

Abstract
Glutamate receptors are associated with various regulatory and cytoskeletal proteins. However, an understanding of the functional significance of these interactions is still rudimentary. Studies in hippocampal neurons suggest that such interactions may be involved in calcium-induced reduction in the open probability of NMDA receptors (inactivation). Thus we examined the role of the intracellular domains of the NR1 subunit and two of its binding partners, calmodulin and α-actinin, on this process using NR1/NR2A heteromers expressed in human embryonic kidney (HEK) 293 cells. The presence of the first 30 residues of the intracellular C terminus of NR1 (C0 domain) was required for inactivation. Mutations in the last five residues of C0 reduced inactivation and produced parallel shifts in binding of α-actinin and Ca2+/calmodulin to the respective C0-derived peptides. Although calmodulin reduced channel activity in excised patches, calmodulin inhibitors did not block inactivation in whole-cell recording, suggesting that inactivation in the intact cell is more complex than binding of calmodulin to C0. Overexpression of putative Ca2+-insensitive, but not Ca2+-sensitive, forms of α-actinin reduced inactivation, an effect that was overcome by inclusion of calmodulin in the whole-cell pipette. The C0 domain also directly affects channel gating because NR1 subunits with truncated C0 domains that lacked calmodulin or α-actinin binding sites had a low open probability. We propose that inactivation can occur after C0 dissociates from α-actinin by two distinct but converging calcium-dependent processes: competitive displacement of α-actinin by calmodulin and reduction in the affinity of α-actinin for C0 after binding of calcium to α-actinin.
Glutamate receptors at central synapses are clustered with a complex of proteins in the postsynaptic density (PSD). This localization presumably is achieved by the association of receptors with other proteins in the PSD. Recent studies have led to the hypothesis that cytoskeletal and regulatory proteins are included in this complex by binding to scaffolding proteins (Sheng and Wyszynski, 1997; O’Brien et al., 1998). In this view, scaffolding proteins not only localize and cluster receptors, but also facilitate the transduction of extracellular signals to specific intracellular signal cascades. Consistent with differential localization of NMDA, AMPA, and metabotropic glutamate receptors, each class of receptor interacts with a different putative scaffolding protein (Kornau et al., 1995; Niethammer et al., 1996; Brakeman et al., 1997; Dong et al., 1997). The subsynaptic scaffold may allow the differential anchoring of enzymes close to their site of action as occurs in the modulation of AMPA receptor by protein kinase A (Rosenmund et al., 1994). Although the evidence provides a rich potential for protein–protein interactions, the influence of these interactions on glutamate channel gating is relatively unknown.
Calcium regulation of NMDA receptors has been postulated to involve a linkage between the receptor and the cytoskeleton (Rosenmund and Westbrook, 1993b) and thus may provide a useful model for exploring interactions between membrane receptors and proteins in the subsynaptic scaffold. Inactivation results from a reduction in the open probability of the NMDA channel after elevation of intracellular calcium (Legendre et al., 1993). Although the molecular mechanisms are not well understood, macroscopic inactivation is not apparent in recombinant NMDA receptors lacking the intracellular C-terminal domains of NR1 (Krupp et al., 1998). Several proteins can bind to the intracellular C-terminal domains of NMDA receptor subunits. PSD-95, the first such protein to be identified (Kornau et al., 1995; Niethammer et al., 1996), binds via a PDZ recognition motif present at the C terminus of all four NR2 subunits and some NR1 splice variants. Calmodulin also binds in a calcium-dependent manner to two distinct sites on the C terminus of the NR1 subunit (Ehlers et al., 1996). The actin-binding protein α-actinin can bind to the C termini of NR2B and NR1 (Wyszynski et al., 1997), providing a potential link between the NMDA receptor and actin filaments. In vitro, α-actinin and calmodulin bind competitively to the initial 30 aa of the C terminus of NR1 (Wyszynski et al., 1997). This segment, called C0 (Ehlers et al., 1996), is common to all NR1 splice variants (Hollmann et al., 1993). Finally, a novel protein (yotaio) (Lin et al., 1998) and the neurofilament subunit NF-L (Ehlers et al., 1998) interact with the alternatively spliced exon cassette C1 of NR1. Except for calmodulin, no data are available on the physiological function of these interactions. Calmodulin reduces the open probability of recombinant NMDA receptors in inside-out patches (Ehlers et al., 1996), and it has been proposed that this interaction underlies inactivation (Zhang et al., 1998).
We examined the role of the intracellular C terminus of NR1 and its binding proteins on inactivation of recombinant NMDA receptors. NR1 constructs were expressed with NR2A in human embryonic kidney (HEK) 293 cells, and inactivation was monitored using whole-cell recording. Our results suggest that the C0 domain directly affects channel open probability in the absence of calmodulin or α-actinin binding. We propose that the calcium-dependence of inactivation can involve calcium binding to both calmodulin and α-actinin.
MATERIALS AND METHODS
Molecular biology. The NMDA subunit cDNAs used were NR1–1a (accession no. U08261), NR1–1b (accession no. U08263), NR1–2a (accession no. U08262), NR1–3a (accession no. U08265), NR1–4a (accession no. U08267) (Hollmann et al., 1993), and NR2A (accession no.D13211) (Ishii et al., 1993). NR1 mutants were generated using the strategy of gene splicing by overlap extension PCR (Horten et al., 1989) using Pfu DNA Polymerase (Stratagene, La Jolla, CA). All NMDA subunit cDNAs and chimeras were cloned into pCDNA1/amp (Invitrogen, San Diego, CA). cDNAs generated by PCR were sequenced. NR1 truncation mutants were generated by replacing the appropriate codon with a stop codon. The aa numbering in all cases refers to the predicted amino acids of the native NR1 sequence according to Ishii et al. (1993). Naming for multiple mutations is as follows: NR1–1aPM1(R-A, K-A, N-A mutations at aa 859, 860, 861); NR1–1aPM2(N-A, L-A, Q-A mutations at aa 861, 862, 863); NR1–1aPM3(R-A, H-A, K-A mutations at aa 839, 840, 841); NR1–1aPM4(R-A, R-A, K-A mutations at aa 844, 845, 846); NR1–1aPM5(R-E, K-E mutations at aa 859, 860); NR1–4aPM1 (R-A, K-A, N-A mutations at aa 859, 860, 861); and NR1–4aPM2 (N-A, L-A, Q-A mutations at aa 861, 862, 863).
The following α-actinin clones were used: human skeletal muscle α-actinin-2 (accession no. M8406) in the pCDNA3 mammalian expression vector (Beggs et al., 1992); chicken nonmuscle α-actinin (accession no. M74143) in the pECE mammalian expression vector (Waites et al., 1992), and chicken smooth muscle α-actinin pKCRC17 (accession no.J03486) in the pKCR3 mammalian expression vector (Baron et al., 1987;Jackson et al., 1989). The truncated α-actinin clone, encoding only the spectrin repeat region of α-actinin, was derived by PCR (Pfu DNA polymerase; Stratagene), using the chicken nonmuscle α-actinin DNA as a template, and then cloned in pCDNA3 (Invitrogen). The resulting cDNA in this clone encodes aa 336–739 of the chicken nonmuscle α-actinin (α-actininm336e-739r), starting with the N-terminal aa sequence (one-letter code) MEINF and ending in the aa sequence QILTRstop. To control for α-actinin expression, the green fluorescent protein (GFP) sequence was tagged to the N terminus of full-length chicken smooth muscle and nonmuscle α-actinins. The resulting cDNAs, identical at the 5′ and 3′ ends and lacking untranslated regions, were generated by PCR amplification and ligated in frame in the EGFP-C1 expression vector. To confirm that the GFP sequence had no direct effect on α-actinin function, a chicken nonmuscle α-actinin cDNA, generated by PCR and cloned in the expression vector EGFP-N1, was engineered with a stop codon after the α-actinin coding region, thus rendering the GFP inactive. The clone for the lymphocyte CD4 receptor was in a JPA vector.
Transfection and handling of HEK293 cells. HEK293 cells were split twice weekly and then plated 3–6 hr before transfection in DMEM plus 10% fetal calf serum (Hyclone, Logan, UT), 1% glutamine (Life Technologies, Gaithersburg, MD), and 1% penicillin–streptomycin (Life Technologies; 37°C, 5% CO2). Cells were plated on polylysine-coated glass coverslips placed in 35 mm dishes. The cDNAs for NR1/NR2A/CD4 were mixed in a 4:4:1 ratio (1 μg cDNA per 35 mm dish for each NMDAR subunit) and added to HEK293 cells as a calcium-phosphate complex (Calcium Phosphate Transfection System, Life Technologies). cDNAs for α-actinin were added to this mixture at the concentration indicated. Kynurenic acid (3 mm; Sigma, St. Louis, MO) and d,l-AP5 (1 mm; Tocris) were routinely added to prevent NMDA receptor-mediated excitotoxic cell death (Cik et al., 1993). The transfection mixture was removed after 10–18 hr by exchanging with fresh culture medium containing 3 mm kynurenic acid and 1 mmd,l-AP5. FUDR (0.2 mg/ml 5′-fluoro-2-deoxyuridine and 0.5 mg/ml uridine, Sigma) was added to inhibit cell proliferation.
Before recording, transfected cells were identified by CD4 receptor antibody-coated beads. For bead coating, Dynabeads M-450 CD4 (1 μl, Dynal, Oslo, Norway) were added to each 35 mm dish and gently swirled for 15–20 min before recording.
Recording, solutions, and drug application. Whole-cell voltage-clamp recordings were performed 12–48 hr after the end of the transfection. The recording chamber was continuously superfused at room temperature (∼20°C) with an extracellular solution of the following composition (in mm): NaCl 162, KCl 2.4, HEPES 10, glucose 10, CaCl2 1, pH 7.25 (NaOH), 325 mOsm. HPLC grade water was used for all solutions to avoid contaminating amounts of glycine or other amino acids. Patch pipettes were pulled from thin-walled borosilicate glass (TW150F-6; World Precision Instruments, Sarasota, FL) and had resistances between 2 and 5 MΩ. The intracellular solution included an ATP-regenerating system (MacDonald et al., 1989;Rosenmund and Westbrook, 1993a) of the following composition (in mm): CsCH4SO3 115.5, HEPES 10, MgCl2 6, Na2ATP 4, phosphocreatine 20, creatine phosphokinase 500 U/ml, leupeptin 0.1, EGTA 0.1, pH 7.2 (CsOH), 320 mOsm (sucrose). In some experiments EGTA was replaced by 10 mm BAPTA plus 1 mm CaCl2 as indicated. Patch solutions were prepared daily from frozen stocks and kept on ice until use. Drugs and peptides were added to the patch solution before the experiment from frozen stock or as powder. Aliquots of frozen drugs were discarded after single use. If the drug vehicle was DMSO, stock solutions were diluted at least 1:1000. For inside-out experiments, electrodes were filled with Ca-free extracellular solution plus 5 mm EGTA, 5 mm EDTA, 100 μmglycine, and 100 μm NMDA, pH 7.2 (NaOH). After patch excision, the solution bathing the cytoplasmic side of the membrane was Ca-free extracellular solution plus 10 mm EGTA, pH 7.2 (NaOH). Peptides were either applied in this solution or with 100 μm CaCl2 and 100 nm calmodulin in extracellular solution without EGTA.
Data were acquired using pClamp6 software in combination with an Axopatch-1B amplifier (Axon Instruments, Foster City, CA). The membrane voltage was clamped at −50 mV unless indicated otherwise. Whole-cell currents were filtered at 5 kHz, low-pass-filtered at 0.2 kHz, and digitized at 1 kHz. Series resistance was routinely compensated (60–90%). Cell input resistances (range: 400–3000 MΩ) were continuously monitored by a short −10 mV voltage step just before each agonist application. Currents from inside-out experiments were low-pass-filtered at 1–2 kHz and digitized at 2–5 kHz. NMDA (Tocris) was applied by a fast microperfusion system described previously (Rosenmund and Westbrook, 1993a). Glycine (50–100 μm) was added to the control and drug solutions to prevent glycine-dependent desensitization (Mayer et al., 1989). Unless noted otherwise, agonist was applied for 5 sec at 30 or 60 sec intervals. The extent of inactivation was measured as the percentage reduction in current amplitude at the end of the 5 sec application as compared with the peak amplitude at the beginning of the application.
Reagents. The following drugs were used (source; stock solvent): calmidazolium (Calbiochem, La Jolla, CA; DMSO), calmodulin binding domain peptide (CaMBD peptide, aa 290–309 of CaM kinase II; Calbiochem; water), KN-93 (Calbiochem; DMSO), calmodulin [Calbiochem and Sigma (St. Louis, MO); water], MK-801 (Tocris; water), and phalloidin (Molecular Probes, Eugene, OR; water). The following peptides with sequences of natural or mutated NR1 C-terminal regions were used: DRKSGRAEPDPKKKATFRAITSTLASSFKRRRSSKDT (C1 peptide); EIAYKRHKDARRKQMQLAFAAVNVWRKNLQ (C0 peptide); EIAYKRHKDARRKQMQLAFAAV (C0 peptide1–22); EIAYKRHKDARRKQMQLAFAAVNVW (C0 peptide1–25); EIAYKRHKDARRKQMQLAFAAVNVWRKN (C0 peptide1–28); and EIAYKRHKDARRKQMQLAFAAVNVWAAALQ (C0 peptider26a,k27a,n28a). Peptides were custom-designed and synthesized by Macromolecular Resources (Colorado State University, Ft. Collins, CO). All peptides were biotinylated at the N terminus and purified by HPLC to >90%.
Binding of C0-derived peptides. Binding of calmodulin or α-actinin to surface-bound peptides derived from the C0 domain was performed on the IAsys optical biosensor (Affinity Sensors, Cambridge, UK) using biotin-coated cuvettes. This method uses the change in resonant angle of the incident light as an indication of specific peptide–protein interactions. Neutravidin was added to link the biotinylated, C0-derived peptides to the surface of the cuvette. The affinity of the bound peptides was tested by adding free calmodulin or α-actinin whereas the concentration of the peptides was kept constant. The cuvettes were first equilibrated with PBST (Dulbecco’s PBS and 0.05% Tween 20) for 10–20 min. Enough neutravidin (∼500 ng) was then added to yield a response of 800 arc-sec. After excess neutravidin was washed off with 5 m NaCl, nonspecific binding (in the absence of peptide) was measured using varying concentrations of free protein (see below). Peptide was then added at a concentration sufficient to obtain a response of 100 arc-sec. Excess peptide was washed out with 5 m NaCl before final measurements were taken.
For all experiments, a baseline was established by incubating the reaction buffer in the cuvette for 2–5 min. Then a small volume of free protein (diluted in PBST from stock solution) was added to the buffer to obtain the desired concentration. Low concentrations of protein were allowed to equilibrate with the peptides (2 min for calmodulin and the CaMBD peptide and 10 min for α-actinin). Reactions were stopped by washing with calcium-free PBST (with 5 mmEGTA) for calmodulin and 5 m NaCl for the CaMBD peptide and α-actinin, followed by excess reaction buffer. At the end of an experiment, the peptides and neutravidin were washed off the cuvette with 12 m KOH, followed by excess deionized water.
In some experiments, a buffer containing calcium-free PBST, 1 μm calmidazolium, or 100 nm CaMBD peptide was pre-equilibrated for a few minutes before saturating concentrations of calmodulin were added. Rabbit skeletal muscle α-actinin was obtained from Cytoskeleton (Denver, CO) as a stock of 5 mg/ml in 20 mm Tris, pH 8.0, 20 mm NaCl, 5 mmβ-mercaptoethanol, and 5% (v/v) glycerol. In preliminary experiments, we found that even the most dilute solutions in PBST resulted in a large initial peak attributable to the refractive properties of the buffer. These buffer “spikes” were subtracted for each dilution of the α-actinin stock. In some experiments, a buffer containing calcium-free PBST, 100 nm CaMBD peptide, or 10–100 nm calmodulin was pre-equilibrated before α-actinin was added.
Measurements were repeated in duplicate or triplicate for each experiment with two experiments performed per peptide–protein pair. Therefore the data are based on four to six total trials per interaction. We used equilibrium binding analysis to compare the affinities of the interactions. The amplitudes of the reactions at equilibrium were measured relative to the starting baselines and normalized to the response of a maximal concentration of free protein. Relative binding at each concentration was equal to A− Ans/Amax −Amaxns, where A is the amplitude of an interaction at a chosen concentration,Ans is the nonspecific binding amplitude,Amax is the amplitude of the maximum concentration, and Amaxns is the nonspecific binding amplitude at the maximum concentration. The normalized values for each concentration were averaged and fitted with the logistic equation Amp = 1/(1 + (Kd/[Protein])n), where Amp is the average relative amplitude of the resonant angle response (normalized to the values at 1 μmcalmodulin or 500 nm α-actinin),Kd is the equilibrium dissociation constant, [Protein] is the concentration of calmodulin or α-actinin, andn is the slope factor.
Data analysis and statistics. Data are expressed as mean ± SEM. For statistical comparisons, Student’s ttest or ANOVA with subsequent Bonferroni test for multiple comparisons was used as appropriate. Statistical significance was set atp < 0.05.
RESULTS
Residues in C0 control calcium-dependent inactivation
The long intracellular C terminus of the NR1 subunit can be divided into three parts (Fig.1A): a 30 residue segment (C0) immediately distal to the end of M4, an alternatively spliced exon cassette C1, and the C-terminal segment C2. Two distinct splice acceptor sites in the gene provide two possible C termini: C2 and C2′. Previous studies indicate that deletion of the entire C terminus of the NR1 subunit prevents inactivation (Krupp et al., 1998). Likewise different binding partners have been identified for each of the three segments (Kornau et al., 1995; Ehlers et al., 1996; Wyszynski et al., 1997; Ehlers et al., 1998; Lin et al., 1998). Thus we first examined whether inactivation differed between NR1 splice variants with four different C termini (Hollmann et al., 1993) expressed as heteromers with the NR2A subunit. Inactivation was measured at the end of a 5 sec application of NMDA (10 μm) in 2 mm extracellular calcium. As shown in Figure1A, inactivation in the most common splice variant NR1–1a (47.6 ± 2.6%; n = 28) did not differ from NR1–4a (51.3 ± 3.6%; n = 11), the splice variant lacking the C1 cassette and containing C2′. NR1–2a (without C1 but with C2) and NR1–3a (with C1 and C2′) also showed similar inactivation (Fig. 1C). Likewise, inactivation was observed in heteromers containing the NR1 splice variant with the N-terminal insert (NR1–1b/2A: 46.4 ± 4.5%; n = 3). Thus the presence of the splice inserts N1 or C1 is not essential for inactivation, and neither is the difference between C2 and C2′. Because C0 is common to all splice variants, we tested an NR1 mutant truncated immediately after C0 (NR1stop863). NR1stop863/2A receptors showed full inactivation (45.3 ± 4.9%; n = 14), whereas inactivation was absent when NR1 was truncated 5 aa after M4 (NR1stop838; 0.3 ± 1.5%; n = 14) (Fig. 1B,C), indicating that inactivation requires the presence of the C0 segment. The absence of macroscopic inactivation indicates that the receptor is either permanently inactivated or has lost the ability to inactivate.

The C0 domain of the NR1 subunit is essential for inactivation. A, NR1 splice variants were coexpressed with NR2A in HEK293 cells. NMDA (10 μm) in the presence of 50–100 μm glycine was applied at a holding potential of −50 mV (2 mm[Ca2+]o). This protocol reveals inactivation of NMDA channels but does not induce macroscopic glycine-independent desensitization (Legendre et al., 1993; Rosenmund and Westbrook, 1993b). Full inactivation of ∼50% was observed with NR1–1a and NR1–4a. The structure of the NR1 splice variants is depicted above the current traces. The four hydrophobic domains are indicated by gray boxes. B, NR1/2A heteromers expressing an NR1 mutant truncated after the C0 domain (NR1stop863) also showed full inactivation. In contrast, inactivation was absent in heteromers with an NR1 truncation of almost the entire C terminus (NR1stop838).C, Pooled data for the degree of inactivation observed with different NR1/2A heteromers. Asterisk indicates significant difference compared with NR1–1a.
To define the critical residues within C0, we constructed a series of C0 truncations and point mutants. Inactivation was reduced when the last 2 aa of C0 were deleted (NR1stop861; 26.0 ± 4.8%; n = 7) and absent after deletion of the last five residues of C0 (NR1stop858; 1.0 ± 1.3%;n = 6) (Fig.2A,B). To further test the role of residues 859–863, we constructed point mutations in the full-length NR1–1a subunit (Fig. 2C,D). Inactivation was reduced by triple alanine mutations in this region (NR1–1aPM1; 25.5 ± 4.1%; n = 5; and NR1–1aPM2; 30.9 ± 4.4%;n = 5). The C1 domain does not affect inactivation, because the same triple mutants in NR1–4a reduced inactivation to the same degree (NR1–4aPM1; 29.3 ± 7.4%;n = 5; and NR1–4aPM2; 29.7 ± 6.0%; n = 5). A double charge inversion of aa 859 and 860 also reduced inactivation (NR1–1aPM5; 29.1 ± 3.4%; n = 5). Triple A mutations in the more N-terminal part of C0 had no effect on inactivation, although they produced a similar reduction in the net charge of the C0 domain (NR1–1aPM3; 41.5 ± 7.2%; n = 5; and NR1–1aPM4; 42.8 ± 4.1%;n = 5). These results indicate that residues 859–863 are essential for full expression of inactivation.

Residues 859–863 within the C0 domain of NR1 are critical for inactivation. A, B, In contrast to the full inactivation (∼50%) observed with NR1 constructs containing the entire C0 domain (NR1stop863), inactivation was completely absent if the last five residues in C0 were deleted (NR1stop858). Deletion of aa 862 and 863 produced a partial reduction of inactivation (NR1stop861). The NR1 subunit structure with the C0 amino acid sequence are indicated at thetop. Arrows point to the last amino acid of each truncation mutant. Asterisks in Bindicate significant differences compared with NR1–1a. C, D, Point mutants within the C0 domain confirmed that residues 859–863 are critical for inactivation. Triple alanine (NR1–1aPM1, NR1–1aPM2) or double glutamate (NR1–1aPM5) mutations in residues 859–863 reduced inactivation, whereas alanine mutations in C0 closer to the fourth membrane region (NR1–1aPM3, NR1–1aPM4) did not affect inactivation. The C1 domain did not affect inactivation because triple alanine mutations in NR1–4a (NR1–1aPM1, NR1–1aPM2) did not differ from the same mutations in NR1–1a.Asterisks in D indicate significant differences compared with NR1–1a for NR1–1aPM1–5 and NR1–4a for NR1–4aPM1+2.
Does calmodulin binding to C0 underlie inactivation?
The above results indicate a critical role for C0 in inactivation. Both calmodulin (Ehlers et al., 1996) and α-actinin (Wyszynski et al., 1997) have been shown to bind to C0 and thus are candidates for mediators of inactivation. We first tested the role of calmodulin. To avoid calmodulin binding to the C1 domain (Ehlers et al., 1996), we used the NR1–4a subunit. Previous studies have reported that calmodulin reduces open probability of recombinant NMDA receptors in inside-out patches (Ehlers et al., 1996), but calmodulin inhibitors had little effect on inactivation of native or recombinant NMDA receptors in whole-cell experiments (Legendre et al., 1993; Rosenmund and Westbrook, 1993a; Krupp et al., 1996). As reported, we found that calmodulin (in the presence of calcium) rapidly and reversibly reduced the NMDA channel activity in inside-out patches from HEK293 cells expressing NR1–4a/2A heteromers (Fig.3A); however, the effect of calmodulin rapidly washed out. The inhibition was >50% after the first application, but the inhibition was markedly reduced during repeated applications (Fig. 3B,C). This suggests that cytosolic factors that are lost in excised patches may be crucial for the full and continued expression of inactivation.

Calmodulin rapidly and reversibly reduces the open probability of NMDA channels in “early” inside-out patches.A, The first application of 100 μm Ca plus 100 nm calmodulin to an inside-out patch from an HEK293 cell expressing NR1–4a/2A heteromers inhibited single-channel activity rapidly and reversibly (top). However, the inhibition was markedly reduced by the fifth application, made 4.5 min after excision of the patch. The overall channel activity also decreased with time of recording (rundown). Patches were exposed to a calcium-free solution containing 10 mm EGTA between calmodulin applications. B, Pooled data for the first five applications of calmodulin to a different inside-out patch than inA. Data are represented as charge per 500 msec and were normalized to the average charge in the 5 sec preceding application of calmodulin. C, Pooled data for applications 6–10 of calmodulin to the same patch as in B. The effect of calmodulin is no longer apparent. Similar results were obtained for four other patches.
If calmodulin binding to C0 is all that is required for inactivation, then calmodulin inhibitors should block inactivation in whole-cell experiments. To assure stable responses, applications of NMDA were made at 5 min intervals. Phalloidin (1 μm) was included in the whole-cell pipette to minimize loss of NMDA current due to actin depolymerization (Rosenmund and Westbrook, 1993b). Under these conditions, the current amplitude and percentage inactivation in NR1–4a/2A heteromers remained stable for 20 min (0 min: −195 ± 51 pA, 43.5 ± 4.5%; 20 min: −167 ± 44 pA, 49.7 ± 2.1%; n = 6; Fig.4A). Inclusion of calmidazolium (100 μm) in the recording electrode did not affect the current amplitude or percentage inactivation (Fig.4E). This concentration of calmidazolium was 100-fold above that necessary to inhibit the interaction of calmodulin (1 μm) with a C0 peptide (see below). Addition of the CaMBD peptide (10 μm) to the pipette also did not produce a reduction in peak current or inactivation. At a higher concentration (20 μm) the peptide produced an apparent reduction in inactivation, but this was caused by a decrease in peak current amplitude (70 ± 4% of initial peak at t = 20 min; n = 7) (Fig. 4B,E) rather than an increase in the steady-state current, as would be expected if the peptide specifically inhibited inactivation.

Calmodulin inhibitors do not block inactivation.A, Under control conditions, both the peak amplitude and percentage inactivation of NR1–4a/2A heteromers remained constant during the first 20 min of whole-cell recording. B, Intracellular perfusion of 20 μm CaMBD peptide reduced the peak current amplitude, but had no effect on the steady-state current at the end of the agonist application. C, The effect of CaMBD peptide did not occur in calcium-free medium with 10 mm BAPTA in the whole-cell pipette. D, The CaM kinase II inhibitor KN-93 (5 μm in intracellular solution) had no effect on the response, suggesting that the CaMBD peptide did not act by inhibiting CaM kinase II. E, The ratio of the peak currents (t = 20 min/t = 0 min) are plotted at leftfor the calmodulin inhibitors and KN-93. The percentage inactivation att = 0 min (black bars) andt = 20 min (white bars) are plotted at right. Asterisks indicate significant differences to respective control.
The reduction of the peak current amplitude by the higher concentration of CaMBD peptide was dependent on calcium influx, because no reduction was observed when NMDA was applied in calcium-free medium using pipettes containing 10 mm BAPTA (103.9 ± 18.0% of initial peak at t = 20; n = 5) (Fig.4C) or at a holding potential of +50 mV (93.1 ± 12.2% of initial peak at t = 20; n = 7). The effect of CaMBD peptide was not caused by inhibition of CaM kinase II, because KN-93 (5 μm) had no effect on peak current amplitude or inactivation (Fig. 4D,E). Furthermore, CaMBD peptide (20 μm) did not reduce the current amplitude of responses from NR1stop838/2A heteromers (107 ± 13% of initial amplitude at t = 20;n = 11), suggesting that the effect of the peptide on current amplitude involves the C terminus. These experiments are inconsistent with the idea that calmodulin binding to C0 is solely responsible for inactivation.
Overexpression of α-actinin can interfere with inactivation
One possible explanation for the lack of effect of calmodulin inhibitors is that another protein is bound to C0 in the intact cell. α-Actinin is an attractive possibility in this regard because it competes with calmodulin for binding to C0 (Wyszynski et al., 1997), and it could provide the proposed link of the NMDA receptor to the actin cytoskeleton (Rosenmund and Westbrook, 1993b). α-Actinin has three functional domains: an N-terminal actin-binding domain, a central rod domain of four spectrin-like repeats, and a C-terminal domain with two EF-hand-like motifs (Bennett, 1990; Hartwig, 1995) (Fig.5A). At least four genes combined with alternative splicing give rise to several α-actinins that diverge primarily in the calcium-binding motif of the EF-hands. The EF-hand motifs can influence binding to actin as α-actinin assembles as an antiparallel homodimer. In nonmuscle isoforms, calcium disrupts the association between α-actinin and actin, whereas this interaction is calcium-insensitive in muscle isoforms. We studied the role of α-actinin in inactivation by co-transfecting NR1–4a/2A heteromers with several α-actinin cDNAs: two putative calcium-insensitive isoforms, human skeletal muscle α-actinin-2 (Beggs et al., 1992), and chicken smooth muscle α-actinin (Baron et al., 1987; Jackson et al., 1989), as well as a putative calcium-sensitive isoform, chicken nonmuscle α-actinin (Waites et al., 1992). The chicken nonmuscle and smooth muscle cDNAs differ only in the first EF-hand and have 90% amino acid similarity to α-actinin-2, the human skeletal muscle isoform that binds to C0 (Beggs et al., 1992). Because α-actinin-2 binds to C0 via its central rod domain, we also tested a truncated chicken nonmuscle α-actininm336e-739r containing only the spectrin repeats.

Overexpression of putative calcium-insensitive α-actinin reduces inactivation. A, Subunits of chicken nonmuscle α-actinin are composed of an N-terminal actin-binding domain, a central rod domain of four spectrin repeats, and a C-terminal domain that contains two EF-hand motifs. B, Cotransfection of chicken nonmuscle α-actinin did not affect inactivation over a 100-fold range of cDNA amount transfected. In contrast, inactivation was reduced with 1 and 2.5 μg of DNA transfected with the chicken smooth muscle α-actinin. These two isoforms are splice variants of one gene, differing only in the predicted functionality of the first EF-hand motif. Cotransfection of a truncated α-actinin, encoding only the central rod domain, produced a dose-dependent block of inactivation. All recordings shown are from cells transfected with 2.5 μg of DNA of the respective α-actinin/35 mm dish. Asterisks indicate significant differences to cells transfected with NR1–4a/2A. C, D, The lack of effect of chicken nonmuscle α-actinin was not caused by low expression levels. C shows an HEK293 cell cotransfected with 2.5 μg DNA/35 mm dish of an N-terminal GFP-tagged chicken nonmuscle α-actinin. The bright fluorescence indicates high expression of the protein. Responses recorded from such cells showed full inactivation. Inset shows the fluorescence intensity (on a scale of 1 to 254) of randomly selected cells from a dish transfected with an N-terminal GFP-tagged chicken nonmuscle α-actinin (average pixel value: 128.0 ± 7.6;n = 106). D shows an HEK293 cell cotransfected with 2.5 μg DNA/35 mm dish of an N-terminal GFP-tagged chicken smooth muscle α-actinin. The distribution and average fluorescence intensity of cells transfected with this clone were similar to cells transfected with the GFP-tagged nonmuscle isoform (seeinset) (average pixel value: 104.3 ± 7.9;n = 102).
Compared with NR1–4a/2A, inactivation was unaffected by cotransfection with chicken nonmuscle α-actinin over a 100-fold range of DNA concentrations (Fig. 5B, left). In contrast, inactivation was significantly reduced in HEK293 cells cotransfected with the chicken smooth muscle α-actinin at DNA concentrations of 1 or 2.5 μg/35 mm dish (Fig. 5B, middle). Similar results were obtained with human skeletal muscle α-actinin-2 (data not shown); inactivation was reduced to 28.6 ± 6.6% (1 μg DNA/35 mm dish, n = 4) and to 19.1 ± 3.6% (2.5 μg DNA/35 mm dish, n = 5). A more prominent effect was seen with coexpression of the spectrin repeats (α-actininm336e-739r), which caused a dose-dependent reduction in inactivation such that inactivation was nearly absent with 2.5 μg DNA/35 mm dish (9.7 ± 6.5%; n = 8) (Fig. 5B, right).
To control for expression levels between different α-actinin cDNAs, we tagged the N terminus of the chicken nonmuscle and smooth muscle isoforms with GFP. The results with these GFP-tagged α-actinins were identical to the untagged isoforms. Inactivation with GFP-tagged chicken nonmuscle α-actinin was unaffected (48.1 ± 6.4%, 2.5 μg DNA/35 mm dish; n = 3) but was significantly reduced with the GFP-tagged chicken smooth muscle isoform (27.6 ± 7.6%, 2.5 μg DNA/35 mm dish; n = 5). The fluorescence intensity of transfected cells was comparable for both isoforms (Fig. 5C,D), indicating that the ineffectiveness of the chicken nonmuscle isoform was not caused by lack of expression. Furthermore, a chicken nonmuscle α-actinin construct in which GFP was silenced by a stop codon after the α-actinin sequence also had no effect, confirming that the presence of GFP did not alter chicken nonmuscle α-actinin. These results demonstrate that α-actinin binding to C0 can interfere with inactivation. The putative calcium-sensitive isoform, chicken nonmuscle α-actinin, had no effect, suggesting that this interaction is calcium-dependent.
Competition between α-actinin and calmodulin
In vitro, calmodulin and α-actinin bind competitively to C0 (Wyszynski et al., 1997). Thus one simple hypothesis is that inactivation occurs when calmodulin is bound to C0, whereas it is prevented when α-actinin is bound. To test this idea, we examined whether calmodulin (20 μm) in the whole-cell pipette could restore inactivation in cells coexpressing chicken smooth muscle α-actinin or α-actininm336e-739r (2.5 μg DNA/35 mm dish). To prevent activation of CaM kinase II or direct effects of calmodulin on the cytoskeleton, 25 μm KN-93 and 1 μm phalloidin were added to the pipette. Exogenous calmodulin had no effect on inactivation in control NR1–4a/2A heteromers (40.9 ± 3.7% at t = 0 min and 40.3 ± 6.5% at t = 10 min;n = 7) (Fig.6C). However, calmodulin rapidly and fully restored inactivation in cells transfected with smooth muscle α-actinin (43.4 ± 8.8% at t= 0 min and 42.9 ± 4.4% at t = 10 min;n = 6) (Fig. 6A,C). Likewise, calmodulin restored inactivation in cells expressing α-actininm336e-739r (45.0 ± 5.2% att = 0 min and 37.0 ± 4.3% att = 10 min; n = 6) (Fig.6B,C). These results suggest that competitive binding of calmodulin and α-actinin to C0 can be involved in inactivation.
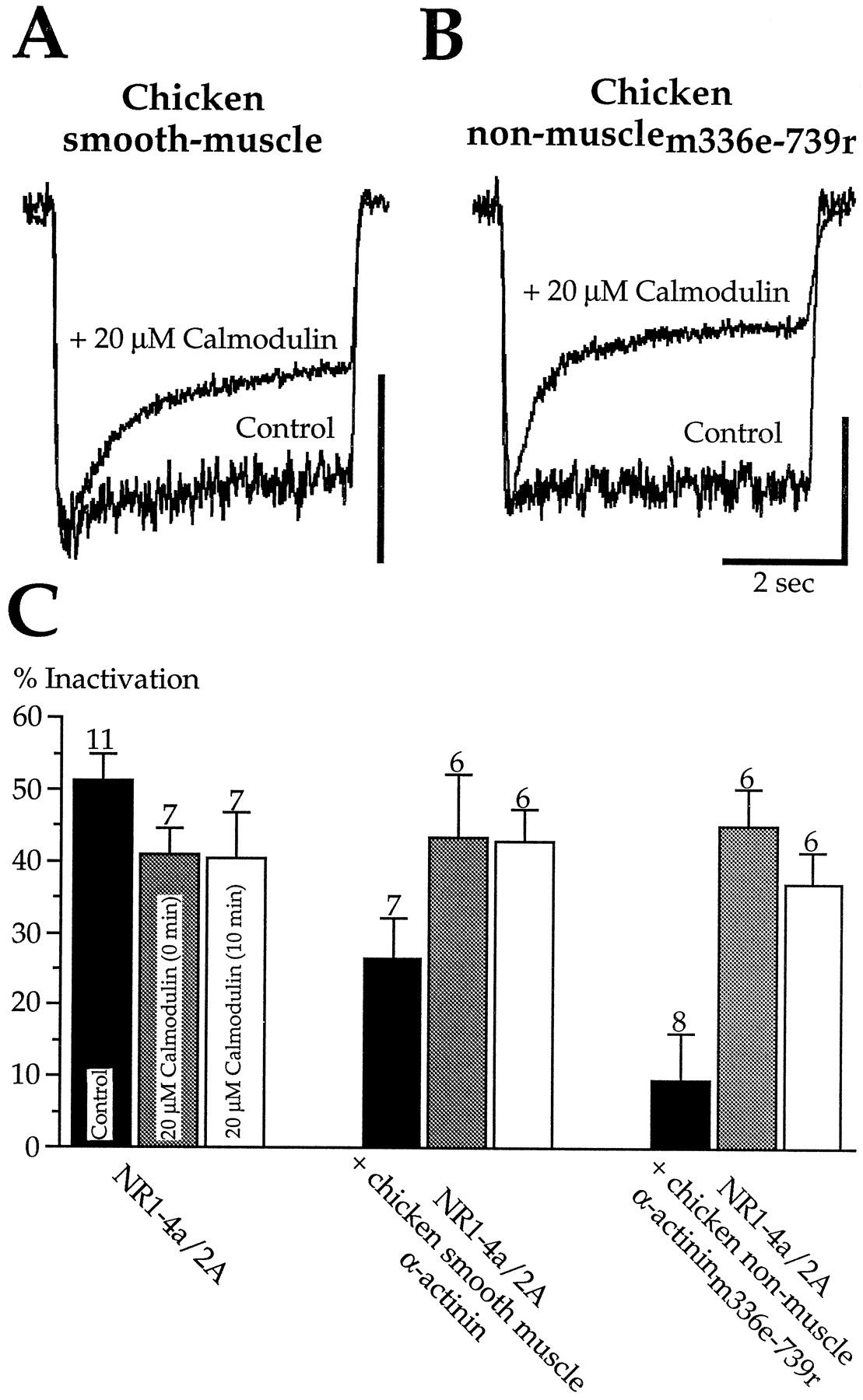
Calmodulin and α-actinin competitively affect calcium-dependent inactivation. A, Inclusion of calmodulin (20 μm) in the whole-cell pipette restored inactivation in HEK293 cells transfected with 2.5 μg DNA/35 mm dish of the putative calcium-insensitive chicken smooth muscle α-actinin. Recordings are from two cells. Scale bar is 100 pA for the control cell and 400 pA for the calmodulin-loaded cell. B, Calmodulin (20 μm) in the pipette also restored inactivation in HEK293 cells transfected with the central rod domain of α-actinin (2.5 μg DNA/35 mm dish). Recordings are from two cells. Scale bar is 100 for the control cell and 250 pA for the calmodulin-loaded cell.C, Pooled data are illustrated from experiments as inA and B.
Binding studies with C-terminal peptides of NR1
Our electrophysiological results indicate a critical role for C0 in inactivation. Because they also suggest a crucial role of α-actinin and calmodulin, the loss or reduction of inactivation seen with the mutated NR1 subunits could be attributable to impaired interactions with these proteins. To test this idea, we examined the binding of calmodulin and α-actinin to peptides corresponding to the C0 domain of several NR1 constructs examined in physiological assays. Biotinylated peptides were immobilized on the surface of biotin-coated IAsys cuvettes through an interaction with neutravidin. Calmodulin or α-actinin binding to the immobilized peptides was detected as a change in the resonant angle of the incident light.
As shown in Figure 7A, increasing amounts of calmodulin (+1 mmCa2+) produced dose-dependent binding to the C0 peptide. Calmodulin concentrations higher than 100 nmproduced saturating responses. The Kd for the calmodulin–C0 peptide interaction was 21 nm, confirming the high-affinity interaction between calmodulin and C0 (Ehlers et al., 1996). This interaction was calcium-sensitive, because it was completely inhibited by EGTA (5 μm, 1 ± 1% of control; n = 3). Calmidazolium (1 μm) also inhibited the interaction of the C0 peptide with 0.1 and 1.0 μm calmodulin to 21.5 and 13.6% of control, respectively. We next tested peptides corresponding to NR1 constructs in which inactivation was reduced or absent. The C0 peptide1–28 and the C0 peptider26a,k27a,n28ahad a lower affinity interaction with calmodulin, withKd values of 79 and 105 nm, respectively (Fig. 7B). The addition of calmodulin (10 μm) to C0 peptide1–22 or the C0 peptide1–25 produced no detectable binding. The responses were 7.2 ± 0.9 and 6.3 ± 0.9 arc-sec in the absence of the C0 peptide1–22 and the C0 peptide1–25, respectively, and 6.3 ± 0.5 and 6.7 ± 1.7 arc-sec in their presence. NR1 constructs with these C0 domains lack inactivation (Fig.2). Thus there was a good correlation between the ability of the peptides to bind calmodulin and the ability of the corresponding C0 domain to support inactivation.

The C0 peptide interacts with α-actinin as well as calmodulin in vitro. A, Protein–protein interactions were assessed by an optical method, as described in Materials and Methods, with binding detected as an increase in the resonant angle of incident light. Addition of calmodulin at the indicated concentrations ([Ca2+] = 1 mm) to C0 peptide-coated cuvettes produced dose-dependent binding to the C0 peptide. Because the change in resonant angle is linear to the mass, the change in resonant angle is a direct measure of the peptide–protein interactions. B, Dose–response curves for the interaction of three different C0 peptides with calmodulin. The amplitude of the resonant angle change was measured after the reaction had reached equilibrium (2 min after addition of calmodulin) and normalized to the response at 1 μm calmodulin. Experiments with C0 peptides corresponding to NR1 constructs producing reduced inactivation had a lower calmodulin affinity as compared with the full-length C0 peptide. C, Rabbit skeletal muscle α-actinin also bound to C0 but showed slower kinetics. D, Estimated dose–response curve for the interaction of the C0 peptide with α-actinin as normalized to the response at 500 nm, the maximal concentration of α-actinin that could be used in our experiments.
We also tested the binding of rabbit skeletal muscle α-actinin to the C0 peptides. As shown in Figure 7C, α-actinin bound to C0 peptide in a dose-dependent manner, although this interaction showed much slower kinetics. Because we could only concentrate α-actinin in the buffer to 500 nm, we were not able to measure higher concentrations of α-actinin. However, theKd, as estimated by normalizing the data to the values at 500 nm α-actinin (Fig. 7D), was 48 nm. The C0 peptide1–22 had no detectable interaction with α-actinin (500 nm), similar to the results with calmodulin for this peptide. The background response was 28.5 ± 6.0 arc-sec in the absence of C0 peptide1–22 and 23.4 ± 6.2 arc-sec in its presence. Binding of α-actinin (100 nm) to the C0 peptide was inhibited to 40% of control (n = 2) by 10 nm calmodulin and to 26.3% of control by 100 nm calmodulin (n = 1). Similar results have been described previously for binding of α-actinin to a glutathioneS-transferase fusion protein containing the C terminus of NR1 (Wyszynski et al., 1997). These results suggest that α-actinin and calmodulin binding depend on common elements in C0. The loss of protein binding to the peptides parallels the loss of inactivation in the NR1 constructs, consistent with the role of both proteins in regulation of channel gating.
The effects of NR1 C-terminal peptides
Although the above experiments provide evidence for a role of C0 and its binding partners in inactivation, they do not provide a molecular model of how this process actually occurs. If one assumes that calmodulin binding to C0 is necessary and sufficient for inactivation, then in the presence of an excess of free C0 peptide the NMDA response should be maintained at peak levels during the agonist application, i.e., there should be no inactivation. To examine this possibility, we tested three NR1 C-terminal peptides in whole-cell experiments. To allow mixing of the peptide with the cytoplasm, the effect of the peptide on inactivation was assessed after 5 min of whole-cell recording. In control experiments, cells expressing NR1–1a/2A heteromers showed a stable degree of inactivation (53.6 ± 6.8% at t = 0 min and 50.4 ± 4.8% att = 5 min; n = 7) without a change in the steady-state current amplitude (98.2 ± 17.3% att = 5 min; n = 7). Low concentrations of C0 peptide (1 μm) had no effect on NR1–1a/2A heteromers (n = 6), whereas high concentrations (100 μm) reduced the initial peak current but had no effect on the steady-state level (64.0 ± 8.2% at t = 5 min of initial peak, 96.4 ± 7.5% at t = 5 min of initial steady-state; n = 8) (Fig.8A).

The C0 peptide reduced the peak current in whole-cell recording but did not affect channel activity in inside-out patches. A, The addition of C0 peptide (100 μm) to the pipette reduced the peak current amplitude in NR1–1a/2A heteromers (10 μm NMDA; 2 mm[Ca2+]o) after 5 min of whole-cell recording. B, In the presence of extracellular calcium, the C0 peptide (100 μm) had no effect on the peak current amplitude when NR1–1a was replaced with the C-terminal truncation mutant NR1stop838. C, Inside-out patches (Vh = +65 mV) from cells expressing NR1stop838/2A heteromers were sequentially exposed to a calcium-free solution (+10 mm EGTA) and a calcium-free solution containing 10 μm C0 peptide. The peptide had no effect on single-channel activity.
The effect of the C0 peptide was dependent on receptor activation, because the peptide (100 μm) did not affect inactivation if the first agonist application was delayed until 5 min of whole-cell recording (51.5 ± 4.3%; n = 5). The C1 peptide (100 μm), which binds calmodulin at a similarly high affinity as C0, had no effect on the peak current amplitude (98.3 ± 28.6% at t = 5 min) or the degree of inactivation (48.5 ± 2.1% at t = 0 min and 43.0 ± 7.3% at t = 5 min; n = 5). However, at 500 μm, the C1 peptide produced a similar effect as high concentrations of the C0 peptide (n = 4). We also tested C0 peptide1–22, which does not bind α-actinin or calmodulin. At low concentrations (1 μm), this peptide reduced the current amplitude (71.5 ± 14.1% of initial peak;n = 4) without affecting the degree of inactivation (54.5 ± 9.9% at t = 0 min and 49.5 ± 8.6% at t = 5 min; n = 4). At 100 μm, the C0 peptide1–22 further reduced the peak amplitude (56.9 ± 6.8% of initial peak; n = 9), resulting in an apparent reduction of inactivation (27.5 ± 5.4% at t = 5 min; n = 9). Thus each of the three peptides reduced the peak amplitude in a manner similar to the CaMBD peptide (Fig. 4), an effect not expected if their mechanism of action is simply to block inactivation. The reduction in peak amplitude could not be attributed to displacement of α-actinin, because the C0 peptide1–22 does not bind α-actinin.
Binding of calmodulin to the C0 peptide requires calcium; thus we were surprised that C0 peptide (100 μm) reduced the current amplitude even in the absence of calcium influx. The amplitude was 68.6 ± 9.1% of control (t = 5 min,n = 5) when agonist was applied in calcium-free medium and in the presence of intracellular BAPTA (10 mm). Likewise, the amplitude was 62.2 ± 12.7% of control (n = 8) at a holding potential of +50 mV. Although substitution of the C-terminal truncation mutant NR1stop838for NR1–1a eliminated the reduction of the peak current by C0 peptide in the presence of calcium influx (106.2 ± 6.4% of control,n = 8) (Fig. 8B), the peptide still reduced the current when calcium influx was prevented (data not shown;n = 14). This raised the possibility that the C0 peptide could have an independent action, perhaps as the ball in a “ball and chain,” as occurs for voltage-dependent potassium channel inactivation (Zagotta et al., 1990). However, even 100 μmof the C0 peptide1–22, which does not bind calmodulin or α-actinin, did not reduce the current amplitude of NR1stop838/2A responses (86.1 ± 14.8% of control;n = 5). We also tested the ball-and-chain hypothesis directly by applying the C0 peptide to inside-out patches expressing NR1stop838/2A heteromers. No change in the single-channel activity was seen on application of 10 μm C0 peptide in calcium-free solution (Fig. 8C). The charge during and after the peptide application was 97.0 ± 5.4% and 95.1 ± 5.8% of control (n = 5 patches). Likewise, no effect was seen with C0 peptide1–22 (10 μm) in calcium-free test solution (n = 4) or with C0 peptide (10 μm) in the presence of 100 μmCa2+ and 100 nm calmodulin (n = 5). Thus the peptide experiments provide no evidence for a ball-and-chain mechanism. NR1stop838/2A heteromers also showed no change in single-channel conductance (data not shown), making it unlikely that deletion of C0 affects the function of the pore or calcium permeability.
Open probability experiments indicate that NR1 residues close to M4 affect the gating of NMDA channels
To understand the influence of calmodulin and α-actinin binding to C0, it is necessary to know whether receptors that lack macroscopic inactivation are “permanently” inactivated or are no longer able to inactivate. Inactivation results from a decrease in channel open probability (Po) (Legendre et al., 1993). Thus we estimated Po for a series of NR1 constructs using the method described in Rosenmund and Westbrook (1993a). This method takes advantage of the irreversible block of NMDA channels by the open channel blocker MK-801. In the continuous presence of agonist and MK-801, channels are blocked as they enter the open state, providing a kinetic means to estimate Po. Responses were evoked by a low concentration of agonist (10 μm NMDA). To avoid contamination of thePo measurement by inactivation, recordings were made with a BAPTA-containing intracellular solution and a calcium-free extracellular solution. After the agonist-induced response reached equilibrium (2 sec), agonist was coapplied with 20 μmMK-801. As shown in Figure 9A, MK-801 produced a complete block of the response in NR1–4a/2A heteromers. The onset of MK-801 block was fitted with two exponentials: τfast and τslow. τfast, reflecting equilibration of MK-801 with already open channels, had a time constant of 124.2 ± 22.4 msec (coefficient 78.4 ± 4.7%; n = 5) that was comparable to hippocampal neurons (Rosenmund and Westbrook, 1993a). τslow is dependent on the rate of entry of receptors into the open state and is directly proportional toPo (Rosenmund and Westbrook, 1993a; Rosenmund et al., 1995). For NR1–4a/2A heteromers (n = 5) (Fig.9B,E), τslow was 1296 ± 325 msec. A lower concentration of NMDA (7.5 μm) produced a twofold reduction in response amplitude and a comparable increase in τslow (3484 + 270 msec; n = 3) (Fig.9B,E), demonstrating that the method was sensitive to the expected change in Po as agonist concentration was decreased.

The C0 domain affects the open probability of NMDA channels and thus has an intrinsic effect on NMDA channel gating.A, Responses from two HEK293 cells expressing either NR1–4a/2A heteromers or NR1stop843/2A heteromers illustrate the protocol used to estimate the open probability (Po). Responses were evoked by a low concentration of NMDA (10 μm). To avoid contamination of the Po measurement by inactivation, recordings were made with a BAPTA-containing intracellular solution, and agonist was applied in Ca-free extracellular solution. After the responses had reached equilibrium, the open-channel blocker MK-801 (20 μm) was coapplied with NMDA. The onset of MK-801 block can be described by two exponentials, with the slower time constant reflecting channels that enter the open state during the presence of MK-801, i.e., it is proportional to Po (see Results for further details). Note that the slow component of MK-801 block is slower for NR1stop843/2A heteromers, indicating a lower Po. Currents are normalized to their steady-state amplitude. B, Semilogarithmic plot of the current decays in MK-801 for responses from NR1–4a/2A heteromers evoked under control conditions (10 μm NMDA), with a lower concentration of agonist (7.5 μm NMDA), or from cells coexpressing the central rod domain of α-actinin (α-actininm336e-739r). The coexpression of the central rod domain did not affect the slow component of MK-801 block, whereas the reduction in agonist concentration slowed the onset of MK-801 block, indicating the expected lower Po with lower agonist concentration.C, Semilogarithmic plot of the current decays in MK-801 for responses from NR1/2A heteromers containing several NR1 truncation mutants. Note that the onset of MK-801 block is slower (i.e., lowPo) with NR1stop858 and NR1stop843, which presumably do not bind calmodulin or α-actinin. The onset of MK-801 block is fast (i.e., highPo) after truncation of almost the entire C0 domain (NR1stop838). D, The ratio between the amount of charge during steady-state current (measured during the 2 sec application preceding MK-801) and the amount of charge during the application of MK-801 is the same for all conditions tested, indicating that differences in MK-801 kinetics between different conditions do not underlie the observed differences in the onset of MK-801 block. E, Pooled data for the slow time constant τslow under all conditions tested demonstrates that the presence of a C0 domain incapable of binding calmodulin and α-actinin results in a low Po.Asterisks indicate significant differences compared with control (NR1–4a/2A; 10 μm NMDA).
Po of NR1–4a/2A heteromers coexpressed with chicken α-actininm336e-739r was as high as in NR1–4a/2A controls, suggesting that receptors with the central rod domain bound to C0 are not inactivated but are still capable of inactivating, consistent with the fact that calmodulin induced inactivation in these heteromers (Fig. 6). In NR1stop863/2A heteromers, τslow was 1657 ± 176 msec (n = 6), similar to NR1–4a/2A heteromers, whereas τslow was approximately twofold larger (i.e., a lowerPo) in NR1 subunits with truncated C0 domains that do not bind calmodulin or α-actinin. Interestingly, receptors lacking C0 (NR1stop838/2A) had a τslow comparable to NR1–4a/2A heteromers (Fig.9C,E). The normalized charge transferred during the MK-801 block was the same for all conditions tested (Fig.9D); thus the differences in τslow reflect differences in Po rather than the number of open channels. Alterations in the length of C0 had no effect on agonist affinity (results not shown). These results demonstrate that NMDA receptors with a C0 domain that is incapable of binding calmodulin or α-actinin have a low Po and thus in effect are permanently inactivated. This implies that the C0 domain has an intrinsic effect on channel gating.
DISCUSSION
The interactions of synaptic receptors with cytoskeletal and regulatory proteins have been the focus of intensive study in the past few years. Previous pharmacological data suggested that inactivation of NMDA receptors might be one example in which channel gating was controlled by such interactions (Rosenmund and Westbrook, 1993b). Biochemical studies have identified some candidate proteins for this effect (Ehlers et al., 1996; Wyszynski et al., 1997). Our results indicate that the C0 domain directly affects the channel open probability and that the regulation of Po via interactions of C0 with calmodulin and the central rod domain of α-actinin is responsible for inactivation.
A molecular model for inactivation
We propose a working molecular model for inactivation as a framework for discussing our results. This model incorporates the intrinsic effects of C0 on the open probability as shown in Figure10. At rest, C0 is bound to the central rod domain of α-actinin. This association is critically dependent on residues 856–863 of NR1, is calcium-independent, and is sufficient to prevent spontaneous channel inactivation, implying that C0 is de facto “latched” to the actin cytoskeleton via α-actinin. Calcium influx during receptor activation or through nearby voltage-dependent calcium channels (Legendre et al., 1993) triggers the dissociation of C0 from the central rod domain. Because our results provide evidence for the involvement of α-actinin and calmodulin, we propose that two processes lead to inactivation: (1) calcium binds to the EF-hands of calcium-sensitive α-actinin isoforms and thereby decreases the apparent affinity of α-actinin for C0, and (2) calcium activates calmodulin, resulting in competitive displacement of α-actinin from C0. In the intact cell, both processes are likely to occur and converge into the same phenotype, in which the resulting “untethered” C0 domain (with or without calmodulin bound) reduces the open probability of the NMDA channel. Residues 834–843 of C0 are necessary for the lowPo conformational state, but the mechanism is not a ball and chain. Although many of the features of this model are consistent with our results, a number of ambiguities remain as discussed below.

A molecular model for inactivation. C0 has an intrinsic effect on channel gating that can shift the open probability between high and low Po states. When the channel has a high Po (top left), C0 is probably attached to actin via α-actinin (for clarity this interaction is not shown). Influx of calcium triggers the dissociation of α-actinin from C0, leading to a lowPo (top right). This effect underlies calcium-dependent inactivation of NMDA channels and can be induced by two mechanisms: calmodulin dependent and calmodulin independent. In the calmodulin-independent mechanism (bottom left), calcium binds to the EF-hand of α-actinin (1), thereby reducing the affinity of α-actinin for C0, resulting in the dissociation of α-actinin from C0 (2). This mechanism incorporates our findings with the overexpressed α-actinins and explains the lack of effect of calmodulin inhibitors on whole-cell inactivation. In the calmodulin-dependent mechanism (bottom right), calcium binds to calmodulin (1). The activated calmodulin then competes with α-actinin for binding to C0 (2), resulting in the dissociation of α-actinin from C0 (3). This mechanism incorporates our competition experiments, in which exogenous calmodulin overcame the effects of overexpressed α-actinins.
Comparison with previous results
Calmodulin has been shown to bind to C0 and in inside-out patches reduces single-channel activity of NR1/2A heteromers (Ehlers et al., 1996). This led to the proposal that inactivation results from calmodulin binding to C0 (Zhang et al., 1998). However, calmodulin inhibitors fail to prevent inactivation (Legendre et al., 1993;Rosenmund and Westbrook, 1993a; Krupp et al., 1996). The lack of effect of calmidazolium, at concentrations that block calmodulin effects in neurons (Zeilhofer et al., 1993), was again confirmed in the present study. Because calmodulin concentrations in kidney cells are reported to be twofold lower than in neurons (Kakiuchi et al., 1982), this concentration of calmidazolium should have been effective. Although calmodulin could be located in a restricted environment inaccessible to inhibitors, exogenous calmodulin, which is much larger than the inhibitors, readily reached the membrane and overcame the effect of overexpressed α-actinin. Nonetheless calmodulin could be concentrated near the membrane by calcium-independent calmodulin binding proteins such as neurogranin (Gerendasy and Sutcliffe, 1997) and unconventional myosins (Porter et al., 1993).
The CaMBD peptide reduced the peak current amplitude without affecting the steady-state response. Such an effect could be misinterpreted as a block of inactivation. However, it indicates either that calmodulin is required for recovery from inactivation or that some calmodulin-inhibitor peptides have unspecific effects. Using a different calmodulin binding peptide, Zhang et al. (1998) also saw a reduction of the peak current amplitude but only a small decrease in the steady-state current (see their Fig. 6A). Zhang et al. (1998)interpreted this as a specific block of inactivation. We disagree with this view because other mechanisms such as channel rundown can give this appearance (Rosenmund and Westbrook, 1993a,b). Although Zhang et al. (1998) did not see this effect with a control peptide, the control peptide differed markedly in total charge from the calmodulin binding peptide. In our experiments, three highly similar peptides, including the C0 peptide1–22 that does not bind calmodulin, reduced the peak current amplitude. This result is most consistent with a nonspecific action, possibly because of the putative amphipathic helical structure of these peptides.
Despite the above issues, there is clear evidence that calmodulin participates in inactivation. We confirmed that calmodulin reduces NMDA single-channel activity in inside-out patches (Ehlers et al., 1996). In intact cells, the loss of inactivation with a series of NR1 truncation mutants matches the affinity of corresponding peptides for calmodulin. The sequence of the last 15 residues of C0 is similar to the 1-5-8-14 consensus motif for calmodulin recognition (Rhoads and Friedberg, 1997). Calmodulin was able to overcome the block of inactivation caused by overexpressed putative calcium-insensitive α-actinins. Thus, the competition between α-actinin and calmodulin seen in vitro(Wyszynski et al., 1997) can be observed in intact cells. This situation may be somewhat analogous to calmodulin regulation of cyclic nucleotide-gated (CNG) channels. In that case, calmodulin binds to the N terminus of the olfactory CNG channel subunits (Liu et al., 1994) and reduces the channel open probability by interfering with a protein–protein interaction between the N terminus and the C-terminal, ligand-binding domain (Varnum and Zagotta, 1997). Interestingly, calmodulin regulates activity of enzymes such as CaM kinase II often through a direct competition (Soderling, 1990).
Effects of α-actinin
The results with overexpressed α-actinins demonstrate that α-actinin can directly alter NMDA channel gating. Consistent with our working model of inactivation, binding of α-actinin to C0 maintainsPo at the top of the twofold range between control and fully inactivated channels. A high level of endogenous α-actinin-2 in HEK293 cells has been reported (Zhang et al., 1998). However, a reduction in inactivation was observed with overexpressed α-actinin-2, inconsistent with expression of saturating endogenous levels of this putative calcium-insensitive isoform. Furthermore, we did not detect antibody staining for α-actinin-2 in untransfected HEK293 cells [data not shown (antibody EA-53, Sigma)], whereas bright staining was present after overexpression of α-actinin-2, suggesting that background levels of α-actinin-2 were low in our experiments.
Consistent with a competition between calmodulin and α-actinin, our peptide binding studies indicate that residues 856–863 of C0 are also essential for α-actinin binding. The highly charged structure of C0 may be involved in the binding site because the interaction of α-actinin with β-integrins (Otey et al., 1993) or the intercellular adhesion molecule-1 (Carpén et al., 1992) is based on charged and aliphatic interactions. Binding to C0 was lost with α-actinin constructs ending within the fourth spectrin repeat (Wyszynski et al., 1997), potentially indicating that the fourth spectrin repeat is the binding site. If true, the EF-hands of an α-actinin bound to C0 would be in close proximity to calcium transients at the intracellular vestibule of the NMDA receptor.
Only the putative calcium-insensitive isoforms reduced inactivation, raising the possibility that the interaction of C0 with α-actinin is itself calcium dependent. Because the calcium-sensitive chicken nonmuscle and calcium-insensitive smooth muscle α-actinins differ only in the first EF-hand region, the two isoforms should, in the absence of calcium, bind C0 with similar affinities. Thus calcium binding to the EF-hand could affect the binding of α-actinin to C0, either by directly reducing the affinity of α-actinin for C0 or through an indirect effect. For example, calcium-dependent dissociation of nonmuscle α-actinin from actin could facilitate other protein–protein interactions. The dependence on the EF-hand domain suggests that inactivation in neurons might be influenced by the expression levels of different α-actinin isoforms. Although putative calcium-insensitive and -sensitive isoforms are present in brain (Waites et al., 1992; Wyszynski et al., 1997), their expression levels and the effects of calcium on their function in intact cells remains to be determined.
Ball and chain, lids, or latches: the question of the molecular mechanism
On release of the C0/α-actinin/actin latch, a conformational change must occur leading to a reduction in the open probability of the channel. Several models of this conformational movement can be imagined. For example, the C0/α-actinin/actin latch could influence gating by exerting tension on the receptor or constraining movements of C0. However, NR1stop838/2A heteromers that completely lack C0 have a high Po as in control. Likewise, coexpression of the spectrin repeats that lack an actin binding site also had a high Po. Thus C0 must dissociate not only from the actin cytoskeleton but also from α-actinin for inactivation to occur. Likewise the high Po in the absence of a C0 domain implies that unlatched C0 interacts with either another receptor domain or another protein. Whether inactivation can occur with calmodulin bound or whether C0 must be free is not clear.
Because amphipathic helices can interact with lipid bilayers (Opella, 1994), unlatched C0 could interact with a transmembrane domain by dipping in and out of the membrane. Such movements occur in the bacteriophage Pf1 coat protein (Shon et al., 1991), but would seem to require the presence of a flexible region between the transmembrane domain (M4 of NR1) and the amphipathic helix interacting with the bilayer (parts of C0). There are no recognizable structural elements such as glycines or prolines in C0 that could fulfill this function. Alternatively, unlatched C0 could interact with an intracellular receptor domain as in the ball-and-chain mechanism for N-type inactivation of Shaker potassium channels (Hoshi et al., 1990; Zagotta et al., 1990); however, C0 peptides did not mimic inactivation in NMDA channels. Furthermore, C0 lacks recognizable structural elements that provide the flexibility of a chain, or an effective chain length, because the critical elements affectingPo involve the first 10 residues of the C0 domain. We favor a hybrid model analogous to the hinged lid model for fast inactivation of the Na+ channel (West et al., 1992). A possible receptor site for a C0 lid could be in the loops flanking M2 in NR1 or NR2, possibly explaining the NR2 subunit specificity of inactivation (Krupp et al., 1996).
Footnotes
This work was supported by National Institutes of Health Grants MH46613 (G.L.W.) and NS28709 (S.F.H.), the McKnight Foundation (S.F.H.), the John Adler Foundation (S.F.H.), fellowships from the Human Frontiers program (J.J.K., B.V.), and the National Health and Medical Research Council of Australia (B.V.). We thank the following for cDNA constructs: Drs. A. H. Beggs and M. Sheng (Harvard Medical School; human α-actinin-2), Dr. D. R. Critchley (University of Leicester, Leicester, UK; chicken α-actinins), and Dr. J. P. Adelman (Vollum Institute; CD4). The excellent technical support by A. Miller is gratefully acknowledged. Dr. M. Faux kindly helped us with the IAsys optical biosensor system.
J.J.K. and B.V. contributed equally to this work.
Correspondence should be addressed to Dr. Gary L. Westbrook, Oregon Health Sciences University-L474, Vollum Institute, 3181 SW Sam Jackson Park Road, Portland, OR 97201.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}