

Abstract
Intracellular β-amyloid 42 (Aβ42) accumulation is increasingly recognized as an early event in the pathogenesis of Alzheimer's disease (AD). We have developed a doxycycline-inducible adenoviral-based system that directs intracellular Aβ42 expression and accumulation into the endoplasmic reticulum of primary neuronal cultures in a regulated manner. Aβ42 exhibited a perinuclear distribution in cell bodies and an association with vesicular compartments. Virally expressed intracellular Aβ42 was toxic to neuronal cultures 24 hr after induction in a dose-dependent manner. Aβ42 expression prompted the rapid induction of stress-inducible Hsp70 protein in neurons, and virally mediated Hsp70 overexpression rescued neurons from the toxic effects of intracellular Aβ accumulation. Together, these results implicate the cellular stress response as a possible modulator of Aβ-induced toxicity in neuronal cultures.
Introduction
The prevailing view that extracellular β-amyloid (Aβ) deposition is the primary toxic insult in Alzheimer's disease (AD) is being modified by an increasing number of studies that suggest intracellular Aβ accumulation may play an early and important pathologic role in the development of the disease (Takahashi et al., 2002a). Although no direct evidence has been provided, it has long been speculated that intraneuronal Aβ accumulation causes neuronal dysfunction, synaptic and neuronal loss, and dementia in AD and related Down's syndrome (LaFerla et al., 1995; Yang et al., 1998; Gouras et al., 2000; D'Andrea et al., 2001; Gyure et al., 2001; Busciglio et al., 2002). Several reports have documented intracellular accumulation of Aβ (Martin et al., 1995; Skovronsky et al., 1998; Gouras et al., 2000; Mochizuki et al., 2000; Walsh et al., 2000; Takahashi et al., 2002b). Aβ42 peptide appears to be the prominent form that accumulates inside neurons in AD brains and in transgenic mice expressing familial AD mutations (Takahashi et al., 2002a). However, only a limited number of reports have specifically studied the intracellular effects of Aβ42 peptide accumulation using either transgenic animals (LaFerla et al., 1995; Link, 1995) or non-neuronal cell lines (Johnstone et al., 1996; Querfurth et al., 2001; Suhara et al., 2003). Recently, delivery of Aβ synthetic peptides into neurons by microinjection has been reported (Zhang et al., 2002). However, the lack of methods allowing the facile transfection of postmitotic cells has hindered the study of mechanisms of intracellular Aβ toxicity in primary neuronal cultures and its role in AD.
Increasing evidence points to a role for molecular chaperones in neurodegenerative diseases (DeArmond and Prusiner, 1995; Bonini, 2002; Sakahira et al., 2002). Recent findings that heat shock proteins (Hsps) can suppress neurotoxicity in animal models of Parkinson's and polyglutamine diseases (Warrick et al., 1999; Chan et al., 2000; Fernandez-Funez et al., 2000; Kazemi-Esfarjani and Benzer, 2000; Cummings et al., 2001; Auluck et al., 2002) suggests potential therapeutic approaches in neurodegeneration associated with abnormal protein folding and toxicity. Whereas Hsp70 and Hsp90 have been implicated in maintaining tau solubility and suppressing tau aggregates (Dou et al., 2003), the involvement of Hsp in Aβ-mediated toxicity has not been evaluated.
In the current study we use primary neuronal cultures and an inducible adenoviral vector system to study the effects of intracellular Aβ42 accumulation. These data suggest that the cellular stress response is an important modulator of intracellular Aβ toxicity in neurons.
Materials and Methods
Antibodies. The following antibodies were used: 6E10 (1:300; Signet Laboratories, Dedham, MA), anti-β-galactosidase (1:500; Promega, Madison, WI), anti-calnexin (1:200; Stressgen Biotechnologies, San Diego, CA), anti-Hsp70 (1:100-5000; Stressgen Biotechnologies), anti-Hsc70 (1:5000; Stressgen Biotechnologies), anti-βIII isoform of tubulin (1:100; Chemicon International, Temecula, CA). Secondary antibodies were used as follows: fluorescent Cy2 and Cy3-conjugated (Jackson ImmunoResearch, West Grove, PA), peroxidase-conjugated (Promega), alkaline phosphatase-conjugated (Dako, Santa Barbara, CA).
Adenoviral vector construction. Recombinant, E1-deleted replication-defective adenoviral constructs were produced by standard techniques (Graham and Prevec, 1995). In brief, the human Aβ42 peptide sequence containing an endoplasmic reticulum signal peptide based on the βAPP751 coding sequence was PCR-amplified from pHSVPrpUC-Aβ42 vector (provided by Dr. R. Neve, McLean Hospital, Belmont, MA) and inserted into HindIII-XhoI sites downstream of the tetracycline response element (TRE) promoter in the plasmid pTRE (Clontech, Palo Alto, CA). To produce a reverse Aβ42 cDNA construct, the same sequence was amplified, but restriction sites were inverted in the generation of the PCR fragment (XhoI-HindIII). The TRE cassette was then subcloned into the multiple cloning site of pShuttle vector, and recombination with pAdEasy-1 vector was achieved following manufacturer's instructions (Quantum Biotechnologies, Montreal, Quebec, Canada). The AdTet-On and AdTRE-LacZ viruses were previously described (Mano et al., 2000). The cDNA of human βAPP751 was digested from NSE-hAPP751 vector (provided by Dr. C. R. Abraham, Boston University, Boston, MA), and inserted into the HindIII site in the plasmid pAdTrack-CMV (Clontech), followed by recombination with pAdEasy-1, as described above. AdHsp70 was provided by Dr. W. H. Dillmann (University of California, San Diego, CA). All viruses were grown to high titer in human embryonic kidney 293 cells and purified by cesium chloride density gradient ultracentrifugation. Viral titer was determined by plaque assay.
Rat primary cortical cultures. Cortices were dissected from neonatal day 1 rat Sprague Dawley brains (Charles River Laboratories, Wilmington, MA), incubated in trypsin and DNase, and then mechanically dissociated in PBS-glucose solution. Neurons were plated into poly-d-lysine (10 μg/ml; Sigma, St. Louis, MO) and laminin (5 μg/ml; Invitrogen, Carlsbad, CA)-coated plates. Mixed cultures were maintained in Neurobasal medium supplemented with B27, 0.5 mm glutamine, 1% penicillin-streptomycin, 4.6 mm NaHCO3, 33 mm glucose, and 1% horse serum (Invitrogen, Gaithersburg, MD). Primary cortical cultures were infected 7 d after plating at multiplicities of infection (moi) ranging from 50 to 100. Unless indicated, AdTet-On and AdTRE-LacZ or AdTRE-Aβ42 were added into culture for 18 hr at a 1:1 ratio, and transgene expression was induced by 1 μg/ml doxycycline (Dox; Sigma) for 24 hr.
Western blot analysis. Cells were washed once in PBS and either scraped directly into sample buffer solution and electrophoresed in 4-12% Bis-Tris gels (Invitrogen, Carlsbad, CA) or harvested in RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 0.25% Na-deoxycholate, and 1% NP-40) containing protease inhibitor cocktail (Roche Biochemicals, Indianapolis, IN), 1 mm sodium vanadate, and 1 mm sodium fluoride. Protein concentration was measured with the bicinchoninic acid (BCA) protein assay reagent (Pierce, Rockford, IL) protocol, and samples were electrophoresed in 7.5% SDS-PAGE gels under reducing conditions. Proteins were transferred onto polyvinylidine difluoride membranes (Millipore, Billerica, MA) and immunoblotted for the indicated proteins as previously described (Magrane et al., 1999). The immunoreaction was visualized by a chemiluminescence system (Amersham Biosciences, Piscataway, NJ). Neither radiolabeling nor immunoprecipitation were required to detect Aβ42 peptide.
Immunocytochemistry and immunogold electron microscopy. Cells were grown on coated coverslips, washed once in PBS, and fixed in 4% paraformaldehyde. For intracellular immunodetection, cells were permeabilized with 0.1% Triton X-100 and then incubated at 37°C for 45 min with the indicated antibodies. Cell preparations were processed for alkaline phosphatase staining or immunofluorescence as described (Magrane et al., 1999) with conjugated secondary antibodies. For TUNEL staining, the in situ cell death detection kit (Roche) was used according to manufacturer's instructions. The percentage of TUNEL-positive cells and pyknotic nuclei were obtained by counting 20 random fields per sample at 20× in each of four different experiments. Nikon Eclipse TE300 and Zeiss LSM 510 laser-scanning confocal microscopes were used. To prepare ultrathin frozen sections, neurons were trypsinized from the dish, fixed with 4% paraformaldehyde, and cryoprotected in 2.3 m sucrose. Ultrathin sections were obtained and processed for immunogold electron microscopy (Griffiths, 1993). Antibodies against Aβ and 10 nm gold-conjugated protein-A (JanSlot, Utrecht, The Netherlands) were used to immunostain sections on carbon-coated hydrophilic grids. Samples were then negatively stained with uranyl acetate and examined on a JEOL1200EX transmission electron microscope (JEOL USA, Peabody, MA).
Statistical analysis. Results are expressed as the mean ± SE. Statistical significance was determined by the Student's two-tailed, unpaired t test, and a P value of <0.05 was considered significant.
Results
Intracellular inducible Aβ42 expression in primary neuronal cultures
We constructed an adenoviral system for inducible Aβ42 expression in neurons using the tetracycline-regulatable Tet-On gene expression system (Gossen et al., 1995). This system uses a co-infection strategy in which one virus (AdTet-On) provides the transactivator, while the other (AdTRE-cDNA) contains a TRE promoter that controls the expression of the transgene. Transcription of β-galactosidase or Aβ42 from the TRE promoter is driven by the transactivator only when the tetracycline analog Dox is added. The absolute levels of gene expression are controlled by the dose of Dox that is added to the medium.
Rat primary cortical neuron cultures were established, and the adenoviral expression system was characterized using TRE-LacZ and TRE-Aβ42 viruses. Levels of β-galactosidase or Aβ42 expression, detected by immunocytochemistry, could be regulated by either varying the number of viral particles used (data not shown) or the amount of Dox added into the medium (Fig. 1a). High-efficiency transduction (>70%) was obtained at 50 moi. The Aβ42 construct was engineered with an N-terminal signal peptide sequence to express the transgene product in the secretory pathway and, thus, mimic the in vivo sites of Aβ production (Takahashi et al., 2002a). The LacZ construct contained no signal peptide. Whereas β-galactosidase immunostaining was typically cytoplasmic, we observed a perinuclear distribution of Aβ42 immunoreactivity and colocalization with calnexin, consistent with routing of Aβ to the endoplasmic reticulum (Fig. 1b,c). Aβ42 expression was observed both in cell bodies and in neuritic processes (Fig. 1b). To further characterize the regulatable properties of this expression system, we performed Western blot analyses on cultures infected with a fixed amount of viral particles (100 moi of AdTet-On and AdTRE-Aβ42, at a 1:5 ratio) and tested Aβ42 expression in response to increasing amounts of Dox (0.3, 0.5, and 1 μg/ml) for 24 hr. We detected a 4-8 kDa band only when Dox was present in the culture media, and levels of Aβ peptide increased with higher doses of Dox (Fig. 1d, top and middle panels). Aβ42 accumulation was detected 6 hr after Dox induction (Fig. 1d, bottom panel). Aβ42 was not detected in the media by ELISA or Western blot techniques in infected cells at any dose of Dox or using higher moi (data not shown). These results demonstrate that this vector system produces regulatable levels of intracellular Aβ expression.

Tet-On system and intracellular inducible Aβ42 expression in primary neuronal cultures. a, Regulatable control of β-galactosidase (top) and Aβ42 expression (bottom) was achieved by varying the amount of Dox added to the culture medium. b, Aβ42 expression (green) revealed a typical intravesicular pattern that filled up the cell bodies (top), including neuritic processes (middle). β-galactosidase immunostaining (bottom) was typically cytoplasmic. c, Confocal images from a cell immunostained for Aβ (green) and calnexin (red), an endoplasmic reticulum marker. Both markers share colocalization in the merged image (bottom). d, Detection of Aβ42 expression with increasing amounts of Dox (top) and time (bottom). Corresponding densitometric quantitation expressed as arbitrary optical density (O.D.) units ± SD (middle). 6E10 antibody was used for Aβ42 detection.
We performed immunogold electron microscopy to determine the subcellular localization of inducible Aβ42 transgene product in the neuronal cultures. Aβ42 appeared to be associated with membrane compartments, mainly the endoplasmic reticulum and associated vesicles (Fig. 2, top panel). Aβ42 was found in both small vesicles (∼45% of identifiable labeling), confined mostly located to the membrane, and in multivesicular bodies (∼35%) (Fig. 2, bottom right panel). Gold particles were present individually or in small groups, but no Aβ fibrils were detected. Non-induced neurons were used as negative controls and confirmed the lack of immunoreactivity (Fig. 2, bottom left panel).

Aβ42 expression within the endoplasmic reticulum and in association with vesicular membranes. Immunogold localization of Aβ42 (arrowheads) using 6E10 antibody in neuronal cultures (top). Lack of immunogold labeling in non-induced controls (bottom left). Aβ42 immunogold particles with small vesicles and multivesicular bodies (bottom right). ER, Endoplasmic reticulum; V, vesicle; MVB, multivesicular body. Scale bars, 200 nm.
Intracellular expression of Aβ is toxic to neurons
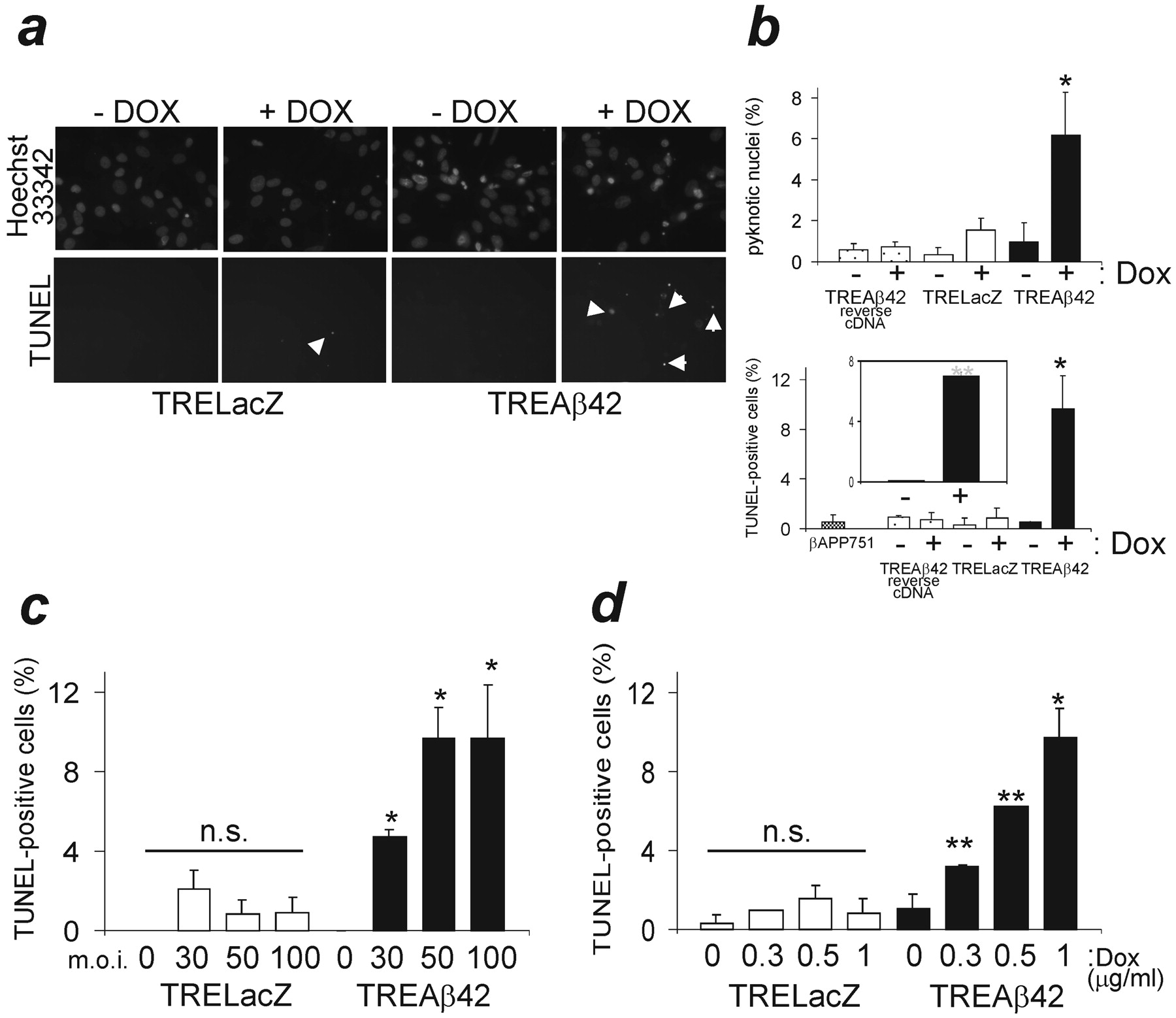
To study the effects of intracellular Aβ expression on primary neuronal culture viability, transgene expression was induced for 24 hr, and cultures were analyzed for the presence of pyknotic nuclei and TUNEL-positive cells as indicators of apoptosis. Expression of a reverse Aβ42 cDNA or β-galactosidase transgene did not result in significant toxicity at 100 moi, whereas Aβ42 expression increased the number of both pyknotic nuclei and TUNEL-positive cells (Fig. 3a,b). In contrast, virally overexpressed βAPP751 had no effect on cell death 24 hr after infection (Fig. 3b). Most of the cells with signs of apoptosis appeared to be neurons. To confirm preferential neuronal vulnerability, we established neuron-enriched cultures (>95% neurons) and observed the same effect when Aβ42 was induced (Fig. 3b, inset). Aβ-induced neuron death could be modulated either by varying the number of viral particles (0-100 moi) or the amount of Dox (0-1 μg/ml, at 50 moi). Thus, a direct correlation was observed between the number of apoptotic cells and the production of the Aβ42 transgene product (Fig. 3c,d). This increase in cell death was not observed in TRELacZ controls (Fig. 3a-d).

Intracellular Aβ42 expression results in apoptosis in primary neuronal cultures. a, Nuclear pyknosis (Hoescht 33342 staining) and TUNEL staining in infected neuronal cultures. LacZ expression and non-induced controls produced similar background levels. b, Quantification of data from four independent experiments is shown. Comparison of induced transgene products, *p < 0.005. Inset, Aβ42 toxicity in neuron-enriched cultures (TUNEL staining), p < 0.001. c, d, The extent of Aβ42 toxicity could be adjusted by varying either the number of viral particles or the amount of Dox (n = 4). Values are subtracted in each experiment with their control (basal) conditions. *p < 0.005; **p < 0.05; n.s., non-significant.
Intracellular Aβ affects the neuronal stress response
Having demonstrated the toxic effects of intracellular Aβ42 accumulation, we analyzed the role of the stress response in these neuronal cultures. The expression of two Hsp family members, Hsp70 and Hsc70, was assessed in cells infected with 50 moi of AdTet-On and AdTRE-transgene (1:5 ratio). Aβ42 expression increased the Hsp70 stress-inducible form as early as 6 hr, and maximal induction was reached by 24 hr. Expression of β-galactosidase in parallel cultures had no effect on Hsp70 expression. Levels of the constitutively expressed form of Hsp70, Hsc 70, did not change at any time point, and similar total protein levels of the β-III isoform of tubulin were detected among all lanes (Fig. 4a,b). In contrast, virally overexpressed βAPP751 had no effect on Hsp70 protein levels (data not shown). We performed immunofluorescence in rat cortical cultures to confirm the upregulation of Hsp70. Aβ42-positive cells showed increased Hsp70 expression, whereas β-galactosidase-expressing cells did not (Fig. 4c,e). Only cells with neuronal morphologies were affected by the stress response (Fig. 4c,e).

Intracellular Aβ42 production induces a cellular stress response in primary neuronal cultures. Cultures were infected, and Aβ42 expression was induced for different durations. a, Protein levels of two Hsp family members were assayed by Western blot. Hsc70 and βIII-tubulin levels were similar among lanes. b, Quantitative data on relative inducible Hsp70 expression levels referred to 0 hr time point (n = 3). *p < 0.02. c, Aβ42 overexpression (green, top panel) in neurons correlated with increased Hsp70 immunostaining (red), whereas β-galactosidase (green, bottom panel) did not. Merged images showing coincidence of staining in yellow (right). d, Quantitation of transgene-expressing cells that show Hsp70 overexpression (n = 3). **p < 0.002. e, Aβ42-positive glial cells (green) did not show Hsp70 upregulation (red).
Hsp70 protects against intracellular Aβ-induced toxicity
To address whether the increase in Hsp70 expression in response to Aβ42 accumulation could be a cellular mechanism to protect against Aβ toxicity, cultures were co-transduced with an adenoviral vector expressing Hsp70. Primary neuronal cultures were infected at 100 moi for 18 hr, after which Dox and 10 moi of AdHsp70 virus or AdLacZ control virus were added for 24 hr. Strikingly, overexpression of Hsp70 (Fig. 5b, inset) protected against the toxic effects of Aβ42 accumulation, as shown by significant reductions in the number of both pyknotic nuclei and TUNEL-positive cells in Aβ42-expressing cultures. Overexpression of β-galactosidase had no effect on Aβ42 neurotoxicity (Fig. 5). The expression levels of Aβ42 were unaltered by co-infection with either AdHsp70 or AdLacZ (Fig. 5c). These data implicate the stress response in a possible role for overcoming Aβ-induced toxicity in neuronal cultures.

Hsp70 overexpression rescues Aβ42-induced toxicity. a, Co-infection with AdHsp70 fully protected against Aβ42 induced neuronal death. Co-infection with the control construct AdLacZ did not effect Aβ42-induced toxicity. *p < 0.0005, n.s., non-significant. b, The neuroprotective effect of Hsp70 overexpression (inset) was also observed by using TUNEL staining in Aβ42-induced neuron cultures. Co-infection with AdLacZ had no neuroprotective effects. **p < 0.005, n.s., nonsignificant. Values from three independent experiments are shown. c, Aβ42 expression levels were not affected by the overexpression of Hsp70 or β-galactosidase.
Discussion
An increasing number of reports suggest that Aβ42 accumulates inside neurons with aging and that intraneuronal Aβ may be a seminal event in the neuronal and synaptic degeneration that is characteristic of AD. Some approaches have been developed (LaFerla et al., 1995; Zhang et al., 2002); however, there is no facile method to date for the intracellular expression of Aβ42 in postmitotic cells and thus, the intracellular effects of Aβ accumulation in neurons have not been fully characterized. Here we describe an inducible viral gene expression system that allows efficient transduction of neurons and provides regulated intracellular expression of the Aβ42 peptide (Harding et al., 1997). Intracellular Aβ42 expression was toxic to neuron cultures from rat cortex, and different Aβ expression levels correlated with levels of toxicity. This toxicity was not associated with peptide secretion into the medium. These data support the hypothesis that neuronal apoptosis in AD and Down's syndrome is associated with intracellular Aβ deposition (Cotman, 1998). Further support is provided by the finding that apoptosis in neurons is indirectly correlated with intracellular Aβ accumulation produced after long-term expression of APP695 (Nishimura et al., 1998; Kienlen-Campard et al., 2002). The recent studies of Oddo et al. (2003) lend strong credence to the notion that synaptic dysfunction correlates with early accumulation of intracellular β-amyloid.
In AD, as in other neurodegenerative diseases, selected cell types and populations are preferentially affected by the accumulation of a toxic product. We observed efficient transduction of both neurons and glial cells in mixed rat cultures, yet neurons appeared to be mainly affected by the virally mediated intracellular Aβ42 expression. Stress responses are also cell type-specific. The observed Aβ42 effect on Hsp70 expression levels in neurons but not in glia seems to be Aβ-specific, because in other stress situations Hsp70 is weakly induced in nerve cells and highly upregulated in glial cells (Nishimura et al., 1991; Satoh and Kim, 1994). Interestingly, the rapid upregulation of Hsp70, as early as 6 hr, corresponded to detectable but low levels of Aβ at this time point. Transduction with Hsp70 reduced Aβ42-induced toxicity, suggesting that the endogenous stress response in neurons was insufficiently protective against Aβ.
Immunofluorescence and ultrastructural studies confirmed Aβ42 localization in the endoplasmic reticulum (Hartmann et al., 1997) and in small vesicles and multivesicular bodies (Takahashi et al., 2002b). Approximately 40-60% of vesicular-associated immunogold labeling was found located on the membrane. No intracellular aggregates of Aβ42 were observed by microscopy techniques, and no higher molecular weight species were detected by Western blot. Taken with results discussed above, these data suggest that soluble Aβ in intracellular compartments is toxic without fibril or aggregate formation. These data are consistent with the growing evidence that toxicity may be dissociated from protein aggregate formation in some neurodegenerative diseases (Takahashi et al., 1994; Cummings et al., 1999; Walsh et al., 2000; Shimura et al., 2001; Ma and Lindquist, 2002).
Neurodegenerative diseases, such as polyglutamine disorders, Parkinson's disease, prion diseases, and AD, appear to be triggered by the progressive intracellular accumulation of specific yet unrelated toxic proteins that target select cell populations. Different mechanisms have been proposed to explain neurodegeneration, for instance the p53, JNK, and PI3K/Akt signaling pathways are reported to be affected by intracellular Aβ (LaFerla et al., 1996; Shoji et al., 2000; Zhang et al., 2002; Suhara et al., 2003). However, increasing evidence points to a role for molecular chaperones in these neurodegenerative processes (Bonini, 2002; Sakahira et al., 2002). The activation of cellular stress pathways has been demonstrated in models of polyglutamine diseases (Chai et al., 1999; Jana et al., 2000; Manning-Bog et al., 2003). However, no clear role of Hsp on Aβ pathology in AD has been established. Our data demonstrates an upregulation of Hsp70 in response to intraneuronal Aβ42 expression in a dose-dependent manner. This finding supports previous observations of increased accumulation of Hsp70 in AD brains (Hamos et al., 1991; Perez et al., 1991). Thus, we hypothesized that an imbalance between the neuronal Hsp capacity and the toxic accumulation of Aβ may be responsible for the observed neuron death.
Our studies identify the neuroprotective role of Hsp70 in a culture model of intracellular Aβ accumulation, and provide the first evidence for the involvement of Hsp in Aβ-mediated toxicity in AD. Although we have no direct evidence of a physical interaction between Aβ42 and Hsp70, Aβ42 peptide may come in contact with Hsp70 if it is reverse translocated to the cytosol by the endoplasmic reticulum quality control system (McCracken and Brodsky, 2003). In support of this hypothesis, Hsp70 family members have been shown to interact with intracellular Aβ when expressed in the cytosolic compartment of a transgenic Caenorhabditis elegans model (Fonte et al., 2002). Because Hsp70 has roles in preventing protein aggregation and promoting protein degradation (Muchowski et al., 2000; Dul et al., 2001; Chan et al., 2002; Dou et al., 2003), it is plausible that Hsp70 is critical in the sequestration of intraneuronal Aβ. Alternatively, Hsp70 may prevent Aβ from interacting with cell survival proteins (Suhara et al., 2003).
Taken together, our findings show that the stress response in neurons is activated by intracellular Aβ42 generation, suggesting that the pathogenesis of AD is mechanistically linked to that of other neurodegenerative disorders. Interventions that further enhance the stress response may have therapeutic value in limiting the neuronal dysfunction and loss that defines these disease states.
Footnotes
This work was supported by National Institutes of Health Grants NS41371 (H.W.Q.) and AG17241, AG15052, and HL66597 (K.W.). We thank K. M. Rosen for many helpful discussions and review of this work, Mark Chafel (Optical Imaging Unit at Center for Brain Imaging, Harvard Center for Neurodegeneration and Repair) for confocal microscope assistance, and M. H. Ericsson (Harvard Medical School Electron Microscopy Facility) for immunogold electron microscopy preparation.
Correspondence should be addressed to Henry W. Querfurth, Caritas St. Elizabeth's Medical Center, Division of Neurology, 736 Cambridge Street, Boston, MA 02135. E-mail: hquerfur{at}opal.tufts.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/241700-07$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}