1 μm vincristine to induce axonal degeneration. Representative images of the AcGFP fluorescence for a single neuron over time are shown in micrographs. Quantification of axonal degeneration is shown in the graph for control, D6, and D9 CypD shRNA-transfected cultures as the average number of neurons clearly showing degeneration ± SEM (n ≥ 36 neurons analyzed for each condition over 3 separate culture experiments; ***p < 0.001 for CypD shRNA vs control shRNA for indicated times by two-way ANOVA with Bonferroni post hoc test). C , To more accurately assess the site of CypD's action, transfected DRG neurons were cultured on a porous membrane that allows for separation of axonal processes from cell bodies and non-neuronal cells (Willis et al., 2005). After 72 h culture, individual neurons were imaged and then the cell bodies and non-neuronal cells were scraped away from the upper membrane surface. The axonal trees of the same neurons were imaged 4 h later. Representative pretransection and posttransection images are shown in the micrographs. Quantification of axonal degeneration is shown in the graph as average percentage of neurons showing axonal degeneration ± SEM (n ≥ 38 and 78 neurons analyzed for each condition over 4 separate culture preparations; ***p ≤ 0.001 for CypD shRNA vs control and for D6 vs D9 CypD shRNAs by two-way ANOVA with Bonferroni post hoc test). Scale bars: (in C ), B , 100 μm; C , 50 μm.
Since these DRG cultures also contain glial cells, the above experiments did not address the site of action for CypD. Even in the severed axons, glial cells closely apposed to the distal severed axon could play a protective role. To directly test the cellular source of the protection, we took advantage of a culture system that physically separates distal axons from glial cells (Willis et al., 2005). In severed axons completely removed from any contact with glial cells, neurons expressing shRNAs against CypD mRNA were significantly protected from axonal degeneration compared with cultures transfected with the control shRNAs (Fig. 7 C). Thus, consistent with previous studies, Wallerian degeneration is intrinsic to the axonal compartment. Moreover, it is neuronal CypD rather than mPTP activation in other cells in the nerve that can trigger Wallerian degeneration.
Discussion
Although axonal degeneration has been extensively studied over the last few years, the molecular pathway connecting the injury event to the destruction of axons and synapses has not been determined. Axonal transport defects, local activation of signaling pathways, mitochondrial dysfunction, and calcium-dependent proteolysis have all been linked to Wallerian degeneration (Ikegami and Koike, 2003). Our data indicate that axonal degeneration triggered by mechanical or chemical stimuli is dependent upon the activation of the mPTP within the axon. Inhibition of axonal degeneration by several mPTP inhibitors and depletion of the mPTP protein CypD show that the axonal degeneration program is executed by an mPTP-dependent mechanism. Furthermore, axonal protection by CsA in both sciatic and optic nerves, points to the mPTP as a common degenerative program for both the PNS and CNS.
Previous studies suggested that in vivo treatment with CsA delays axonal degeneration in the PNS (Sunio and Bittner, 1997; Taskinen and Röyttä, 2000), however this was attributed to immunosuppression rather than any direct effects on the neuronal mitochondria. Genetic inactivation of CypD was also shown to reduce axonal damage and disease severity in experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (Forte et al., 2007). Although these in vivo models are compelling, they do not provide significant insight into the site of action for CsA and CypD. The ex vivo nerve explants and neuronal cultures with isolated axons that were used here clearly show that CypD's function in axonal degeneration is autonomous to the neuron and specifically occurs within the axon. Together with ultrastructural observations of mitochondrial membrane disruption and volume alterations, the axon protective effects of CsA and CypD knockdown point to mitochondrial permeability transition activation as a central mediator of axonal degeneration.
In vivo axonal degeneration in the mouse PNS takes place after a latent phase of 1–1.5 d postaxotomy (Beirowski et al., 2005; Kerschensteiner et al., 2005). This is likely the time period required for activation of signaling pathways, loss of axonal ionic homeostasis and/or extinction of cellular mechanisms that prevent mPTP activation. Several lines of evidence point to impaired axonal transport as a common event leading to axonal degeneration (Morfini et al., 2009), but the mechanism(s) by which transport deficits trigger axonal degeneration remains unclear. The recent observation that continuous transport of endogenous NMNAT2 along axons delays axonal degeneration in wild-type neurons suggests that NMNAT activity can also suppress these events (Gilley and Coleman, 2010). In the Wlds mice, long-lived Wlds protein could substitute for the more labile NMNAT2 in severed axons that do not have a source of cell body-derived proteins (Coleman and Freeman, 2010). Since activation of mPTP with ATR triggered axonal degeneration in axons from Wlds mice (Figs. 5, 6), the protective effects of Wlds protein, and likely of NMNAT2 in WT animals probably lie upstream of mPTP activation. Our results using purified brain mitochondria from WT and Wlds mice shown comparable lost of ΔΨm with increasing [Ca2+] (Fig. 6 D), suggesting that the Wlds protein or endogenous NMNAT2 is not directly inhibiting mPTP in the mitochondria, but rather acting in a regulatory cascade upstream mPTP, which probably includes the activation of JNK as our results demonstrate (supplemental Fig. S5, available at www.jneurosci.org as supplemental material).
Several nonexclusive mechanisms could be proposed for the events that occur after mPTP activation leading to axonal degeneration, which will require detailed temporal and spatial examination. The final stages of axonal degeneration are characterized by granular disintegration of the axoplasm, and these are dependent upon intra-axonal calcium overload with activation of calcium-dependent proteases (George et al., 1995). Mitochondria dynamically participate in calcium homeostasis by buffering intracellular calcium (Gunter et al., 2004). Mitochondrial calcium overloading leads to mPTP opening, and unregulated calcium release through mPTP and ROS generation act as a positive feedback loop further activating the mitochondrial permeability transition (Bernardi et al., 2006; Halestrap, 2009). This would be consistent with the catastrophic cycle for axonal degeneration that has been suggested for both central and peripheral axons (Beirowski et al., 2005; Kerschensteiner et al., 2005; O'Brien et al., 2009). Furthermore, a very early calcium rise in axonal mitochondria after injury precedes axoplasmic increase of this ion (LoPachin and Lehning, 1997), and might be one of the triggering stimuli for mPTP activation. This is temporally consistent with our results demonstrating an early and defined stage of mitochondrial swelling before disintegration of the axonal cytoskeleton (Fig. 3 L).
As a consequence of mPTP activation, the mitochondrial electrochemical proton gradient is dissipated, inhibiting ATP production (Halestrap, 2009). The decrease in axonal energy metabolism has been proposed to reduce Na+/K+ ATPase activity, Na+ influx, and reverse action of the Na+-Ca2+ antiporter, resulting in calcium influx from the extracellular milieu (Stys, 2005), which can further activate calcium-dependent degeneration pathways in the axoplasm. Such a loss of energy production with subsequent activation of the mPTP helps to explain why supplementing severed axons with NAD and pyruvate has sometimes been protective from injury and vincrinstine-induced degeneration (Araki et al., 2004; Wang et al., 2005). In addition to the role of NAD and pyruvate in mitochondrial energetic metabolism, each have been shown to inhibit activation of mPTP in other cellular systems (Fontaine and Bernardi, 1999; Kerr et al., 1999).
Our studies together with published data suggest a cascade of events from diverse stimuli that culminates with activation of mPTP and subsequent destruction of the axon. In our model (Fig. 8), transport of NMNAT2, or the Wlds protein in axons, is essential for inhibiting mPTP activation (Fig. 8 A) and proteasome-dependent degradation of NMNAT2 (Gilley and Coleman, 2010) accounts for the effects of proteasome inhibitors in delaying axonal degeneration (Zhai et al., 2003; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The axon's volume can represent >99% of the neuron's volume; this effectively makes the axon particularly vulnerable to defects in transport. In addition, other insults that locally target mitochondrial function will make them more susceptible to mitochondrial permeability transition activation (Fig. 8 B). Therefore, mitochondrial permeability transition likely represents a central pathway upon which the downstream effects of a variety of stimuli converge, acting as a decision node for axonal degeneration. For example, the local calcium overload in neurites adjacent to Aβ plaques in mice that model Alzheimer disease could effectively make these processes vulnerable for a localized activation of the mPTP (Kuchibhotla et al., 2008). Drugs affecting the polymerization state of microtubules induce axonal degeneration (Fig. 7 B) (Singer and Steinberg, 1972; Röyttä and Raine, 1986), and accumulation of misfolded proteins in several neurodegenerative diseases with axonal degeneration decreases axonal transport efficiency (Stokin et al., 2005; Morfini et al., 2009). Axonal JNK activation, which leads to axonal transport defects (Stagi et al., 2006) promotes degeneration of axotomized axons (Miller et al., 2009), and our results suggest its activation lies upstream mitochondrial permeability transition induction and downstream Wlds-dependent axonal protection (Fig. 3 D,E; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). After axonal mPT activation, diverse downstream events can be potentially involved in axonal destruction, including intra-axonal calcium overload, mitochondrial metabolic failure, ROS production and release of prodegenerative molecules from swelling mitochondria (Fig. 8 B).

Model of mPTP-dependent degeneration of the axonal compartment. A , Schematic representation of neuron with soma, axon, and terminals. Mitochondria are transported along the axon by a microtubule-dependent mechanism; function of the mitochondria largely requires transport of nuclear-encoded proteins from the cell body, including proteins that seem to inhibit mPTP activation (depicted by minus sign). Defects in axonal transport (star), which could be complete (e.g., nerve transection) or partial (e.g., toxic agents, protein aggregates, or genetic disorders), disturb the physiological equilibrium between the nuclear-encoded mPTP inhibitors and locally produced activators of the mPTP. B , Proposed molecular species involved in mPTP activation and axonal degeneration. In axons, mPTP formation is inhibited by NMNAT2, which is delivered from the cell body and is a target for the proteasome in injured axons (Coleman and Freeman, 2010). Wlds mutation, CsA, and reduction of CypD expression all prevent mPTP opening. mPTP activation will lead to calcium overload in the axon, decrease in ATP production, increase in ROS generation, and liberation of prodegenarative factors, which are potentially involved in degeneration of axonal components as well as triggering further mitochondrial dysfunction.
Importantly, mPTP inhibition by genetic or pharmacologic means does not seem to have overt secondary effects in animal models and extensive studies for pharmacological inhibition of this process are underway due to its involvement in a variety of pathological conditions, including neurodegenerative, myocardial ischemia-reperfusion, liver and muscle diseases (Bernardi et al., 2006). We surmise that identification of the mPTP as a convergence point to execute axonal degeneration from multiple signaling pathways presents a new target for therapeutic strategies in neurodegenerative conditions characterized by mitochondrial failure and axonal degeneration.
Footnotes
This work was supported by grants from Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) (no. 1070377) and Millennium Nucleus (no. P-07-011-F) (to F.A.C.); National Institutes of Health Grants R01-NS049041 and R01-NS041596 (to J.L.T.); and FONDECYT no. 1070444, Fondo de Financiamiento de Centros de Excelencia en Investigación (FONDAP) no. 15010006, Millennium Nucleus no. P-07-048-F, The Muscular Dystrophy Association, CHDI Foundation Inc., M. J. Fox Foundation for Parkinson Research, and the International Centre for Genetic Engineering and Biotechnology (to C.H.). We thank Sebastian Bernales and Ivan Alfaro for advice on mitochondrial experiments, and Monica Perez for excellent EM processing.
- Correspondence should be addressed to Dr. Felipe A. Court, Department of Physiology, Faculty of Biology, Pontificia Universidad Católica de Chile, Av. B. O'Higgins 340 / Casilla 114-D, Santiago 8331150, Chile. fcourt{at}bio.puc.cl; court.felipe{at}gmail.com





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}