

Abstract
Recent studies have suggested a role for phosphatidylinositol (PI) 3-kinase in cell survival, including the survival of neurons. We used rat sympathetic neurons maintained in vitro to characterize the potential survival signals mediated by PI 3-kinase and to test whether the Akt protein kinase, a putative effector of PI 3-kinase, functions during nerve growth factor (NGF)-mediated survival. Two PI 3-kinase inhibitors, LY294002 and wortmannin, block NGF-mediated survival of sympathetic neurons. Cell death caused by LY294002 resembles death caused by NGF deprivation in that it is blocked by a caspase inhibitor or a cAMP analog and that it is accompanied by the induction of c-jun, c-fos, andcyclin D1 mRNAs. Treatment of neurons with NGF activates endogenous Akt protein kinase, and LY294002 or wortmannin blocks this activation. Expression of constitutively active Akt or PI 3-kinase in neurons efficiently prevents death after NGF withdrawal. Conversely, expression of dominant negative forms of PI 3-kinase or Akt induces apoptosis in the presence of NGF. These results demonstrate that PI 3-kinase and Akt are both necessary and sufficient for the survival of NGF-dependent sympathetic neurons.
The survival of developing neurons requires extracellular signals that actively prevent programmed cell death. These signals are provided, in part, by neurotrophic factors such as nerve growth factor (NGF) (Oppenheim, 1991). Sympathetic neurons from the rat superior cervical ganglion (SCG) provide a well characterized model for studying NGF-mediated neuronal survival. If NGF is withdrawn from cultured sympathetic neurons, they undergo apoptosis (Deckwerth and Johnson, 1993; Edwards and Tolkovsky, 1994). Similarly, the SCG from mice lacking NGF (Crowley et al., 1994) or its receptor, TrkA (Smeyne et al., 1994), fail to develop because of massive neuronal death. In contrast, adding exogenous NGF in vivo prevents the naturally occurring death of sympathetic neurons during development (Hendry and Campbell, 1976). Thus, cultures of dissociated sympathetic neurons provide a useful in vitromodel for studying neurotrophic factor dependence.
Although the function of NGF as a survival-promoting factor is well established, the mechanisms responsible for NGF-mediated survival remain, in large part, uncharacterized. NGF affects neuronal survival and differentiation by binding to and activating the TrkA tyrosine kinase receptor (Barbacid, 1994). Once activated, TrkA autophosphorylates specific tyrosine residues within its intracellular domain (Kaplan et al., 1991; Klein et al., 1991). The phosphorylated tyrosines serve as protein interaction sites for several signaling molecules, including SHC, phospholipase C-γ, and phosphatidylinositol (PI) 3-kinase (Ohmichi et al., 1991; Carter and Downes, 1992; Raffioni and Bradshaw, 1992; Soltoff et al., 1992; Obermeier et al., 1993). The consequences of TrkA activation include Shc/Grb2/Sos-dependent activation of Ras and the subsequent activation of mitogen-activated protein (MAP) kinases, phospholipase C-γ-mediated production of diacylglycerol and inositol trisphosphate, and PI 3-kinase-mediated production of 3′-phosphorylated phosphoinositides (Kaplan and Stephens, 1994).
Although previous studies have focused on Ras and MAP kinases in NGF-mediated survival (Borasio et al., 1989, 1993; Ferrari and Greene, 1994; Nobes and Tolkovsky, 1995; Xia et al., 1995; Yao and Cooper, 1995; Creedon et al., 1996; Virdee and Tolkovsky, 1996), several recent reports suggest that PI 3-kinase functions in the survival pathways initiated by certain growth factors and survival-promoting agents. Using the PI 3-kinase inhibitors wortmannin and LY294002, Yao and Cooper (1995) reported that the inhibition of PI 3-kinase activity induces apoptosis in PC12 cells in the presence of NGF. This observation since has been extended to other immortalized cell lines, particularly those dependent on insulin-like growth factor-1 (IGF-1) for survival. For example, in Rat-1 fibroblasts PI 3-kinase inhibitors block IGF-1-mediated protection from apoptosis induced by UV irradiation (Kulik et al., 1997) and serum- or IGF-1-mediated protection from c-Myc-induced apoptosis (Kauffmann-Zeh et al., 1997;Kennedy et al., 1997). Similarly, PI 3-kinase inhibitors block IGF-1-mediated survival of PC12 cells (Parrizas et al., 1997). Consistent with these results, a function for PI 3-kinase has been demonstrated recently in the survival of cerebellar granule neurons mediated by IGF-1 or by potassium depolarization (D’Mello et al., 1997; Dudek et al., 1997; Miller et al., 1997).
To characterize further the role of PI 3-kinase in the survival of primary neurons, we have used NGF-dependent sympathetic neurons to compare the cell death caused by inhibitors of PI 3-kinase with that caused by NGF withdrawal. We also have tested whether a putative effector of PI 3-kinase, the serine/threonine protein kinase Akt (also known as protein kinase B or Rac protein kinase) (Burgering and Coffer, 1995; Franke et al., 1995; Kohn et al., 1995), functions in the survival of NGF-dependent neurons.
MATERIALS AND METHODS
Cycloheximide, actinomycin D, wortmannin, and chlorophenylthio-cAMP (cpt-cAMP) were purchased from Sigma (St. Louis, MO); boc-aspartyl(OMe)-fluoromethylketone (BAF) was obtained from Enzyme Systems Products (Dublin, CA); LY294002 was obtained from Biomol Research Laboratories (Plymouth Meeting, PA). Flavopiridol was provided by Drs. J. Johnson and E. Sausville (Drug Synthesis and Chemistry Branch, National Cancer Institute, Bethesda, MD).
Cell culture. Primary cultures of sympathetic neurons were prepared from SCG of embryonic day 21 rats as previously described (Martin et al., 1992) except that a preplating step was included to minimize non-neuronal cells. For preplating, SCG neurons were dissociated and resuspended in NGF-containing media (AM50 medium) consisting of 90% Minimum Essential Media (MEM; Life Technologies, Gaithersburg, MD), 10% FBS (Sigma), 2 mm glutamine, 20 μm uridine, and 20 μm fluorodeoxyuridine (to inhibit the proliferation of non-neuronal cells), 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml NGF (Harlan Bioproducts, Madison, WI). The cell suspension was filtered through a Nitex filter (size 3–20/14; Tetko, Briarcliff Manor, NY) and plated onto Primaria tissue culture dishes (Becton Dickinson, Lincoln Park, NJ) for a period of 1–2 hr. Nonadherent cells were collected and concentrated by centrifugation (10 min at 450 × g) and plated onto 60 mm collagen-coated dishes for reverse transcription (RT)-PCR (25,000 cells/dish) and kinase assays (125,000 cells/dish), onto collagen-coated two-well chamber slides (Nalge Nunc, Naperville, IL) for viability measurements (3000 cells/well), or onto poly-l-ornithine-coated (Sigma) and laminin-coated (Collaborative Biomed, Bedford, MA) 35 mm glass-bottomed dishes (MatTek, Ashland, MA) for microinjection (3000 cells/dish).
For NGF deprivation studies the neurons were switched into a medium identical to that described above except that it lacked NGF and contained neutralizing anti-NGF antiserum (AM0 medium). For potassium depolarization treatments, neurons cultured for 5 d in AM50 were switched into high K+ medium consisting of MEM supplemented to 50 mm KCl, 10% FBS, 2 mmglutamine, 20 μm uridine, 20 μmfluorodeoxyuridine, 100 U/ml penicillin, 100 μg/ml streptomycin, and neutralizing anti-NGF antiserum for 2 d before drug treatment.
Terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end labeling (TUNEL) analysis. Neurons plated on collagen-coated two-well chamber slides were treated with 100 μm LY294002 in AM50 for 72 hr. The cells were fixed in fresh 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.3% Triton X-100/PBS for 10 min, rinsed in PBS, and subjected to the TUNEL assay (Gavrieli et al., 1992). For the terminal deoxynucleotidyl-transferase reaction, neurons were overlaid with a reaction solution containing 1× terminal deoxynucleotidyl-transferase reaction buffer, 1 mm CoCl2, 0.25 U/μl terminal deoxynucleotidyl-transferase (Boehringer Mannheim, Indianapolis, IN), and 6 μm digoxigenin-11-dUTP (Boehringer Mannheim) and then incubated for 90 min at 37°C. Neurons were rinsed in PBS, incubated in blocking buffer (2% BSA and 5% goat serum in PBS) for 1 hr, and incubated overnight at 4°C with FITC-conjugated anti-digoxigenin antibodies (Oncor, Gaithersburg, MD) diluted 1:2 in blocking buffer. Then the neurons were rinsed in PBS and stained with 2 μg/ml Hoechst 33342 (Molecular Probes, Eugene, OR) in PBS for 5 min to visualize the nuclei of cells. After two additional rinses with PBS, the slides were covered with glass coverslips, using a mounting solution of 50% glycerol and 0.1% phenylenediamine in PBS, and then visualized by fluorescence microscopy with a Nikon Diaphot 300 inverted microscope.
Quantitation of neuronal viability. Equal numbers of neurons plated on collagen-coated two-well chamber slides were subjected to the appropriate treatments and then were fixed with fresh 4% paraformaldehyde/PBS overnight at 4°C. Neurons were rinsed in PBS and stained briefly with 0.1% crystal violet (EM Science, Gibbstown, NJ). The neurons were destained in H2O, dehydrated in increasing ethanol concentrations, transferred to xylene (Fisher Scientific, Pittsburgh, PA), and finally coverslipped by using Pro-Texx mounting media (Baxter Diagnostics, Deerfield, IL). Neurons staining darker than debris with a clearly defined cellular outline and a well defined nucleus were scored as viable. For each experimental treatment four fields of cells from each of three to four wells were counted under a 20× objective, and the average number of viable cells per field was determined. This was normalized to the average number of viable neurons in parallel nontreated control cultures. The results reported for each condition represent the means and errors (where appropriate) obtained from two to four independent experiments.
RT-PCR analysis. Preparation of cDNAs and analysis of gene expression in SCG neurons treated with LY294002 were essentially the same as those described for NGF-deprived neurons (Freeman et al., 1994). Preplated cultures (25,000 neurons plated per time point) were maintained in AM50 for 5 d and then treated with LY294002 diluted to a final concentration of 100 μm in AM50 for the indicated intervals. Polyadenylated RNA was isolated by direct hybridization to oligo-dT-cellulose beads, as described by the manufacturer (QuickPrep Micro mRNA Purification Kit, Pharmacia Biotech, Piscataway, NJ). One-half of the recovered mRNA was reverse-transcribed by using Moloney murine leukemia virus reverse transcriptase (Superscript II RT; Life Technologies) and random hexamers (16 μm) as primers in 20 μl reactions containing 50 mm Tris, pH 8.3, 40 mm KCl, 10 mmDTT, 6 mm MgCl2, 20 U RNAsin (Promega, Madison, WI), and 500 μm each dATP, dCTP, dGTP, and dTTP (Boehringer Mannheim). After a 1 hr incubation at 42°C, the reaction was terminated by adding 80 μl of H20 and heating the reaction for 5 min at 95°C. Specific cDNAs were amplified in 30 μl PCR reactions containing the appropriate primer pairs (0.6 μm each), 1× Taq polymerase buffer, 1 UTaq polymerase, 1.5 mm MgCl2, 50 μm dCTP, 100 μm each dATP, dGTP, and dTTP, 6 μCi [α-32P] dCTP (DuPont NEN, Boston, MA), and 0.6 μl cDNA synthesized in the RT reaction. PCR parameters were 1 min at 94°C, 1 min at 60°C, and 2 min at 72°C for 16–28 cycles, followed by a final 10 min incubation at 72°C. Reaction products were separated by electrophoresis and analyzed by autoradiography and PhosphorImager analysis (Molecular Dynamics, Sunnyvale, CA). Control experiments to determine the linear range of PCR amplification and to verify the identity of amplified products were as described previously, as were the sequences of oligonucleotide primers (Estus et al., 1994;Freeman et al., 1994).
Plasmid expression vectors. Expression vectors for theEscherichia coli β-galactosidase (LacZ) gene, p110*, and p110*Δkin under the control of the human cytomegalovirus immediate early gene promoter have been described previously (Greenlund et al., 1995a; Hu et al., 1995). Myr-Akt and A2myr-Akt cDNAs (Kohn et al., 1996b) were cloned behind the cytomegalovirus promoter in the plasmid pcDNA3 (Invitrogen, San Diego, CA) by inserting the KpnI toXbaI fragments from pECE-myr-Akt or pECE-A2myr-Akt between the KpnI and XbaI sites of pcDNA3. The Δp85 cDNA was removed from pGEX-Δp85 (Kotani et al., 1994) as aBamHI to EcoRI fragment and inserted between theBamHI and EcoRI sites in pcDNA3. The rat AH-Akt·Flag construct, encoding amino acids 1–148 of rat Akt followed by the Flag epitope, was generated by pfu polymerase (Stratagene, La Jolla, CA) amplification from rat SCG cDNA, using a 5′ primer (5′-GCG GAT CCA CCA TGA ACG ACG TAG CCA TTG TG-3′) containing aBamHI site, a consensus transcription start site, and nucleotides 1–21 of the rat Akt open reading frame (Konishi et al., 1994). The 3′ primer (5′-GCG AAT TCT CAC TTG TCA TCG TCG TCC TTG TAG TCG TTC ATG GTC ACA CGG TG-3′) consisted of an EcoRI site, nucleotides 427–444 of the rat Akt open reading frame, and sequences corresponding to the Flag epitope. The PCR-generated fragment was ligated into the BamHI and EcoRI sites of pcDNA3. DNA sequencing revealed that the construct was correct. AktK179A–pcDNA3 was constructed by ligating anEcoRI/Klenow-filled–BglII fragment from pSG5 HA-PKB K179A (Burgering and Coffer, 1995), containing an Akt kinase-inactive mutant cDNA, into the EcoRI andEcoRV sites of pcDNA3.
Intracellular microinjections. Neurons plated on poly-l-ornithine and laminin-coated glass-bottomed 35 mm dishes were microinjected by using a Nikon Diaphot 300 inverted microscope equipped with a PLI-100 picoinjector (Medical Systems, Greenvale, NY) and a Narishige micromanipulator (Nikon-Narishige, Tokyo, Japan). Microinjection needles were pulled from glass capillaries with a horizontal micropipette puller (Sutter Instruments, Novato, CA). For experiments in which neuronal survival was evaluated, expression plasmids were diluted to a final concentration of 50–100 μg/ml in KPi buffer (100 mm KCl and 10 mm potassium phosphate, pH 7.4) containing 4 mg/ml rhodamine-dextran (10 KD; Sigma) to mark the injected cells. For microinjections, neurons were maintained in AM50 for 5–6 d and then transferred to Leibovitz’s L-15 medium (Life Technologies) immediately before injection. Approximately 100–125 neurons per dish were injected directly into the nucleus. After microinjection, neurons were returned to AM50 for 12–15 hr to allow for the expression of the cDNA. Then the number of injected (rhodamine-positive) neurons was determined before NGF deprivation was initiated. Approximately 48 hr later the cells were stained with the DNA-binding dye Hoechst 33342 in L-15 medium and evaluated for survival. Neuronal viability was assessed by counting the number of rhodamine-positive cells that were phase-bright with smooth and intact neurites, a discernible nucleus, and diffuse and homogenous chromatin. A small percentage of injected neurons did not regain membrane integrity and died within a few hours of injection; these neurons did not affect subsequent analyses. The percentage of survival was equal to the number of viable cells remaining after NGF deprivation (determined as described above) divided by the number of rhodamine-positive cells counted before NGF withdrawal. For each plasmid the results reported were derived from at least three independent experiments involving a minimum of 200 injected neurons per plasmid per experiment. In all microinjection experiments a blinded observer accessed cell viability.
Immunofluorescence. Expression of p110*, p110*Δkin, Δp85, myr-Akt, A2myr-Akt, AH-Akt·Flag, and AktK179A in injected neurons was confirmed by using indirect immunofluorescence. In each case the neurons were microinjected with solutions containing 50 μg/ml expression vector DNA and 2 mg/ml lysine-fixable tetramethylrhodamine-dextran dye (Molecular Probes) in KPibuffer. Injected neurons were incubated ∼15 hr in AM50 and then fixed in fresh 4% paraformaldehyde in PBS for 15 min at room temperature. After being permeabilized for 10 min in fixative containing 0.3% Triton X-100, the neurons were incubated for 45–60 min in blocking buffer containing 1% BSA, 5% goat serum, and 0.05% Tween-20 in PBS. Then the cells were incubated with the appropriate primary antibody diluted in blocking buffer for 2 hr at room temperature. For the detection of p110 molecules, mouse monoclonal antibody 9E10 (Sigma) against the c-Myc epitope was used at a 1:500 dilution. Myr-Akt and A2myr-Akt were detected with the T7·Tag mouse monoclonal antibody (Novagen, Madison, WI) diluted 1:100. AktK179A was detected with anti-Akt CT serum (Franke et al., 1995) diluted 1:400, whereas AH-Akt·Flag was detected with the anti-Flag M2 monoclonal antibody (Kodak IBI, New Haven, CT) at a 1:300 dilution. An anti-rat PI 3-kinase polyclonal antibody that recognizes the p85 subunit (Upstate Biotechnology, Lake Placid, NY) was used to detect Δp85. FITC-conjugated goat anti-mouse (for detecting p110*, p110*Δkin, myr-Akt, A2myr-Akt, and AH-Akt·Flag molecules) or goat anti-rabbit (for detecting AktK179A and Δp85) secondary antibodies (Jackson Laboratories, Bar Harbor, ME), diluted 1:100 in blocking buffer, were applied to the neurons for 1 hr at room temperature. After being rinsed in PBS, the neurons were mounted under glass coverslips and viewed by fluorescence microscopy. For each DNA tested, 85–90% of microinjected neurons expressed the appropriate protein. Use of the lysine-fixable dye in these experiments allowed for the identification of injected cells after fixation but resulted in significant toxicity to neurons after 2–3 d. Therefore, this dye was not used in survival experiments.
Akt kinase assays. SCG neurons were plated on collagen-coated 60 mm dishes (125,000 neurons/dish) and maintained in AM50 medium for 6–8 d before being deprived of NGF by incubation in AM0 medium. After 10 hr of NGF deprivation, the medium was replaced with either fresh AM0 (“minus NGF” samples) or AM50 (“plus NGF” samples) for an additional 15 min. For testing the effects of PI 3-kinase inhibitors on Akt activation, we pretreated neurons with LY294002 or wortmannin during the final 45 min of NGF deprivation before exposing them to AM50 medium containing the same inhibitor. Cleared cell lysates were prepared by incubating cells in NP-40 lysis buffer [containing (in mm) 20 Tris, pH 7.4, 137 NaCl, 1 EDTA, 20 NaF, 1 Na4P2O7, 1 Na3VO4, and 1 PMSF with 1% NP-40, 10% glycerol, 5 μg/ml aprotinin, and 5 μg/ml leupeptin] for 30 min at 4°C and then by centrifuging the lysates (14,000 ×g) for 5 min at 4°C. Lysates were preabsorbed for 20 min with 20 μl of a 50% slurry of protein A–Sepharose (Pharmacia Biotech), centrifuged briefly to pellet the protein A beads, and then incubated with a 1:300 dilution of anti-Akt-CT serum and 40 μl of protein A–Sepharose beads for 3 hr at 4°C with constant rotation. The immune complexes were washed three times with lysis buffer, once with ice-cold H2O, and twice with kinase buffer [containing (in mm) 20 HEPES, pH 7.4, 10 MgCl2, 10 MnCl2, 1 DTT, and 0.2 EGTA with 5 μm ATP] before being incubated for 30 min at 30°C in 30 μl of kinase buffer containing 1 μm PKA inhibitor (Sigma), 0.1 mg/ml histone H2B (Boehringer Mannheim), and 10 μC [γ-32P]ATP (DuPont NEN). Reactions were terminated by briefly pelleting the immune complexes, adding SDS-PAGE sample buffer to the supernatant, and boiling the samples for 5 min. Phosphorylation of histone H2B was analyzed by 15% SDS-PAGE, followed by autoradiography and PhosporImager analysis.
RESULTS
Characterization of the death of NGF-dependent sympathetic neurons caused by inhibitors of PI 3-kinase
Postmitotic sympathetic neurons isolated from neonatal rat SCG and maintained in vitro are homogenous in their requirement for NGF such that all neurons die within 48–72 hr after NGF withdrawal (Martin et al., 1988; Deckwerth and Johnson, 1993). Cell death can be induced in the presence of NGF by treating neurons with the selective PI 3-kinase inhibitor LY294002 (Fig. 1). LY294002-treated neurons have shrunken cell soma and fragmented neurites and frequently contain one or more compact spheres of condensed chromatin in their nuclei, in contrast to the uniformly dispersed chromatin present in the nuclei of nontreated neurons (Fig.1B,E). Many of the nuclei are labeled by the TUNEL assay, indicating the presence of DNA strand breaks (Fig. 1F). These characteristics of LY294002-treated neurons are indistinguishable from those that typify apoptosis caused by NGF deprivation (Deckwerth and Johnson, 1993;Edwards and Tolkovsky, 1994).
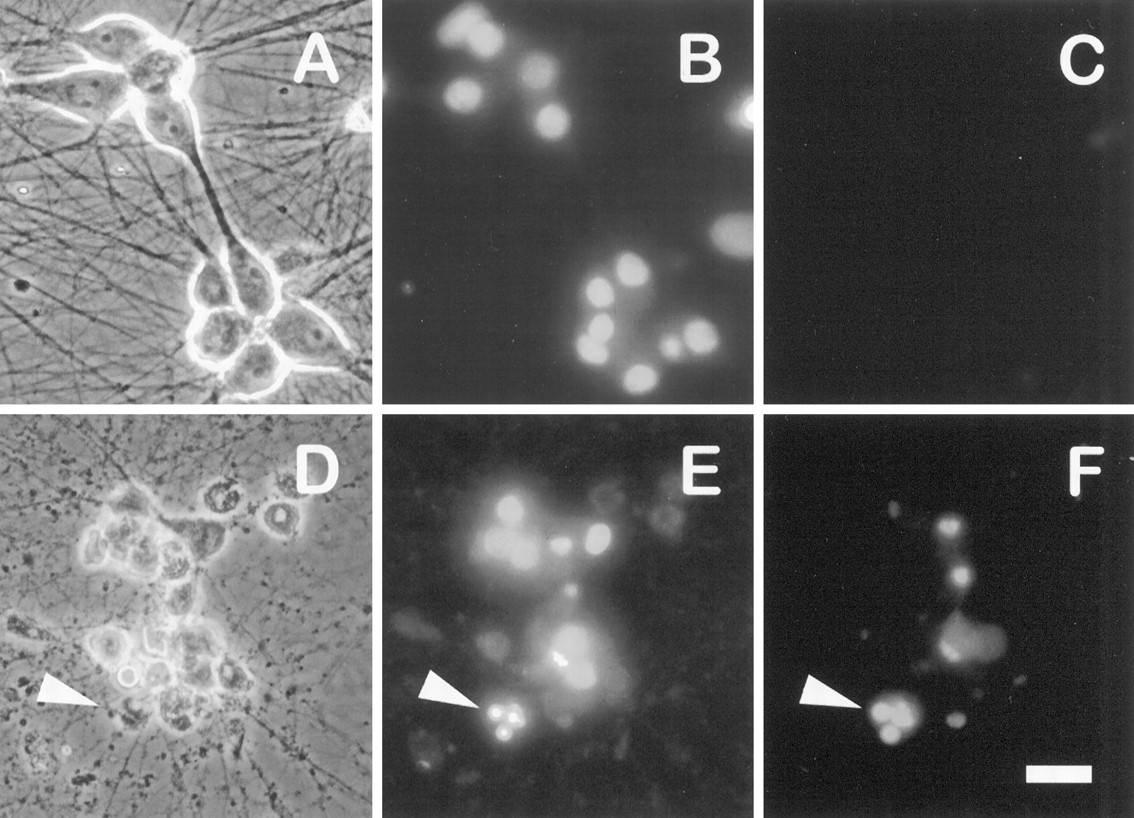
LY294002 kills sympathetic neurons in a manner that morphologically resembles NGF deprivation. The 5 d neuronal cultures were treated with or without 100 μm LY294002 in AM50 media. Approximately 72 hr later the neurons were fixed, processed for TUNEL analysis and Hoechst staining, and photographed.A–C show phase-contrast, Hoechst-stained chromatin, and TUNEL-labeled views, respectively, from the same field of NGF-maintained (nontreated) control cultures. D–F show parallel views of LY294002-treated neurons. Arrowheadspoint to a representative apoptotic neuron with a degraded soma containing a condensed, fragmented nucleus labeled by TUNEL. Scale bar, 30 μm.
Despite these morphological similarities, SCG neurons treated with LY294002 die more slowly than neurons deprived of NGF (Fig.2A). In control cultures the removal of NGF caused ∼60% death by 24 hr and >75% death by 48 hr. In contrast, 34% of neurons treated with 100 μm LY294002 in the presence of NGF died by 48 hr, with 85% dying by 96 hr. Moreover, death of LY294002-treated neurons commences only after at least 24 hr, as compared with a lag period of 15–18 hr for NGF deprivation-induced death (Deckwerth and Johnson, 1993). In contrast to these results, death of cerebellar granule neurons caused by LY294002 treatment occurs at the same rate as death caused by the withdrawal of survival factors (Miller et al., 1997). Thus, the increased rate of death caused by NGF withdrawal relative to LY294002 treatment may indicate that factors other than the inactivation of PI 3-kinase are rate-determining for the death of sympathetic neurons after NGF removal.

Death of sympathetic neurons by LY294002 is time- and dose-dependent. A, The 5 d neuronal cultures were treated with 100 μm LY294002 in AM50 or were deprived of NGF. Survival was assayed at 24, 48, 72, and 96 hr after a single application of LY294002 or after 24 and 48 hr of NGF deprivation by counting Nissl-stained neurons. Results represent mean ± SEM from three independent experiments. B, The 5 d cultures were treated with a single dose of 10, 30, or 100 μm LY294002 in AM50. Survival was assayed after 4 d by counting Nissl-stained neurons, as described in Materials and Methods. The 100 μm LY294002 treatment data are the same as those shown in A. Results represent the mean ± SEM from three independent experiments and are presented as a percentage of the survival in control NGF-maintained cultures.
Under our experimental conditions LY294002 inhibited NGF-mediated survival at concentrations as low as 10 μm, with a 50% inhibitory concentration (IC50) of ∼30 μm (Fig. 2B). Although the IC50 of LY294002 for blocking PI 3-kinase activity in vitro is 1.4 μm (Vlahos et al., 1994), concentrations ranging from 10 to 100 μm often are necessary to inhibit PI 3-kinase in intact cells (Vlahos et al., 1994;Yao and Cooper, 1996; Miller et al., 1997). Wortmannin, another PI 3-kinase inhibitor (Yano et al., 1993), also blocked the survival-promoting effects of NGF on sympathetic neurons (data not shown); the time course was similar to that of LY294002 and death was virtually complete at a concentration (100 nm) previously shown to be necessary for efficiently blocking PI 3-kinase activity and inducing DNA fragmentation in NGF-treated PC12 cells (Yao and Cooper, 1995). The ability of two structurally distinct inhibitors of PI 3-kinase to block NGF-mediated survival strongly implicates PI 3-kinase, or a PI 3-kinase-related enzyme, as a necessary transducer of the survival signals initiated by NGF in primary neurons.
A variety of pharmacological agents can inhibit the death of NGF-deprived sympathetic neurons. These include the protein synthesis inhibitor cycloheximide and the RNA synthesis inhibitor actinomycin D (Martin et al., 1988), cell-permeable cAMP analogs (Rydel and Greene, 1988), the cyclin-dependent kinase inhibitor flavopiridol (Park et al., 1996), membrane-depolarizing concentrations of extracellular potassium (Koike et al., 1989), and the nonselective caspase inhibitor BAF (Deshmukh et al., 1996). We tested several of these agents for their ability to inhibit LY294002-induced death (Fig.3). The addition of either BAF (100 μm) or cpt-cAMP (300 μm) in large part prevented the death of LY294002-treated neurons. The addition of actinomycin D also provided protection from cell death, albeit to a lesser extent. Although the cell bodies of neurons rescued by BAF, cpt-cAMP, or actinomycin D remained phase-bright with clearly discernible nuclei and nucleoli, significant neuritic degeneration continued to occur in these cultures (data not shown), suggesting that the mechanisms that maintain neurite integrity may be distinct from those that control cell survival. In contrast to the above reagents, flavopiridol (1 μm) provided little protection against LY294002-induced death, whereas LY294002 treatment of potassium-depolarized neurons resulted in even greater cell death than exposure to LY294002 in the presence of NGF. The ability of cpt-cAMP, BAF, and actinomycin D to prevent death caused either by NGF withdrawal or LY294002 suggests that both treatments activate a similar cell death pathway.

BAF, cpt-cAMP, and actinomycin D inhibit LY294002-induced death. The 5 d cultures maintained in AM50 were treated with 100 μm LY294002 alone (LY) or in the presence of each of the following: 1 μm flavopiridol (Flavo), 0.1 μg/ml actinomycin D (ActD), 100 μm BAF (BAF), or 300 μm cpt-cAMP (cAMP). For one set of treatments the neurons maintained in depolarizing concentrations of potassium without NGF were treated with 100 μm LY294002 (High K+). Neuronal survival was assayed after 80 hr by counting Nissl-stained neurons. In control experiments each agent effectively prevented the death of NGF-deprived neurons (data not shown). Results represent the mean ± range from two independent experiments and are presented as a percentage of the survival in control NGF-maintained cultures.
Withdrawal of NGF from sympathetic neurons results in increased mRNA expression of a select subset of genes, including c-jun,c-fos, and cyclin D1 (Estus et al., 1994; Freeman et al., 1994). RT-PCR analysis of mRNAs isolated from LY294002-treated and nontreated cultures demonstrated that the expression ofc-fos, c-jun, and cyclin D1 increases during LY294002-induced death (Fig. 4). Whereas c-jun expression exhibited a relatively constant prolonged elevation, c-fos expression increased sharply between 25 and 30 hr. cyclin D1 message levels exhibited a sustained elevation (three- to fourfold), peaking after 30 hr of treatment. As expected, LY294002 treatment led to a reduction in the abundance of the ubiquitously expressed cyclophilin mRNA and in the neuronally expressed tyrosine hydroxylase and p75 neurotrophin receptor mRNAs. Unlike NGF deprivation (Freeman et al., 1994), LY294002 treatment resulted in a steady decrease in the mRNA level of the Schwann cell marker S100β, suggesting that LY294002 also may be detrimental to certain non-neuronal cells present at low levels in these cultures. The induction of c-fos, c-jun, and cyclin D1 during neuronal death induced by inhibiting PI 3-kinase or by the withdrawal of NGF provides further evidence that LY294002-induced death and NGF deprivation-induced death share a common mechanism.

Expression of cyclin D1,c-jun, and c-fos increase during LY294002-induced death. Neurons cultured in AM50 for 5 d received no treatment (0 hr) or were treated with 100 μm LY294002 in AM50 for the indicated time intervals. Relative changes in the mRNA levels of specific genes were measured by semiquantitative RT-PCR analysis, as outlined in Materials and Methods. Shown are representative results from one of three independent time courses, all of which yielded similar results. PCR cycle numbers for each of the genes were as follows: cyclophilin, 18 cycles; tyrosine hydroxylase (TOH), 18 cycles; p75 neurotrophin receptor (p75), 18 cycles; c-fos, 24 cycles; c-jun, 24 cycles; cyclin D1, 25 cycles; S100β, 28 cycles.
NGF-stimulated Akt protein kinase activity is blocked by PI 3-kinase inhibitors
The Akt protein kinase is activated by a variety of growth factors via a PI 3-kinase-dependent pathway (Burgering and Coffer, 1995; Cross et al., 1995; Franke et al., 1995; Kohn et al., 1995; Klippel et al., 1996). Akt has been implicated in transducing growth factor and extracellular matrix-dependent survival signals in fibroblast, epithelial, and lymphoid cell lines (Ahmed et al., 1997; Kauffmann-Zeh et al., 1997; Kennedy et al., 1997; Khwaja et al., 1997; Kulik et al., 1997). Recently, a role for Akt also has been defined in the survival of rat cerebellar granule neurons (Dudek et al., 1997). To assess a potential function for Akt in NGF-mediated survival of sympathetic neurons, we first tested whether endogenous Akt protein kinase activity is stimulated in neurons treated with NGF, using histone H2B as anin vitro substrate (Fig. 5). After 15 min of NGF stimulation, Akt protein kinase activity increased approximately threefold above the basal level observed in the absence of NGF. Treatment with 100 μm LY294002 (or 100 nm wortmannin; data not shown) reduced the NGF-stimulated kinase activity to the level observed in NGF-deprived neurons. In contrast, treatment of neurons with 10 μm LY294002 caused only a partial (50–60%) reduction in NGF-stimulated Akt kinase activity (data not shown). Thus, NGF treatment leads to the activation of endogenous Akt protein kinase activity in sympathetic neurons, which can be blocked by inhibitors of PI 3-kinase at concentrations similar to those that block survival.

NGF stimulates Akt protein kinase activity in a PI 3-kinase-dependent manner. Akt protein kinase activity was analyzed in immune complex kinase assays prepared from SCG neurons treated as follows: lane 1, neurons deprived of NGF for 10 hr;lane 2, neurons deprived of NGF for 10 hr and then stimulated with NGF for 15 min; lane 3, neurons deprived of NGF for 10 hr and then treated with NGF for 15 min in the presence of 100 μm LY294002; lane 4, mock kinase reaction containing only histone H2B, [γ-32P]ATP, and protein A beads. Reaction products were analyzed by SDS-PAGE, followed by autoradiography and PhosphorImager analysis. Akt protein kinase activity increased an average of 3.1-fold after NGF stimulation (n = 4).
Expression of activated PI 3-kinase or activated Akt prevents the death of NGF-deprived neurons
The results described above indicate that NGF stimulates a PI 3-kinase-regulated pathway that leads to Akt activation. To test whether the activation of this pathway in the absence of NGF would be sufficient to promote neuronal survival, we microinjected sympathetic neurons with plasmid DNAs expressing either a constitutively active form of PI 3-kinase (p110*) or a kinase-inactive mutant (p110*Δkin) (Hu et al., 1995; Kulik et al., 1997) (Fig.6). Indirect immunofluorescence analysis of microinjected neurons verified that p110* and p110*Δkin were overexpressed successfully in 85–90% of injected cells. After 48 hr of NGF deprivation, most neurons injected with p110* maintained a phase-bright cell soma with uniformly dispersed chromatin and intact neurites (Fig. 7A–C). In contrast, the majority of neurons injected with β-galactosidase (LacZ) or p110*Δkin and then deprived of NGF were morphologically indistinguishable from uninjected NGF-deprived cells and either contained condensed chromatin or lacked detectable chromatin (Fig.7D–F). Quantifying the cell survival (see Materials and Methods) revealed that expression of p110* resulted in the survival of 78% of injected neurons after 48 hr of NGF deprivation (Fig.8). Survival of p110*Δkin or LacZ-injected neurons was 17 and 25%, respectively, which was similar to the survival of uninjected NGF-deprived neurons. These data indicate that PI 3-kinase activity is sufficient to promote the survival of sympathetic neurons in the absence of NGF.

Constitutively active PI 3-kinase (p110*) is expressed efficiently in microinjected sympathetic neurons. Neurons were injected with p110* plasmid and lysine-fixable tetramethylrhodamine-dextran. At 15 hr after microinjection the neurons were fixed and stained with the 9E10 monoclonal antibody, which recognizes the myc epitope attached to p110*. The 9E10 antibody was detected by FITC-conjugated anti-mouse antibodies. Ashows rhodamine-labeled (injected) cells. B, Immunofluorescence analysis shows that all of the injected neurons inA overexpress p110*. Overall, p110* was detected in 90% of the injected neurons. Scale bar, 30 μm.

Overexpression of constitutively active PI 3-kinase is sufficient to promote the survival of NGF-deprived sympathetic neurons. Shown are photomicrographs of NGF-deprived neurons injected with p110* (A–C) or p110*Δkin (D–F) expression vectors. Neurons were microinjected with the appropriate expression vectors, allowed to recover in AM50 for 12–15 hr, and then deprived of NGF for 48 hr. Neurons were stained with Hoechst dye and photographed. Photographs show rhodamine (injected cells) (A, D), Hoechst-stained nuclei (B, E), and phase-contrast views (C, F) of one field of cells for each injected DNA. Cells scored as alive in these experiments retained spherical, phase-bright cell bodies, intact neurites, and uniformly dispersed chromatin (A–C). Dead cells were characterized by phase-dark somas, fragmented neurites, and nuclear remnants devoid of stained chromatin or nuclei containing highly condensed chromatin (D–F). The arrowhead inD points to the remnant of a cell devoid of a nucleus. The arrowheads in E and Findicate phase-dark cells containing condensed chromatin. Scale bar, 40 μm.

Quantitation of neuronal survival mediated by constitutively active PI 3-kinase. Neurons cultured in AM50 for 5–6 d were microinjected with expression vectors for p110*, p110*Δkin, and LacZ and then deprived of NGF for 48 hr. Survival was assayed by a blinded observer in accordance with the criteria described above. Error bars represent the mean ± SEM from four independent experiments. Survival for each of the following injections was p110*, 78.3 ± 4.3%; p110*Δkin, 17.3 ± 2.5%; and LacZ, 25.7 ± 5.9%. Survival with p110*-injected cells was significantly greater than p110*Δkin-injected or LacZ-injected cells (two-tailedp values = 0.0001 and 0.0003, respectively). Survival between p110*Δkin and LacZ was not significantly different (p > 0.35).
Akt activation involves the binding of D3 phosphoinositides, the products of PI 3-kinase, to the N-terminal pleckstrin homology (PH) domain of Akt. This is thought to localize Akt to the cell membrane where subsequent phosphorylation of regulatory serine and threonine residues occurs (Alessi et al., 1996; Kohn et al., 1996b; Franke et al., 1997; Klippel et al., 1997). Accordingly, a form of Akt lacking its PH domain but containing a membrane-targeting Src myristoylation sequence at its N terminus (myr-Akt) is constitutively active in the absence of stimuli normally required for PI 3-kinase activation (Kohn et al., 1996a,b). To examine whether activated Akt is sufficient for neuronal survival, we expressed myr-Akt or a version of myr-Akt containing an inactive myristoylation signal (A2myr-Akt) in SCG neurons and then removed NGF from the culture medium (Fig.9). Expression of myr-Akt resulted in the survival of 77% of injected neurons after 48 hr of NGF deprivation, as compared with 23% survival for control injections. A2myr-Akt provided an intermediate, but significant, level of protection (46% survival) against NGF withdrawal. Thus, overexpression of activated forms of either PI 3-kinase or Akt protein kinase effectively prevents the death of NGF-deprived neurons. These results raise the possibility that NGF promotes the survival of sympathetic neurons, at least in part, via a PI 3-kinase-regulated pathway that involves Akt.

Overexpression of activated Akt (myr-Akt) is sufficient to promote the survival of NGF-deprived sympathetic neurons. Neurons cultured in AM50 for 5–6 d were microinjected with myr-Akt, A2myr-Akt, or p110*Δkin (used as a negative control) expression vectors and then deprived of NGF for ∼48 hr. Then the injected cells were evaluated for survival. Error bars represent the mean ± SEM from three independent experiments. Survival for each of the following injections was myr-Akt, 77.4 ± 1.3%; A2myr-Akt, 46.5 ± 0.26%; and p110*Δkin, 23.2 ± 2.4%. Survival with myr-Akt-injected cells was significantly greater than for both A2myr-Akt-injected and p110*Δkin-injected cells (two-tailed p values = 2.1 × 10−5 and 3.8 × 10−5, respectively). Survival between A2myr-Akt and p110*Δkin was also significantly different (p = 0.0006).
Dominant negative forms of PI 3-kinase and Akt induce neuronal death in the presence of NGF
Although LY294002 and wortmannin induce apoptosis in NGF-maintained neurons, the specific targets of these drugs, at the concentrations used here, cannot be identified. As an independent means of addressing whether PI 3-kinase is required for NGF-mediated survival, we microinjected NGF-maintained neurons with a plasmid expressing a dominant negative form of PI 3-kinase. The PI 3-kinase enzyme activated by tyrosine kinase receptors such as TrkA consists of a p110 catalytic subunit and a p85 regulatory subunit. The dominant negative mutant (Δp85) contains a deletion within the inter-SH2 domain of p85 that abolishes its binding to p110, but not to growth factor receptors (Dhand et al., 1994; Hara et al., 1994). Thus, Δp85 competes with wild-type p85 for binding to activated receptors, but it is unable to induce p110 catalytic activity. Expression of Δp85 in neurons maintained in the presence of NGF resulted in a significant decrease in survival, as compared with control neurons microinjected with a LacZ expression vector (Fig.10A). These data, together with the results obtained by using PI 3-kinase inhibitors, indicate that PI 3-kinase is required for the survival of sympathetic neurons by NGF.

Expression of dominant negative PI 3-kinase (Δp85), kinase-inactive Akt (AktK179A), or a truncated dominant negative Akt (AH-Akt) inhibits NGF-mediated neuronal survival. Neurons cultured in AM50 for 5–6 d were microinjected with LacZ or Δp85 (A) or LacZ, AH-Akt·Flag, or AktK179A expression vectors (B) and then maintained in AM50 media for 3 d. Then the injected cells were evaluated for survival.A, Survival was 60.2 ± 5.2% and 27.9 ± 2.5% for LacZ and Δp85, respectively (two-tailed pvalue = 0.001). Error bars represent the mean ± SEM from four independent experiments. B, Survival for the following injections were LacZ, 81.6 ± 6.3%; AH-Akt, 28.4 ± 6.7%; and AktK179A, 24.1 ± 4.9%. Error bars represent the mean ± SEM from four experiments for LacZ and AH-Akt and three experiments for AktK179A. Survival of AH-Akt-injected or AktK179A-injected neurons was significantly less than the survival of LacZ-injected cells (two-tailed p value = 0.001). Survival between AH-Akt and AktK179A was not significantly different (p = 0.65).
Expression of either a truncated form of Akt consisting of residues 1–147 (AH-Akt) or a kinase-inactive Akt mutant (AktK179A) induces cell death in insulin-treated cerebellar granule cells and adherent epithelial cells (Dudek et al., 1997; Khwaja et al., 1997). In these studies the dominant negative nature of the mutants was demonstrated by a reduction in growth factor-stimulated Akt kinase activity that occurred in cells cotransfected with mutant and wild-type Akt. To test whether Akt is necessary for NGF-promoted survival, we microinjected NGF-maintained neurons with expression plasmids containing either an epitope-tagged version of AH-Akt or AktK179A (Fig.10B). Expression of either AH-Akt or AktK179A in the presence of NGF substantially increased the number of apoptotic neurons, as compared with control neurons expressing LacZ. The extent of cell death in neurons expressing AH-Akt or AktK179A in the presence of NGF was slightly greater than that observed in neurons treated with LY294002 for 3 d (see Fig. 2). In control experiments the expression of AktK179A did not affect cell death caused by NGF deprivation. These results show that Akt, in addition to PI 3-kinase, is required for NGF-mediated neuronal survival.
DISCUSSION
Recent studies have implicated PI 3-kinase as an intracellular transducer of survival signals initiated by various growth factors. We began this study by asking whether PI 3-kinase also functions in the NGF-dependent survival of primary neurons. We found that neuronal death caused by inhibition of PI 3-kinase shares several features with death caused by NGF deprivation, suggesting that PI 3-kinase may function as an essential mediator of survival in NGF-dependent neurons. Consistent with this, expression of activated PI 3-kinase rescues NGF-deprived neurons from cell death, whereas expression of dominant negative PI 3-kinase blocks survival in the presence of NGF. NGF treatment of neurons activates Akt, a putative effector of PI 3-kinase, and Akt activation is dependent on PI 3-kinase. Expression of dominant negative forms of Akt block NGF-promoted survival, whereas expression of activated Akt is sufficient for survival in the absence of NGF. Taken together, these results identify Akt as an important mediator of PI 3-kinase-dependent survival signals initiated by NGF in sympathetic neurons.
The neuronal death caused by PI 3-kinase inhibitors resembles, in part, the death caused by NGF withdrawal, suggesting that both stimuli may activate similar cell death pathways. In both cases the death exhibits features characteristic of apoptosis, including cellular atrophy, chromatin condensation, TUNEL reactivity, and neurite fragmentation. The caspase inhibitor BAF and the RNA synthesis inhibitor actinomycin D block death caused by either treatment, suggesting a shared requirement for caspase activation and certain transcriptional events. Likewise, cpt-cAMP treatment blocks death caused by PI 3-kinase inhibition or NGF withdrawal, indicating the presence of a cAMP-mediated survival pathway that is independent or downstream of PI 3-kinase. Death caused by either treatment is accompanied by increased expression ofc-jun, c-fos, and cyclin D1, with maximal gene expression persisting longer after PI 3-kinase inhibition, as compared with NGF withdrawal; this is consistent with the delayed time course of LY294002-induced death (see below).
Although LY294002 and wortmannin are selective inhibitors of PI 3-kinase, the use of these drugs does not permit positive identification of their target in sympathetic neurons. The concentrations of LY294002 and wortmannin that are required to induce apoptosis efficiently in this and other systems are also capable of inhibiting the activity of several members of the PI 3-kinase superfamily. Among these are the tyrosine kinase-activated p85/p110 PI 3-kinases, heterotrimeric G-protein-stimulated p110γ, and the mammalian target of rapamycin (mTOR) kinase (Stephens et al., 1994;Hartley et al., 1995; Brunn et al., 1996). Of these, only p85/p110 PI 3-kinase is known to be activated by NGF (Carter and Downes, 1992;Raffioni and Bradshaw, 1992). Moreover, rapamycin (0.01–100 nm) does not block the survival of NGF-maintained sympathetic neurons (R. Freeman, unpublished observation), suggesting that mTOR is not necessary for NGF-mediated survival. Finally, the expression of a dominant negative mutant of p85 blocked NGF-promoted survival. Thus, although we cannot exclude the possibility of other effects, the inhibitors used in this study most likely inhibit the activation of p85/p110 PI 3-kinase by NGF.
Expression of activated PI 3-kinase or activated Akt is sufficient to keep neurons alive in the absence of NGF. The degree of saving conferred by activated PI 3-kinase and activated Akt is similar to that obtained by overexpressing the Bcl-2 protein in sympathetic neurons (Garcia et al., 1992; Greenlund et al., 1995b) (R. Crowder, unpublished observation). The form of PI 3-kinase that we used consists of the p110 catalytic subunit of PI 3-kinase fused at its N terminus to the inter-SH2 domain of the p85 regulatory subunit. Although expression of this and similar forms of activated PI 3-kinase can suppress apoptosis under certain conditions (Kauffmann-Zeh et al., 1997; Kennedy et al., 1997; Khwaja et al., 1997; Kulik et al., 1997), the mechanism by which this occurs is poorly characterized. When the activated form of PI 3-kinase used in our experiments was transiently expressed in COS-7 cells, Akt and pp70S6 kinase were activated constitutively in the absence of growth factor stimulation (Hu et al., 1995; Klippel et al., 1996). Although it is not known whether the expression of activated PI 3-kinase in neurons leads to activation of Akt, our data indicating that Akt is sufficient for survival in the absence of NGF and necessary for NGF-dependent survival suggest that Akt activation may be a critical event.
The activated form of Akt used in our experiments lacks its phospholipid-binding PH domain but is targeted to the cell membrane via a myristoylation sequence added to its N terminus (Kohn et al., 1996b). In fibroblasts, COS-7 cells, and cerebellar granule cells the overexpression of Akt or membrane-targeted forms of Akt also inhibit cell death (Dudek et al., 1997; Kauffmann-Zeh et al., 1997; Kennedy et al., 1997; Kulik et al., 1997). In these studies full-length Akt (with its PH domain intact) was expressed rather than the PH domain-minus form used in our experiments. Assuming that the survival that occurs after expression of myr-Akt (lacking the PH domain) is not dependent on endogenous wild-type Akt, then the binding of phospholipids or other interactions mediated by the PH domain would not appear to be necessary for myr-Akt to promote survival in neurons.
The ability of activated Akt to prevent apoptosis after NGF withdrawal, taken together with the increase in Akt kinase activity that occurs after NGF stimulation, suggests that Akt may function to transduce NGF-initiated survival signals in neurons. To demonstrate more directly a role for Akt in NGF-mediated survival, we tested the effects of functionally inhibiting Akt with dominant negative mutants of Akt. Expression of either the N-terminal AH domain of Akt or a kinase-inactive Akt mutant blocked survival promotion by NGF. Because activation of Akt by NGF is prevented by LY294002 and wortmannin, these results identify Akt as a downstream target of PI 3-kinase and TrkA in the survival-promoting pathway initiated by NGF.
Although our studies demonstrate a role for Akt and PI 3-kinase in survival, they do not rule out the possibility that NGF activates additional survival pathways. The delayed onset of cell death, the slower rate of death, and the prolonged expression of c-junand cyclin D1 caused by inhibitors of PI 3-kinase (relative to NGF withdrawal) may indicate that other survival pathways not affected by LY294002 or wortmannin might be downregulated after the removal of NGF. Turning off such pathways by withdrawing NGF could lead to faster cell death by (1) more completely inactivating the PI 3-kinase pathway than is possible with LY294002 or wortmannin in this system or (2) inactivating additional or alternative survival pathways. Studies in PC12 cells suggest that certain MAP kinases, in particular the extracellular signal-regulated kinases (ERKs), may be important for survival mediated by NGF or other growth factors (Xia et al., 1995). However, recent studies using sympathetic neurons demonstrate that inhibitors of MAP kinase kinase-1, an upstream activator of ERKs, do not block NGF-mediated survival (Creedon et al., 1996; Virdee and Tolkovsky, 1996), suggesting that ERK activation may be dispensable for survival promotion by NGF. Previous studies have implicated a Ras-dependent pathway in the NGF-mediated survival of chick sensory neurons (Borasio et al., 1989, 1993) and rat sympathetic neurons (Nobes and Tolkovsky, 1995; Nobes et al., 1996). In both cases the introduction of Ras-neutralizing antibodies into dissociated neurons blocks survival in the presence of NGF, whereas the introduction of activated Ras results in NGF-independent survival. Known or putative effectors of Ras include the Raf/MAP kinase pathway, PI 3-kinase, Ral-GDS, and the Ras-related small GTPases, Rac and cdc42 (Marshall, 1996). Because both Ras and PI 3-kinase now have been implicated in survival promotion by NGF and because PI 3-kinase is a possible effector of Ras (Rodriguez-Viciana et al., 1994, 1997), these two proteins may lie within the same survival pathway.
Besides inactivating additional survival molecules, NGF withdrawal may lead to the activation of proapoptotic pathways that either are not activated or are activated incompletely after PI 3-kinase inhibition. JNK and the related kinase p38 are critical mediators of apoptosis in NGF-deprived PC12 cells (Xia et al., 1995). JNK also is activated after NGF withdrawal in sympathetic neurons, and c-Jun, a target of JNK and p38 phosphorylation, is required for neuronal death caused by NGF withdrawal (Estus et al., 1994; Ham et al., 1995; Virdee et al., 1997). Activation of JNK or p38 by NGF withdrawal, but not after inhibition of PI 3-kinase, could contribute to the increased rate of death caused by withdrawal of NGF.
In summary, our results suggest that survival of sympathetic neurons mediated by NGF is dependent on a PI 3-kinase and Akt-regulated pathway. Other relevant participants in this survival pathway are unknown but could include Ras, phosphatidylinositol-dependent kinase-1 (Alessi et al., 1997), and two downstream targets of Akt—glycogen synthase kinase-3 (Cross et al., 1995) and Bad (Datta et al., 1997; del Peso et al., 1997).
Footnotes
This work was supported in part by a Lucille P. Markey Charitable Trust award to the University of Rochester, the Paul Stark Endowment at the University of Rochester, and National Institutes of Health Grant NS34400. R.J.C. was supported by a National Institutes of Health predoctoral training grant. We thank Anke Klippel for generously providing the p110* and p110*Δkin expression plasmids, Richard Roth for the gift of pECE-myr-Akt and pECE-A2myr-Akt plasmids, Jill Johnson and Edward Sausville for flavopiridol, and Thomas Franke and David Kaplan for anti-Akt-CT serum. We also thank Paul Coffer for the pSG5-HA-PKB K179A plasmid, Masato Kasuga for plasmids containing Δp85 cDNAs, and Eugene Johnson and Patricia Osborne for generously providing neutralizing anti-NGF antiserum. We are grateful to Robert Gross for the use of his micropipette puller and Leah Larocque for the preparation of collagen and tissue culture reagents.
Correspondence should be addressed to Dr. Robert S. Freeman, Department of Pharmacology and Physiology, University of Rochester, School of Medicine, 601 Elmwood Avenue, Rochester, NY 14642.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}