

Abstract
Tumor necrosis factor-α (TNF-α), a proinflammatory cytokine, is now emerging as an important modulator of the function of the CNS. Methamphetamine (METH) is a widely abused psychostimulant that causes euphoria, hyperactivity, and drug dependence. High doses of METH cause long-term neurotoxicity in dopaminergic neurons. In this study, we investigated a role of TNF-α in METH-induced dependence and neurotoxicity. Repeated treatment with METH (2 mg/kg for 5 d) in rats induced a significant increase in TNF-α mRNA and protein expression in the brain. Exogenous TNF-α (1-4 μg) blocked locomotor-stimulating and rewarding effects of METH, as well as METH (4 mg/kg; four times at 2 hr intervals)-induced dopaminergic neurotoxicity in mice. To examine a role of endogenous TNF-α in behavioral and neurochemical effects of METH, we used mice with targeted deletions of the TNF-α gene. TNF-α-(-/-) mice showed enhanced responses to the locomotor-sensitizing, rewarding, and neurotoxic effects of METH compared with wild-type mice. We also examined the role of TNF-α in METH-induced dopamine (DA) release and uptake in vitro and in vivo in C57BL/6 mice. Exogenous TNF-α (4 μg) attenuated the METH-induced increase in extracellular striatal DA in vivo and potentiated striatal DA uptake into synaptosomes in vitro and in vivo. Furthermore, TNF-α activated vesicular DA uptake by itself and diminished the METH-induced decrease in vesicular DA uptake. Our findings suggest that TNF-α plays a neuroprotective role in METH-induced drug dependence and neurotoxicity by activating plasmalemmal and vesicular DA transporter as well as inhibiting METH-induced increase in extracellular DA levels.
Introduction
Methamphetamine (METH) is a drug of abuse that has steadily gained in popularity (Anglin et al., 2000; National Institute on Drug Abuse, 2001). Repeated use of METH can cause neurological deficits that may lead to serious psychiatric and neurological signs and symptoms in users (Lan et al., 1998). Neuroimaging (Sekine et al., 2001) and postmortem studies (Wilson et al., 1996) have documented neurochemical deficits in the brains of METH abusers. In addition, it has been demonstrated that high doses of METH produces long-term neurotoxicity to dopaminergic and serotonergic nerve terminals within the neostriata, as well as to serotonergic terminals in multiple forebrain regions of rats, mice, and monkeys (Ricaurte et al., 1980; Miller and O'Callaghan, 1994). These effects of METH are associated with an increase in extracellular dopamine (DA) levels in the brain, by facilitating DA release from presynaptic nerve terminals and inhibiting reuptake (Heikkila et al., 1975; Kalivas and Stewart, 1991; Seiden et al., 1993; Giros et al., 1996). Furthermore, METH and the amphetamines redistribute DA from synaptic vesicles to the cytosol through vesicular monoamine transporter-2 (VMAT-2) (Sulzer et al., 1995).
It has recently become accepted that multidirectional communication exists among the immune, autonomic, hormonal, and central nervous systems (Maier and Watkins, 1998). Tumor necrosis factor-α (TNF-α) plays an important role in a variety of infections and inflammatory and autoimmune conditions (Vassali, 1992). TNF-α also affects the CNS directly or indirectly through stimulation of vagal afferents (Maier and Watkins, 1998). Thus, this cytokine is emerging as a modulator of CNS function. Brain TNF-α levels are typically increased in a wide range of CNS disorders, including trauma (Goodman et al., 1990), ischemia (Liu et al., 1994), and multiple sclerosis (Rieckmann et al., 1995). Although TNF-α has been suggested to be toxic to neurons (Gelbard et al., 1993; Westmoreland et al., 1996) and glia (Robbins et al., 1987), recent studies have also demonstrated neuroprotective effects of this cytokine. For instance, TNF-α prevents cell death in vitro after exposure of neurons to β-amyloid (Barger et al., 1995) and in vivo after administration of excitotoxins (Bruce et al., 1996) and cerebral ischemia (Nawashiro et al., 1997).
Recent studies have suggested a role of these cytokines in the effects of psychostimulants such as METH and amphetamine. For instance, interleukin-6 (IL-6) increases sensitivity to the locomotor-stimulating effects of amphetamine (Zalcman et al., 1999). With regard to TNF-α, it has been reported that transgenic mice expressing high levels of TNF-α in the brain showed reduced tyrosine hydroxylase (TH) immunoreactivity in the caudate-putamen without neuronal cell death, and several changes in exploratory activity and emotional behavior (Aloe and Fiore, 1997). In contrast, mice with targeted deletions of the TNF-α gene [TNF-α-(-/-) mice] show anxiogenic-like behavior accompanied by an increase in serotonin (5-HT) metabolism (Yamada et al., 2000).
In the present study, we examined the role of TNF-α in the behavioral and neurochemical effects of METH. Here, we present several lines of evidence that TNF-α plays an inhibitory role in METH dependence and neurotoxicity by modulating DA responses.
Materials and Methods
TNF-α-(-/-) mice
Male C57BL/6-TNF-α-(-/-) and C57BL/6 mice, 9-12 weeks of age, were used in the present experiment. Briefly, TNF-α-(-/-) mice derived from the TT2 ES cell line (established from C57BL/6 × CBA/JNCrj Fi blastocyte) were backcrossed to C57BL/6 more than eight generations (Taniguchi et al., 1997). Homozygous TNF-α-(-/-) mice were obtained by interbreeding of heterozygotes and confirmed by Southern blot analysis for the TNF-α allele. The wild-type C57BL/6 mice were obtained from Charles River Japan (Yokohama, Japan). The animals were housed in groups of 10 in a temperature-, humidity-, and light-controlled room (23 ± 1°C; 50 ± 5% humidity; 12 hr light/dark cycle starting at 8:00 A.M.) and had ad libitum access to food and water, except during behavioral experiments. All animal care and use was in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee of Nagoya University School of Medicine.
Reagents
Human recombinant TNF-α was kindly provided by Dainippon Pharmaceutical Co. (Osaka, Japan). TNF-α was dissolved in PBS and injected in a volume of 0.25 ml. Methamphetamine HCl (METH) was purchased from Dainippon Pharmaceutical Co. (Osaka, Japan).
Behavioral testing
Locomotor activity. Locomotor activity was measured using an infrared detector (Neuroscience, Tokyo, Japan) in a plastic box (32 × 22 × 15 cm high). C57BL/6 mice were administered TNF-α (1 or 4 μg, i.p.) or vehicle, and the locomotor activity was measured for 1 hr. METH (1 mg/kg, s.c.) was administered 1 hr after TNF-α treatment, and the locomotor activity was measured for an additional 1 hr. The 1 hr interval between the TNF-α and METH administration was chosen on the basis of a study showing that behavioral variations of TNF-α were marked at this time (Hayley et al., 1999; Brebner et al., 2000). TNF-α and METH were injected once a day for 7 d (i.e., days 1-7). After an 8 d withdrawal of METH treatment (i.e., on day 16), the mice were administered TNF-α or vehicle, followed by METH (1 mg/kg, s.c.) injection. On day 21, the mice were administered only METH (1 mg/kg, s.c.). TNF-α-(-/-) mice were administered METH (1 mg/kg, s.c.) as described above.
Place conditioning paradigm. The apparatus used for the place-conditioning task consisted of two compartments: a black Plexiglas box and a transparent Plexiglas box (both 15 × 15 × 15 cm high). To enable mice to distinguish easily the transparent box from the black one, the floor of the transparent and black boxes were covered with white plastic mesh and black frosting Plexiglas, respectively. Each box could be divided by a sliding door (10 × 15 cm high). The place-conditioning paradigm was performed according to the method of Noda et al. (1998). In the preconditioning test, the sliding door was opened, and the mouse was allowed to move freely between both boxes for 15 min for 3 d. On the third day of the preconditioning test, we measured pre-value. Conditioning was performed during 6 successive days. On days 4, 6, and 8, a mouse was given METH and put in its nonpreferred side for 20 min. On days 5, 7, and 9, the mouse was given saline and placed opposite the drug-conditioning side. In the postconditioning test, the sliding door was opened, and we measured post-value. Place-conditioning behavior was expressed as post-value minus pre-value.
Microinjection of TNF-α into the nucleus accumbens
C57BL/6 mice were anesthetized with sodium pentobarbital and placed in a stereotaxic apparatus. A pair of metal guide cannulas were implanted bilaterally and stereotaxically into the nucleus accumbens (+1.1 mm anteroposterior, +0.8 mm mediolateral from bregma, and -3.0 mm dorsoventral to dura) (Franklin and Paxinos, 1997), and mice were allowed to recover for 14 d. On day 14, an infusion cannula was inserted through the guide cannula until they protruded 1 mm beyond the respective inner ends. Mice received microinjections at a rate of 0.25 μl/min for 2 min (final volume, 0.5 μl) with either vehicle or 10 ng of TNF-α. METH (1 mg/kg, s.c.) was administered 5 min after microinfusions, and then locomotor activity was measured for 1 hr as described above. Determination of the location of the infusion cannula placements was assessed at the completion of the experiments. Brains were removed by decapitation and cut into coronal sections on ice. Infusion cannula placements were examined to verify that their location was within the nucleus accumbens.
METH discrimination
Male Sprague Dawley rats (7 weeks old; Charles River Japan) weighing 230 ± 10 gm at the beginning of experiments were used in the study. The body weights were reduced gradually to ∼80% of free-feeding weight by limiting daily access to food. Water was available ad libitum. Experiments were conducted in a standard operant-conditioning chamber (Neuroscience) set in ventilated and sound-attenuated box. The chambers were equipped with two response levers, spaced 16 cm apart, with a food pellet trough mounted midway between levers. A houselight was located over the trough. Reinforcement consisted of a 45 mg food pellet (Bio Serv, Frenchtown, NJ). Scheduling of reinforcement contingencies, reinforcement delivery, and data recording were controlled by a computer system. Rats were trained initially to press each of the two levers under a fixed ratio (FR)-1 schedule of food reinforcement. The FR response requirement for food delivery was gradually increased from 1 to 20. After response under the FR-20 schedule of food reinforcement had stabilized, drug discrimination training was begun (Mori et al., 2001). Rats received injections 10 min before the session with either saline or METH (0.5 mg/kg, s.c.). In drug discrimination training sessions, METH or saline was administered randomly to ensure that no olfactory cues associated with the two levers would bias the discrimination. After administration of METH, 20 consecutive responses (FR-20) on one lever produced a food pellet, whereas after saline administration, 20 consecutive responses on the other lever produced a food pellet. Responding on the incorrect lever reset the FR requirement for the correct lever. For half the rats, the right lever was the drug lever, and for the other half, the left lever was the drug lever. Each session ended after 20 food pellets were delivered or 20 min had elapsed. The criteria for learning the discrimination were five consecutive sessions with (1) >85% correct-lever responding before the first reinforcement, and (2) >90% correct-lever responding throughout the session. Discrimination training sessions were conducted 6 d per week under a double alternation schedule (i.e., MMSSMMSS, etc.; M = METH, S = saline). Once the rats reliably discriminated METH from saline, dose-response effects of METH were tested. Test sessions were conducted once per week, and the rats that fulfilled the criteria in a training session for three consecutive training sessions were used. Test sessions were identical to training sessions, except that 20 consecutive responses on either lever resulted in delivery of a food pellet. Lever selection was examined after the administration of various doses of METH (0.1-0.5 mg/kg). To examine the effects of TNF-α on the discriminative stimulus effects of METH and the discrimination-induced Fos expression, rats received injections of TNF-α (25-100 μg/kg) or vehicle 1 hr before METH (0.2 mg/kg) treatment and then were subjected to the drug discrimination test.
Fos immunohistochemistry
Five rats received injections of TNF-α (25 μg/kg), whereas the others (n = 4) received injections of the vehicle before METH treatment, and all animals were examined in the METH (0.2 mg/kg) discrimination test. Two hours after METH discrimination test, rats were deeply anesthetized with pentobarbital (50 mg/kg) and perfused transcardially with ice-cold saline, followed by 4% paraformaldehyde in phosphate buffer. The brains were removed, postfixed in the same fixative for 2 hr, and then cryoprotected in 30% sucrose in phosphate buffer. The brains were cut into 50 μm coronal sections on a cryostat, and free-floating sections were used for Fos immunohistochemistry (He et al., 2002). The sections were incubated with 5% goat serum and 0.3% Triton X-100 in 0.1 m phosphate buffer and then incubated with rabbit anti-Fos antibody (1:2000; sc-52; Santa Cruz Biotechnology, Santa Cruz, CA) for 48 hr at 4°C with constant rotation. Sections were then washed with phosphate buffer containing 0.3% Triton X-100 and incubated with biotinylated goat anti-rabbit antibody at room temperature for 2 hr. Sections were washed and processed with avidin-biotinylated HRP complex (Vector ABC kit; Vector Laboratories, Burlingame, CA), and the reaction was visualized using DAB. To quantify the number of Fos-stained cells in the brain, we examined the sections, blind to the animal's treatment, with a computer-assisted image analysis system (C. Imaging Systems; Compix, Mars, PA) attached to a light microscope (Olympus BX60-FLB-3; Olympus, Tokyo, Japan), as described previously (He et al., 2002). Both right and left hemispheres of four sequential sections for selected brain area, located according to the atlas of Paxinos and Watson (1982), were examined for the counting of Fos-positive cells. Selected brain areas were: the core (0.31 mm2) and shell (0.31 mm2) of the nucleus accumbens in sections at a level of +1.60 mm from bregma; ventral tegmental area (0.31 mm2) in sections at a level of -5.30 from bregma.
Double immunostaining for TNF-α and microtuble-associated protein-2
Male Wistar rats (Charles River Japan) were killed 4 hr after repeated administration of METH (2 mg/kg, s.c., for 5 d). The brains were sectioned at 20 μm in a cryostat. Sections were rinsed briefly in PBS containing 0.01% Triton X-100, blocked for 30 min at room temperature using 5% rabbit serum containing 0.01% Triton, and incubated with primary antibodies overnight at 4°C. After washing, sections were incubated for 30 min with secondary antibodies. Polyclonal goat anti-TNF-α antibody (1:100; Santa Cruz Biotechnology) and monoclonal mouse anti-microtuble-associated protein-2 antibody (1:200; Upstate Biotechnology, Lake Placid, NY) served as primary antibodies. Affinity-purified fluorescein (FITC)-conjugated rabbit anti-mouse IgG and rhodamine-conjugated donkey anti-goat IgG served as secondary antibodies.
Monoamine levels
TNF-α-(-/-) mice and the wild-type mice were killed by focused microwave irradiation for 1.5 sec at 5 kW. The brains were removed and dissected according to the method of Yamada et al. (2000). The contents of DA and 5-HT were determined using an HPLC system with an electrochemical detector (Eicom, Kyoto, Japan), as described by Miyamoto et al. (2001).
Synaptosomal [3H]DA uptake
Striatal synaptosomal [3H]DA uptake was determined as described by Fleckenstein et al. (1997). Striatal tissue was homogenized in ice-cold 0.32 m sucrose and centrifuged at 1000 × g for 10 min at 4°C. The supernatant fractions were removed and centrifuged at 22,000 × g for 15 min. The resulting pellet was resuspended in an ice-cold modified Krebs'-Ringer's solution composed of (in mm) 125 NaCl, 4.8 KCl, 25 NaHCO3, 1.2 KH2PO4, 1.3 MgCl2, 1.2 CaCl2, 10 glucose, and 0.57 ascorbic acid gassed with 95% O2 and 5% CO2. Each assay tube contained 1 μm pargyline (Sigma). After incubation for 10 min at 37°C, assays were initiated by the addition of [3H]DA (60.0 Ci/mmol; NEN Life Science Products, Boston, MA). Samples were incubated at 37°C for 4 min, and then ice-cold Krebs'-Ringer's solution containing 10 μm GBR12909, a specific DA uptake inhibitor, was added. Samples were filtered through Whatman GF/B filters (Brandel, Gaithersburg, MD). Nonspecific values were determined in the presence of 100 μm GBR 12909. The radioactivity trapped on filters was measured using a liquid scintillation counter. Protein levels were determined using the Dc Protein Assay kit (Bio-Rad, Hercules, CA).
Vesicular [3H]DA uptake
Vesicular [3H]DA uptake was determined as described by Erickson et al. (1990). Synaptosomes were prepared as described above. Synaptosomes were then resuspended and homogenized in cold distilled deionized water. Osmolarity was restored by the addition of HEPES and potassium tartrate (final concentration of 25 and 100 mm, respectively; pH 7.0). Samples were centrifuged for 20 min at 20,000 × g (4°C). MgSO4 (1 mm final concentration) was added to the supernatant, which was then centrifuged for 45 min at 100,000 × g (4°C). The resulting pellet was gently resuspended in the assay buffer immediately before use. Based on published reports using similar protocols for vesicle preparation (Kadota and Kadota, 1973; Teng et al., 1997; Sandoval et al., 2002), we believe vesicles isolated in these studies to be of the small synaptic vesicle size, the predominant type found in dopaminergic terminals in the striatum (Nirenberg et al., 1997). Vesicular [3H]DA uptake was performed by incubating 100 μl (8-10 μg of protein) of synaptic vesicle samples at 30°C for 4 min in assay buffer (final concentration (in mm): 25 HEPES, 100 potassium tartrate, 1.7 ascorbic acid, 0.05 EGTA, 0.1 EDTA, and 2 ATP-Mg2+, pH 7.0) in the presence of [3H]DA (30 nm final concentration, except in the kinetic analyses in which 0.03-1 μm [3H]DA was used). The reaction was terminated by the addition of 1 ml of cold wash buffer (assay buffer containing 2 mm MgSO4 substituted for the ATP-Mg2+, pH 7.0) and rapid filtration. Filters were washed three times with cold wash buffer. Nonspecific values were determined by measuring vesicular [3H]DA uptake at 4°C.
Striatal microdialysis
C57BL/6 mice were anesthetized with sodium pentobarbital before stereotaxic implantation of a probe into the left striatum (+0.0 mm anteroposterior, +2.5 mm mediolateral from bregma, and -4.4 mm dorsoventral to dura). Probes were secured to the skull using stainless steel screws and dental acrylic and were perfused continuously with artificial CSF (147 mm NaCl, 4 mm KCl, and 2.3 mm CaCl2) at a rate of 2 μl/min. Dialysate was collected in 20 min fractions and injected into the HPLC system for measurement of DA levels. Three samples were taken to establish baseline levels of extracellular DA before the administration of TNF-α and METH.
Western blotting
TH protein levels were determined by the method of Mizuno et al. (2003). The striatum was dissected out on ice-cold glass plate, according to the method of Yamada et al. (2000), and was lysed at 4°C in a lysis buffer composed of 50 mm Tris-HCl, 150 mm NaCl, 10 mm EDTA, 1% Triton X-100, 1 mm sodium orthovanadate, 10 mm sodium diphosphate decahydrate, 0.5 mm DTT, 0.2 mm PMSF, 4 μg/ml pepstatin, 4 μg/ml aprotinin, and 4 μg/ml leupeptin, pH 7.4. For analysis of TH levels, 50 μg of protein was boiled in a sample buffer [0.25% bromophenol blue/0.25% xylene cyanol/30% glycerol/20% 2× Tris-borate EDTA (90 mm Tris/64.6 mm boric acid/2.5 mm EDTA, pH 8.4)] and a 7.5% polyacrylamide gel and subsequently transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA) and blocked with a Detector Block kit (KPL, Gaithersburg, MD). Membranes were incubated with a goat anti-TH antibody (1:1000; Santa Cruz Biotechnology). The membranes were then washed with TBST (10 mm Tris-HCl, pH 7.4, and 150 mm NaCl, 0.1% Tween 20) three times for 10 min each. After incubation with a 1:1000 dilution of HRP-conjugated IgG for 2 hr, membranes were washed with TBST three times for 10 min each. The immune complex was detected by a chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ) and exposed to x-ray film. The band intensities on the film were analyzed by densitometry.
Quantative mRNA analysis by real-time PCR
Male Wistar rats (Charles River Japan) were used in this study. Total RNA was extracted by a method described previously (Mizuno et al., 2000). cDNA was synthesized from total RNA by a reverse transcriptase reaction using oligo(dT) 12-18 primer (Life Technologies, Tokyo, Japan). Levels of TNF-α and TNF receptor-1 (TNFR-1) mRNA were determined after reverse transcription by real-time PCR using an ABI PRISM 7700 Sequence Detection System (Perkin-Elmer). The TaqMan Pre-Developed Assay Reagent kit for rat TNF-α and TNFR-1 (Perkin-Elmer) was used. The expression levels were calculated as described previously (Wada et al., 2000).
Enzyme-linked immunoadsorbent assay
Male Wistar rats (Charles River Japan) were killed 4 hr after repeated administration of METH (2 mg/kg, s.c., for 5 d). Tissue was homogenated for 1 min in 10 mm HEPES-KOH, pH 7.9, buffer containing 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT, 1.0 mm PMSF, 10 mm leupeptin, 10 mm pepstatin, 10 mm aprotinin, and 0.1% Nonidet P-40. Homogenates were centrifuged at 12,000 × g for 4 min at 4°C. The resulting supernatants were removed. TNF-α levels were assessed in duplicate 50 μl aliquots using a specific ELISA kit (Bio Track RPN2734; Amersham Biosciences).
METH concentration in plasma and brain tissues
The concentrations of METH were determined using the method of Hayakawa et al. (1989). Fifty microliters of plasma sample or homogenated brain tissue were vortexed adding 350 μl of acetonitrile containing β-phenyletylamine (PEA; 0.5 μg/ml) as an internal standard, 50 μl of saturated salt, and 10 μl of 10% NaOH. After deproteinization by centrifugation at 12,000 × g for 5 min, the top acetonitrile-rich layer was collected and evaporated to dryness under a nitrogen gas at 45°C. The dried residues were reconstituted with 100 μl of 10 mm sodium carbonate-sodium bicarbonate buffer, pH 9.0, and 100 μl of 2 mm dansyl (DNS) chrolide. Samples were heated at 45°C for 1 hr in the dark to derive DNS-METH and DNS-PEA from METH and PEA, respectively. Samples were analyzed by HPLC with fluorescence detection (emission, 580 nm; excitation, 475 nm).
Statistical analysis
All data were expressed as means ± SEM. Statistical differences between two groups were determined with Student's t test. Statistical differences among more than three groups were determined by a one-way ANOVA, followed by the Student-Newmann-Keuls multiple comparison test. p < 0.05 was regarded as statistically significant.
Results
METH alters the expression of TNF-α and its receptor TNFR-1
As an initial step in assessing the relationship between TNF-α and METH, we examined whether acute or chronic METH exposure altered the expression of TNF-α or its receptor. The function of TNF-α is mediated through two distinct cell surface receptors, TNFR-1 and TNFR-2. The majority of TNF functions are mediated primarily through TNFR-1, whereas TNFR-2 seems to play a role in only a limited number of TNF responses (Hsu et al., 1995). It has been reported that immunoreactivity for TNFR-1 was found in cell bodies and processes of dopaminergic neurons (Boka et al., 1994).
We measured mRNA levels of TNF-α and TNFR-1 in the frontal cortex, nucleus accumbens, striatum, hippocampus, ventral tegmental area, and amygdala in rats by a real-time reverse transcription-PCR method. Although acute METH treatment at a dose of 2 mg/kg did not change TNF-α gene expression in any regions examined, TNF-α mRNA levels were elevated by 400-800% in the nucleus accumbens, striatum, hippocampus, and amygdala after repeated daily treatment with METH (2 mg/kg for 5 d) (Fig. 1A). Because it is well known that DA is involved primarily in the pharmacological effects of METH, we examined whether METH-induced increase in TNF-α mRNA levels is mediated by the activation of DA neurotransmission. The increase in TNF-α mRNA expression in the nucleus accumbens was inhibited by pretreatment with either the DA D1 receptor antagonist SCH23390 or the D2 receptor antagonist raclopride (Fig. 1C), suggesting that the activation of both DA D1 and D2 receptors are attributable to METH-induced gene expression of TNF-α. In contrast, TNFR-1 gene expression was decreased to 15-20% of control in the ventral tegmental area and amygdala after acute METH treatment, although repeated treatment did not affect TNFR-1 mRNA levels (Fig. 1B).

Changes in the expression of TNF-α and TNFR-1 after METH treatment. TNF-α (A) and TNFR-1 (B) mRNA expression in the brain after METH treatment. Rats were killed 2 hr after a single or repeated METH treatment (2 mg/kg, s.c., for 5 d). Values are means ± SEM (n = 7). *p < 0.01 versus control; #p < 0.01 versus acute treatment. C, The effects of the DA D1 receptor antagonist SCH23390 and D2 receptor antagonist raclopride on METH-induced TNF-α mRNA expression in the nucleus accumbens. SCH23390 (0.5 mg/kg) or raclopride (2 mg/kg) was administered intraperitoneally 30 min before daily METH (2 mg/kg) treatment for 5 d. Values are means ± SEM (n = 6). *p < 0.05 versus control; #p < 0.05 versus METH alone. D, TNF-α protein levels in the nucleus accumbens after repeated METH treatment. Values are means ± SEM (n = 6). *p < 0.05 versus control. E, TNF-α immunostaining in the nucleus accumbens after repeated METH treatment. Scale bar, 50 μm. F, Double immunostaining for TNF-α and microtuble-associated protein-2 in the nucleus accumbens reveals TNF-α expression in neurons. Scale bar, 20 μm.
We confirmed that the protein level of TNF-α was increased in the nucleus accumbens after repeated METH treatment by ELISA and immunocytochemistry (Fig. 1D,E). Double immunostaining for TNF-α and microtuble-associated protein-2, a neuronal marker, revealed the neuronal localization of TNF-α (Fig. 1F). These results suggest that METH induces TNF-α mRNA and protein expression in the nucleus accumbens, a brain area closely associated with drug dependence (Pierce and Kalivas, 1997), through the activation of both DA D1 and D2 receptors.
Role of TNF-α in METH-induced hyperlocomotion and sensitization in mice
We studied the role of TNF-α in behavioral responses to METH. At first, we examined the effect of systemic administration of exogenous TNF-α on METH-induced hyperactivity and sensitization. Exogenous TNF-α (1 or 4 μg, i.p.) did not affect spontaneous locomotor activity in mice (data not shown). As shown in Figure 2A, acute METH treatment in mice at a dose of 1 mg/kg caused a marked increase in locomotor activity, and the repeated administration for 7 d resulted in an enhancement of locomotor-stimulating effect of METH (sensitization) (F(6,13) = 8.9977; p < 0.0001; repeated ANOVA). This sensitization was observed on days 16 and 21 after challenge administration of METH (1 mg/kg) (F(2,39) = 27.748; p < 0.01; one-way ANOVA). TNF-α dose-dependently inhibited acute METH-induced hyperlocomotion on day 1 (Fig. 2A) (F(2,30) = 5.9048; p < 0.01; one-way ANOVA). A low dose of TNF-α (1 μg/mouse, i.p.) inhibited the locomotor-stimulating effect of METH on days 1-3 but did not affect it on days 4-7. A high dose of TNF-α (4 μg/mouse, i.p.) inhibited METH-induced hyperactivity on days 1-7 (Fig. 2A). From these results, it raises a possibility that TNF-α may inhibit both acute locomotor-stimulating effect of METH and the development of sensitization induced by repeated administration of METH. To test this possibility, we assessed the locomotor-stimulating effect of METH in the sensitized mice in the presence (day 16) or absence (day 21) of TNF-α pretreatment. On day 16, TNF-α, both at 1 and 4 μg, inhibited METH-induced hyperlocomotion in the sensitized mice (F(2,30) = 9.4667; p < 0.05; one-way ANOVA). On day 21, METH-induced locomotor activity was significantly lower in chronic (METH and 4 μg of TNF-α) treated mice than that in the vehicle-treated control animals, although TNF-α was absent (Fig. 2A) (F(2,30) = 9.4149; p < 0.05; one-way ANOVA). Taken together, these results suggest that systemic administration of TNF-α not only inhibits the locomotor-stimulating effect of METH but also the development of sensitization induced by repeated administration of METH.

Role of TNF-α in METH-induced hyperactivity and sensitization. A, Effects of exogenous TNF-α on the sensitization of METH-induced hyperactivity in wild-type mice. METH (1 mg/kg, s.c.) was administered repeatedly once per day 1 hr after TNF-α (1 or 4 μg, i.p.) treatment. Values are means ± SEM (n = 9-14). Locomotor activity at several individual time points are inhibited by TNF-α treatment (*p < 0.05 vs vehicle-treated group; one-way ANOVA). B, Locomotor activity in response to acute METH treatment after microinjection of TNF-α into the nucleus accumbens. Vehicle or TNF-α (10 ng/site) was administered 5 min before METH (1 mg/kg) treatment. Values are means ± SEM (n = 6-9). *p < 0.05 versus respective control group; #p < 0.05 versus vehicle+METH group. C, The location of the cannula placements within the nucleus accumbens are illustrated in diagrams of coronal sections. Circles indicate the location and diffusion of TNF-α. All drawings are based on the atlas of Franklin and Paxinos (1997). D, Sensitization of METH-induced hyperactivity in TNF-α-(-/-) mice. METH (1 mg/kg, s.c.) was administered repeatedly once per day, and then locomotor activity was measured for 1 hr. Values are means ± SEM (n = 7-8). An ANOVA with repeated measures revealed a significant difference in locomotor activity (strain × days interaction: F(1,13) = 4.077; p = 0.003). Locomotor activity on days 7, 9, and 16 was also augmented in TNF-α-(-/-) mice. *p < 0.05 versus wild-type mice.
To examine whether TNF-α directly affects brain function and METH-induced hyperlocomotion and sensitization, we investigated the effect of microinjection of TNF-α into the nucleus accumbens, because it is well known that this brain region plays an important role in METH-induced hyperlocomotion (Pierce and Kalivas, 1997). TNF-α (10 ng/site) was microinjected bilaterally into the nucleus accumbens of mice 5 min before a single injection of METH, which elicited a significant increase in locomotor activity. METH-induced hyperlocomotion was markedly inhibited by the treatment (Fig. 2B). Mice receiving TNF-α alone showed no alteration in spontaneous locomotor activity compared with vehicle-injected control mice (Fig. 2B). Histological analyses revealed that all animals had evidence of cannula placements and infusion tracts that were within the nucleus accumbens as depicted in Figure 2C. The cannula placements within the nucleus accumbens were localized medial to the anterior commissure in the posteromedial region of the nucleus accumbens. The infusion sites ranged approximately between 0.6 and 1.0 mm mediolateral and between 0.9 and 1.3 mm anteroposterior. Damage to the overlying regions appeared to be minimal. These results support the hypothesis that TNF-α inhibits the locomotor-stimulating effect of METH, at least in part, through the action in the nucleus accumbens. Our data, however, does not exclude a possibility that other brain areas are involved in the effect of TNF-α.
We next examined the role of endogenous TNF-α in METH-induced hyperlocomotion and the development of sensitization induced by repeated METH treatment in TNF-α-(-/-) mice. TNF-α-(-/-) mice showed a decreased locomotor response when first placed in the locomotor chamber, compared with wild-type mice, but an equivalent level of locomotor activity after a 2 d habituation period (data not shown). After a 4 d habituation, mice received injections of METH (1 mg/kg), and the locomotor response was measured. As shown in Figure 2D, wild-type and TNF-α-(-/-) mice showed an equivalent locomotor activity in response to the first injection of METH. However, TNF-α-(-/-) mice showed a significantly greater increase in locomotor activity after repeated exposure to METH (sensitization), compared with wild-type mice (F(1,13) = 4.077; p < 0.01; repeated ANOVA). The alterations of locomotor response to METH in TNF-α-(-/-) mice are consistent with the changes in TNF-α gene expression after METH treatment: single METH treatment had no effect, but repeated METH treatment resulted in an increase in the gene expression.
Although the magnitude of the sensitized response after repeated METH treatment in TNF-α-(-/-) mice was significantly different from the level in wild-type mice, TNF-α-(-/-) mice exhibited an initial locomotor response to acute METH, a pattern of initiation of stimulant-induced sensitization, and a pattern of expression of sensitization, identical to wild-type mice. Thus, one might argue a role of endogenous TNF-α in acute METH-induced hyperlocomotion and development of the sensitization. Rather, endogenous TNF-α may play a modulatory role in the expression of the sensitized locomotor-stimulating effect of METH.
Role of TNF-α in the rewarding effects of METH in mice
The role of TNF-α in the rewarding effects of METH was examined in a place-conditioning paradigm, in which animals learned to associate an environment paired with drug exposure. Place conditioning is, therefore, considered a measure of the rewarding properties of drugs of abuse. As shown in Figure 3A, METH (2 mg/kg, s.c.) significantly produced place preference in C57BL/6 mice. When TNF-α was pretreated 1 hr before METH treatment, METH-induced place preference was attenuated in a dose-dependent manner (Fig. 3A) (F(5,85) = 2.6752; p < 0.05; one-way ANOVA).

Role of TNF-α in the rewarding effects of METH. A, Effects of exogenous TNF-α on METH-induced place preference in wild-type mice. C57BL/6 mice were trained for conditioned place preference to METH (2 mg/kg, s.c.). TNF-α (1 or 4 μg, i.p.) was administered 1 hr before METH treatment. Values are means ± SEM (n = 9-20). *p < 0.05 versus saline-treated mice; #p < 0.05 versus METH-treated mice. B, Facilitated conditioned place preference in TNF-α-(-/-) mice treated with METH (1 mg/kg, s.c.). Wild-type and TNF-α-(-/-) mice were trained for conditioned place preference to METH (1 mg/kg, s.c.). Values are means ± SEM (n = 10-14). *p < 0.05 versus saline-treated TNF-α-(-/-) mice; #p < 0.05 versus METH-treated wild-type mice.
The role of endogenous TNF-α in the rewarding effects of METH was also examined in TNF-α-(-/-) mice. As shown in Figure 3B, although a low dose of METH (1 mg/kg) did not establish place preference in wild-type mice, TNF-α-(-/-) mice developed place conditioning at the same dose of METH (F(3,47) = 4.1435; p < 0.05; one-way ANOVA). Both types of mice developed place conditioning at a higher dose of METH (4 mg/kg) (data not shown). These results suggest that TNF-α acts to negate the rewarding effects of METH.
Effect of TNF-α on discriminative stimulus effects of METH and the associated Fos expression in rats
The discriminative stimulus effects of psychostimulants in experimental animals are related to their subjective effects in humans (Schuster and Johanson, 1988; Kamien et al., 1993). Therefore, the drug discrimination procedure has been used to elucidate the mechanism of action underlying the unique properties of addictive drugs such as amphetamines and cocaine (Callahan et al., 1997; Munzar and Goldberg, 2000). We, therefore, investigated the effect of TNF-α on discriminative stimulus effects of METH and the associated Fos expression in rats trained to discriminate METH from saline under a two-lever FR-20 schedule of food reinforcement. Quantification of the changes in the expression of the immediate early gene c-fos has proven to be a very useful method by which the distribution of neurons that are activated by physiological and pharmacological stimuli may be mapped (Sagar et al., 1988; Morgan and Curran, 1991). Immunohistochemistry has indicated that METH produces Fos-like immunoreactivity in a wide variety of brain area of the rat and that it dose-dependently increases the density of Fos-positive cells (Umino et al., 1995).
Rats reliably discriminated METH from saline after an average of 60 training sessions (range, 45-70). Once the training criterion was reached, METH-saline discrimination stabilized and was maintained with a high degree of accuracy (>95%) in all the subjects for the remainder of the investigation. In the dose-response test, METH produced a dose-related increase in METH-appropriate responding, whereas the response rate was stable at the doses examined (Fig. 4A). Rats that received injections of TNF-α (25-100 μg/kg) 1 hr before METH (0.2 mg/kg) treatment showed a decrease in METH-appropriate responding without any changes in the response rate (Fig. 4B), suggesting that the inhibitory effect of TNF-α on the discriminative stimulus effect of METH is not attributable to the motor impairment. In contrast, saline-appropriate responding was not affected by the pretreatment with TNF-α (50 μg/kg) (data not shown). These findings indicate that TNF-α specifically attenuates the discriminative stimulus effects of METH in rats. Next, we attempted to clarify the brain sites of action of TNF-α. For this end, we examined the effect of TNF-α on METH discrimination-associated Fos expression in the brain. It has been suggested that DA D1 and D2 receptors in the mesocorticolimbic system are involved in modulating the discriminative stimulus properties of psychostimulants (Callahan et al., 1997). We observed that Fos expression in the rats that can discriminate METH from saline was significantly increased in the nucleus accumbens and the ventral tegmental area but not in other brain areas including the cerebral cortex, caudate putamen, substantia nigra, hippocampus, amygdala, and habenulla, compared with the expression in control animals that were maintained under the FR-20 schedule (A. Nakajima, K. Yamada, J. He, N. Zeng, A. Nitta, T. Nabeshima, manuscript in preparation). Thus, we chose the nucleus accumbens and ventral tegmental area for Fos staining in this study. Representative photomicrographs of Fos staining in the nucleus accumbens core and shell are shown in Figure 4C, and summaries of Fos expression are shown in Figure 4D. The number of Fos-positive cells in the nucleus accumbens core and shell of TNF-α-pretreated rats was significantly lower than that of vehicle-pretreated rats (p < 0.05), whereas no alteration of Fos expression was observed in the ventral tegmental area (Fig. 4D), suggesting that the inhibitory effect of TNF-α on discriminative stimulus effects of METH is related to the modulation of neural activity in the nucleus accumbens.

Effects of TNF-α on the discriminative stimulus effects of METH. A, Dose-dependent discriminative stimulus effects of METH in rats. Values are means ± SEM (n = 9). B, Effects of TNF-α on discriminative stimulus effects of METH. Rats received injections of TNF-α (25-100 μg/kg) or vehicle 1 hr before METH (0.2 mg/kg) treatment and then were subjected to the drug discrimination test. Values are means ± SEM (n = 9). *p < 0.05 versus vehicle treatment. C, Representative photomicrographs of Fos immunostaining of the nucleus accumbens core and shell in rats subjected to the drug discrimination test for METH. aca, Anterior commissure anterior part; mfba, medial forebrain bundle. Scale bar, 100 μm. D, Summaries of Fos expression in the core and shell of the nucleus accumbens and ventral tegmental area. Values are means ± SEM (n = 4-5). *p < 0.05 versus vehicle treatment.
Effects of TNF-α on neurotoxicity of high doses of METH in mice
Administration of high doses of METH has been shown to result in damage to DA terminals and 5-HT terminals, and perhaps in cell loss, in the brains of rodents and nonhuman primates (Ricaurte et al., 1980). The evidence for DA axon terminal damage includes a long-term decrease in DA content, the depletion of DA uptake sites, and a decrease in TH levels (Hayashi et al., 2001). We examined the effects of TNF-α on the METH-induced depletion of DA and 5-HT in C57BL/6 mice. A neurotoxic dose of METH (4 mg/kg, s.c.; four times at 2 hr intervals) caused a marked depletion in DA contents in the frontal cortex (-45%) and striatum (-55%), but not the hippocampus (-6%), 3 d after METH treatment (Fig. 5A). In contrast, consistent with a previous report (Kita et al., 1998), this dose regimen of METH did not affect 5-HT levels (data not shown), suggesting that this dose regimen did not induce serotonergic neurotoxicity in C57BL/6 mice. Pretreatment with TNF-α (4 μg, i.p.) 1 hr before the first and third injections of METH completely blocked METH-induced depletions of DA in the frontal cortex and striatum (Fig. 5A). To confirm the protective effect of TNF-α against METH neurotoxicity, we also examined TH protein levels by Western blotting in mice killed 3 d after METH treatment. METH caused ∼70% loss of TH protein in the striatum (Fig. 5B). Pretreatment with TNF-α provided almost complete protection against the loss of striatal TH protein (Fig. 5B). These results are parallel to those obtained for DA contents, as reported above. Body temperature has been found to influence METH-induced DA neurotoxicity. In particular, hyperthermia consistently exacerbates METH-induced DA neurotoxicity, whereas hypothermia is neuroprotective (Bowyer et al., 1994). As shown in Figure 5D, METH (4 mg/kg, s.c.; four times at 2 hr intervals) significantly increased the rectal temperature. TNF-α (4 μg, i.p.) by itself did not affect rectal temperature (data not shown). However, when mice were given TNF-α 1 hr before the first and third injections of METH, rectal temperatures decreased significantly, but temporally, compared with mice treated with METH alone (Fig. 5D).
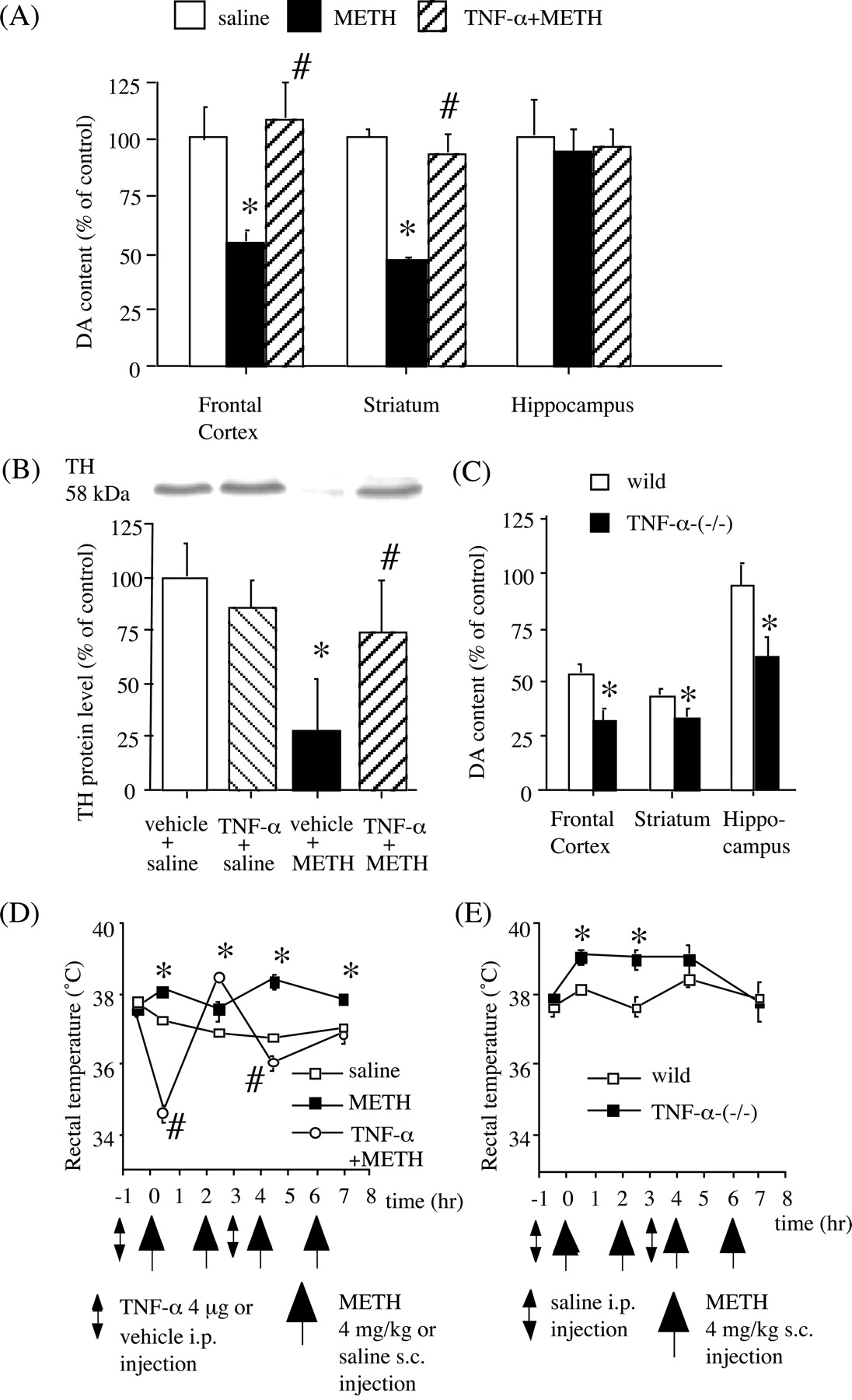
Role of TNF-α in METH-induced neurotoxicity. Effects of TNF-α on METH-induced depletion of DA (A) and striatal TH (B) protein levels in wild-type mice. METH (4 mg/kg, s.c.) was administered four times at 2 hr intervals in C57BL/6 mice. Vehicle or TNF-α (4 μg, i.p.) was administered 1 hr before the first and third injections of METH. DA content and TH protein levels were measured 3 d after METH treatment. Values are means ± SEM (n = 8-9). *p < 0.05 versus saline-treated group; #p < 0.05 versus METH-treated group. C, METH-induced DA depletion in TNF-α-(-/-) mice. METH (4 mg/kg, s.c.) was administered four times at 2 hr intervals in wild-type and TNF-α-(-/-) mice. DA content was measured 3 d after METH treatment. Values are means ± SEM (n = 7-9). *p < 0.05 versus wild-type mice. METH-induced hyperthermia in wild-type (D) and TNF-α-(-/-) (E) mice. METH (4 mg/kg, s.c.) was administered four times at 2 hr intervals. The recorded ambient temperature was 21°C. D, Effects of TNF-α on METH-induced changes in rectal temperature in wild-type mice. Vehicle or TNF-α (4 μg, i.p.) was administered 1 hr before the first and third injections of METH. Values are means ± SEM (n = 8-9). *p < 0.05 versus vehicle+saline-treated group; #p < 0.05 versus vehicle+METH-treated group. E, Effects of METH on rectal temperature in TNF-α-(-/-) mice. Values are means ± SEM (n = 7-9). *p < 0.05 versus wild-type mice.
We also examined the role of endogenous TNF-α in METH-induced neurotoxicity in TNF-α-(-/-) mice. As shown in Figure 5C, multiple injections of METH produced a 46 and 54% decrease in DA levels in the frontal cortex and striatum, respectively, in wild-type mice. METH caused a greater reduction (-68 and -66% in the frontal cortex and striatum, respectively) in TNF-α-(-/-) mice. In the hippocampus, no significant reduction in DA levels was observed in wild-type mice, whereas DA levels were significantly decreased by 34% in TNF-α-(-/-) mice (Fig. 5C). TNF-α-(-/-) mice showed significantly higher rectal temperatures than wild-type mice after the first and second METH treatment (Fig. 5E), whereas there was no difference in rectal temperature between the two groups when saline was administered (data not shown).
Blood and brain concentrations of METH
To exclude a possibility that the inhibitory effects of TNF-α on the behavioral effects of METH are attributable to the changes in pharmacokinetics of METH, blood and brain concentrations of METH were determined by HPLC in wild-type and TNF-α-(-/-) mice after METH treatment. There were no differences in levels of METH in plasma and brain between the two genotypes at any time points and doses examined, suggesting no change in the pharmacokinetics of METH in TNF-α-(-/-) mice (Table 1). We also examined whether pretreatment with exogenous TNF-α influenced blood and brain concentrations of METH in C57BL/6 mice. No differences were observed between the saline- and TNF-α-pretreated groups in the levels of METH in plasma and brain (Table 1). These results indicate that the inhibitory effects of exogenous TNF-α on the neuropsychopharmacological effects of METH are not attributable to the changes in the pharmacokinetics of METH.
METH levels in plasma and brain in wild-type and TNF-α-(−/−) mice
Effects of TNF-α on METH-induced DA responses
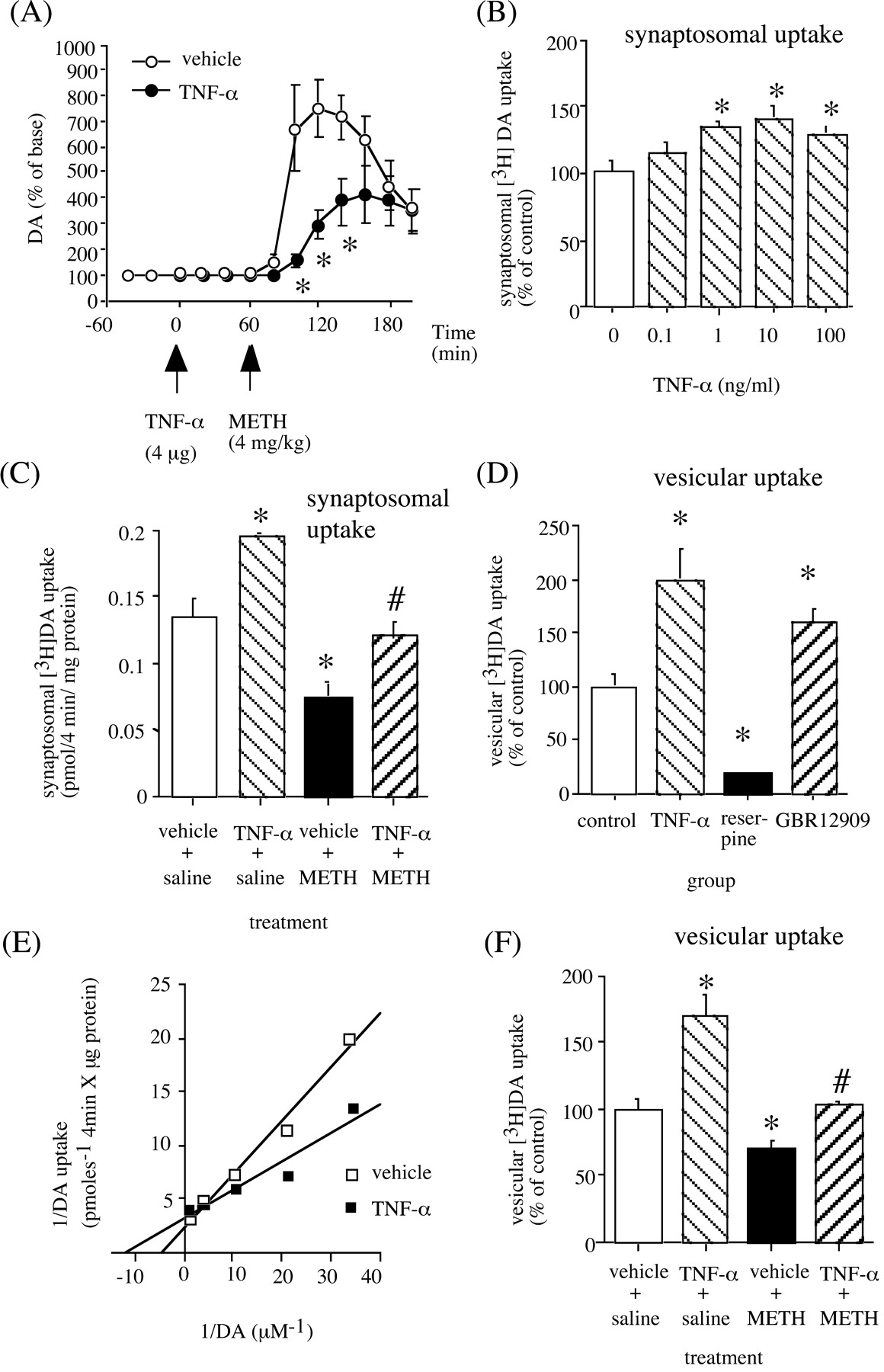
The pharmacological effects of METH are linked to its capacity to elevate extracellular DA levels by releasing DA from presynaptic nerve terminals and inhibiting the reuptake (Heikkila et al., 1975; Seiden et al., 1993). In addition, METH and the amphetamines redistribute DA from synaptic vesicles to the cytosol and promote reverse transport (Sulzer et al., 1995). Accordingly, to explore the mechanisms by which TNF-α inhibits the neuropsychopharmacological effects of METH, we examined the in vivo effects of TNF-α on the METH-induced increase in extracellular DA in the striatum of C57BL/6 mice, by using a microdialysis technique. Results showed that METH (4 mg/kg, s.c.) caused a marked increase in extracellular DA levels in the striatum. Peak extracellular DA was increased 7.5-fold above the baseline. Pretreatment with TNF-α (4 μg, i.p.) 1 hr before the METH treatment significantly inhibited METH-induced increase in extracellular DA levels (F(1,12) = 5.706; p < 0.05) (Fig. 6A).

Effects of TNF-α on DA release and uptake. A, In vivo effects of TNF-α on METH-induced increase in extracellular DA levels. TNF-α (4 μg, i.p.) was injected 1 hr before METH (4 mg/kg, s.c.) treatment. Extracellular levels of DA were measured in the striatum by in vivo microdialysis. Basal extracellular DA levels were 6.6 ± 0.9 pg/40 μl and 8.3 ± 1.4 pg/40 μl for the vehicle- and TNF-α-treated groups, respectively. Values are means ± SEM (n = 7). ANOVA with repeated measures revealed a significant differences in extracellular DA levels (F(1, 12) = 5.706; p < 0.05). *p < 0.05 versus vehicle-treated mice. B, In vitro effects of TNF-α on [3H]DA uptake. The [3H]DA uptake in the control was 0.141 ± 0.022 pmol/mg protein/4 min. Values are means ± SEM (n = 4). *p < 0.05 versus control. C, In vivo effects of TNF-α on METH-induced inhibition of striatal [3H]DA uptake in wild-type mice. TNF-α (4 μg, i.p.) was administered 1 hr before METH (4 mg/kg, s.c.) treatment in C57BL/6 mice, and the mice were killed 1 hr after METH treatment. Values are means ± SEM (n = 4). *p < 0.05 versus vehicle+saline group; #p < 0.05 versus vehicle+METH group. D, Effects of TNF-α on vesicular [3H]DA uptake. Reserpine (5 mg/kg, i.p.) was administered 18 hr before decapitation. TNF-α (4 μg, i.p.) or GBR12909 (20 mg/kg, i.p.) was administered 1 hr before decapitation. The mean [3H]DA uptake value from control was 55.1 ± 4.5 fmol/μg protein/4 min. Values are means ± SEM (n = 4). *p < 0.05 versus vehicle-treated group. E, Lineweaver-Burk plots of vesicular [3H]DA uptake obtained from vehicle- and TNF-α-treated mice. TNF-α (4 μg, i.p.) was administered 1 hr before decapitation. The mean Vmax values were 435.3 ± 66.4 and 304.7 ± 14.8 fmol/μg protein for 4 min for vehicle- and TNF-α-treated mice, respectively. The mean Km values were 193.0 ± 39.8 and 72.7 ± 9.4 nm for vehicle- and TNF-α-treated mice, respectively; these values differed significantly (p < 0.05). F, Effects of TNF-α on the METH-induced decrease in vesicular [3H]DA uptake. METH (4 mg/kg, s.c.) was administered four times at 2 hr intervals in C57BL/6 mice. Vehicle or TNF-α (4 μg, i.p.) was administered 1 hr before the first and third injections of METH. Mice were killed 1 hr after the final METH treatment. The mean [3H]DA uptake value from control was 59.2 ± 4.6 fmol/μg protein/4 min. Values are means ± SEM (n = 5). *p < 0.05 versus control group.
Next, we examined the in vitro effect of TNF-α on [3H]DA uptake into striatal synaptosomes. As shown in Figure 6B, TNF-α (1-100 ng/ml) significantly increased striatal [3H]DA uptake by ∼35%. We examined the kinetics of [3H]DA uptake in the absence or presence of TNF-α (10 ng/ml). Lineweaver-Burk plots showed that TNF-α potentiated [3H]DA uptake by increasing the affinity (Km) (control, 326.8 ± 2.6 nm; TNF-α, 55.3 ± 22.7) accompanied by reducing the maximum number of [3H]DA transporters (Vmax) (control, 9.9 ± 2.4 pmol/4 min/tube; TNF-α, 1.9 ± 0.5) (data not shown). We also examined the in vivo effects of TNF-α on [3H]DA uptake into striatal synaptosomes. We used TNF-α (4 μg, i.p.) and METH (4 mg/kg) to examine the METH-induced neurotoxicity; thus, here we treated TNF-α (4 μg, i.p.) 1 hr before METH (4 mg/kg, s.c.) treatment, and [3H]DA uptake was measured 1 hr later. Treatment with TNF-α increased striatal [3H]DA uptake by 50%, whereas METH decreased it by 40%, compared with the vehicle-treated group (Fig. 6C). Furthermore, pretreatment with TNF-α completely blocked the METH-induced inhibition of striatal [3H]DA uptake into synaptosomes (Fig. 6C).
Finally, we examined the effects of TNF-α on [3H]DA uptake into synaptic vesicle preparations, because redistribution of DA from synaptic vesicles to cytoplasmic compartments through interaction with VMAT-2, and consequent elevation of oxidizable DA concentrations, has been postulated to be primarily responsible for DA terminal injury by METH and amphetamines (Cubellus et al., 1994; Liu and Edwards, 1997; Wrona et al., 1997; Uhl, 1998). As shown in Figure 6D, TNF-α (4 μg, i.p.) significantly increased vesicular [3H]DA uptake as assessed in purified striatal synaptic vesicles obtained from C57BL/6 mice 1 hr after the treatment. This increase was attributable to an increase in the affinity (Km) (control, 193.0 ± 39.8 nm; TNF-α, 72.7 ± 9.4) with little change in the Vmax of vesicular [3H]DA uptake (control, 435.3 ± 66.4 fmol/4 min/μg protein; TNF-α, 304.7 ± 4.8) (Fig. 6E). In accordance with a previous report (Metzger et al., 2002), reserpine, which inhibits the vesicular transporter in monoaminergic neurons, inhibited vesicular [3H]DA uptake 18 hr after treatment (Fig. 6D). In contrast, GBR12909, an inhibitor of the plasmalemmal DA transporter, increased vesicular [3H]DA uptake 1 hr after treatment (Fig. 6D), the data being consistent with a previous report that cocaine, as well as other agents that inhibit the DA transporter (i.e., GBR12935 and amfonelic acid), increases vesicular [3H]DA uptake as assessed in purified vesicles prepared from treated rats (Brown et al., 2001). It has been reported that multiple high-dose administrations of METH as well as a single METH administration produce a rapid (within 1 hr; Brown et al., 2001, 2002) and prolonged (persisting at least for 24 hr Brown et al., 2000; Hogan et al., 2000) decrease in vesicular [3H]DA uptake. To examine the effect of TNF-α on the METH (4 mg/kg, s.c.; four times at 2 hr intervals)-induced decrease in vesicular [3H]DA uptake, TNF-α (4 μg, i.p.) was administered 1 hr before the first and third injections of METH. As illustrated in Figure 6F, multiple administrations of METH (4 mg/kg, s.c.; four times at 2 hr intervals) resulted in a significant decrease in [3H]DA uptake by 29%. TNF-α by itself increased [3H]DA uptake into purified synaptic vesicles and negated the METH-induced decrease in vesicular [3H]DA uptake. These results suggest that both the inhibition of METH-induced increase in extracellular DA levels and the activation of DA uptake into synaptosomes and synaptic vesicles induced by TNF-α may be plausible mechanisms by which TNF-α inhibits the METH-induced hyperlocomotion, sensitization, rewarding and discriminative stimulus effects as well as dopaminergic neurotoxicity.
Discussion
It has been suggested that neurotrophic factors and cytokines are involved in the effects of psychostimulants. For example, basic FGF and BDNF enhance the effects of psychostimulants (Flores et al., 2000; Horger et al., 1999). Glial cell line-derived neurotrophic factor inhibits biochemical and behavioral changes caused by drugs of abuse (Messer et al., 2000). METH-induced neurotoxicity was attenuated in mice with a null mutation for IL-6 (Ladenheim et al., 2000). Lipopolysaccharide, which activates endogenous production of TNF, blocks reinforcing effects of cocaine (Suzuki et al., 1994). More recently, it is reported that a high dose of METH (10 mg/kg; single injection) induces TNF-α mRNA and protein expression in the brain of C57BL/6 mice (Flora et al., 2002).
Here, we showed that chronic low doses of METH treatment (2 mg/kg for 5 d), but not acute treatment, increased the expression of TNF-α (Fig. 1A). Expression of the TNF-α gene is induced through activation of transcription factors such as activator protein-1 (AP-1) and nuclear factor-κB (NFκB) (Guha et al., 2000; Rahman and MacNee, 2000). Changes in transcription factors may result in long-term changes in gene expression, thereby contributing to neuronal adaptations that underlie behavioral sensitization by chronic psychostimulant treatment (Nestler, 2001). Although it is reported that chronic cocaine treatment desensitizes the response of Fos immunoreactivity (Rosen et al., 1994), recent studies suggest a prolonged induction of ΔFosB, which can activate transcription at AP-1 sites, after chronic psychostimulant treatment (Chen et al., 1997; McClung and Nestler, 2003).
Evidence indicates that acute high doses of METH can activate AP-1 and NFκB DNA-binding in mouse brain. For example, Lee et al. (2002) reported that a single injection of METH 10 or 20 mg/kg increased AP-1 DNA-binding activity in the striatum, frontal cortex, hippocampus, and cerebellum. Asanuma and Cadet (1998) suggested that METH injections (10 mg/kg; four times at 2 hr intervals) activated NFκB DNA binding. In contrast, an acute low dose of METH (1 mg/kg) has a limited effect on AP-1 and NFκB DNA binding (Lee et al., 2002). Collectively, it is plausible that acute high doses as well as repeated low doses of METH can induce activation of transcription factors such as AP-1 and NFκB, followed by upregulation of TNF-α, whereas an acute low dose of METH has a limited effect. We showed the neuronal localization of TNF-α in the nucleus accumbens after METH treatment (Fig. 1F). Because METH-induced TNF-α gene expression was antagonized by DA D1 and D2 receptor antagonists, it is likely that activation of DA neurotransmission is necessary for METH-induced TNF-α expression.
We next showed that exogenous TNF-α blocked the locomotor-stimulating, rewarding, and discriminative stimulus effects of METH (Figs. 2, 3, 4). It is not clear whether the inhibitory effect of TNF-α is mediated directly by penetration into the brain via passive or active transport (Gutierrez et al., 1993) or indirectly through stimulation of afferent neural circuits (e.g., vagal or spinal projections) (Maier and Watkins, 1998) or enzymatic alterations at peripheral target sites, such as the liver (Libert et al., 1991). However, we provided evidence that microinjection of TNF-α directly into the nucleus accumbens inhibited the locomotor-stimulating effect of METH (Fig. 2B), suggesting that TNF-α inhibits the locomotor-stimulating effects of METH, at least in part, through the action in the nucleus accumbens.
We also investigated a role of endogenous TNF-α in METH-induced behavioral changes in TNF-α-(-/-) mice. TNF-α-(-/-) mice exhibited a significantly enhanced locomotor response after repeated METH treatment compared with wild-type mice and showed a marked conditioned place preference at a low dose of METH, which failed to establish place preference in wild-type mice. These findings continue to support a role of TNF-α in METH-induced behavioral changes. TNF-α-(-/-) mice, however, exhibited an initial locomotor response to acute METH, a pattern of initiation of stimulant-induced sensitization, and a pattern of expression of sensitization, identical to wild-type mice. Therefore, although exogenous TNF-α inhibited both acute locomotor-stimulating and long-lasting sensitizing effects of METH, it seems that the role of endogenous TNF-α in acute locomotor response to METH and in the initiation of sensitization is minimal.
Repeated treatment with drugs of abuse produces structural adaptations. Exposure to amphetamine produced a long-lasting increase in the length of dendrites and the number of branched spines on medium spiny neurons (Robinson and Kolb, 1997). In contrast, cultured neurons exhibited reduced outgrowth and branching of neurites after the addition of TNF-α (Neumann et al., 2002). Furthermore, several reports have suggested that TNF-α influences synaptic strength and transmission (Albensi and Mattson, 2000; Beattie et al., 2002). Collectively, these observations lead to the hypothesis that TNF-α may have inhibitory effects on long-lasting behavioral changes induced by repeated METH treatment, by inhibiting the drug-induced changes of neuronal morphology and synaptic function. This hypothesis may explain why the magnitude of the sensitized response after repeated METH treatment was significantly increased in TNF-α-(-/-) mice. It is not clear, at present, whether the acute effects of exogenous TNF-α on METH-induced behavior and the striatal dopaminergic response to METH may be related to the long-term compensatory adaptations to the drug treatment.
TNF-α may also play an inhibitory role in METH-induced neurotoxicity. This seems to be consistent with the role of TNF-α in the parkinsonian drug 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxication, as described by Rousselet et al. (2002), which demonstrated that MPTP-induced decrease in DA content was enhanced in mice lacking TNF receptors. We showed that exogenous TNF-α inhibited the METH-induced depletion of DA and decrease in TH protein levels (Fig. 5A,B) and that METH-induced depletions of DA were potentiated in TNF-α-(-/-) mice (Fig. 5C). Treatment with TNF-α before METH induced hypothermia, whereas METH-induced hyperthermia was exacerbated in TNF-α-(-/-) mice (Fig. 5D,E). Increased body temperature has been reported to play a role in METH neurotoxicity (Miller and O'Callaghan, 1994). A broad range of pharmacological agents that protect against METH-induced neurotoxicity seems to do so by producing hypothermia (Bowyer et al., 1994). However, it is reported that treatment with reserpine, which produces hypothermia, does not prevent the neurotoxic effects of METH (Albers and Sonsalla, 1995) and that MDL27777, a selective 5-HT uptake inhibitor, does not alter methylenedioxymethamphetamine-induced hyperthermia but completely prevents the neurotoxicity to serotonergic neurons (Schmidt et al., 1990). These findings suggest that hyperthermia might contribute to, but cannot be the sole cause of, the neuropathology produced by amphetamines. Taken together, it is suggested that although TNF-α has a neuroprotective effect against METH-induced dopaminergic neurotoxicity, we cannot exclude a possibility that the neuroprotective effect of TNF-α is attributable to changes in body temperature.
The pharmacological properties of METH are linked to its capacity to elevate extracellular DA levels through redistribution of DA from synaptic vesicles to the cytosol and promotion of reverse transport (Sulzer et al., 1995). We demonstrated that TNF-α attenuated METH-induced increase in extracellular DA levels in the striatum of living mice (Fig. 6A). Furthermore, we showed that TNF-α potentiated striatal DA uptake into synaptosomes (Fig. 6B,C) and negated METH-induced inhibition of DA uptake (Fig. 6C). We also showed that TNF-α potentiated DA uptake into synaptic vesicles and blocked METH-induced decrease in DA uptake into synaptic vesicles (Fig. 6D,F). Both the inhibition of METH-induced increase in extracellular DA levels and activation of DA uptake into synaptosomes and synaptic vesicles induced by TNF-α may be one plausible mechanism by which TNF-α inhibits the METH-induced acute behavioral changes and neurotoxicity (Fig. 7). One might consider that if TNF-α activates DA uptake and thereby attenuates METH-induced increase in extracellular DA levels, this cytokine could attenuate the rewarding effects of other drugs of abuse. In agreement with the hypothesis, our preliminary results showed that TNF-α inhibited the conditioned place preference induced by morphine.

A model for the modulation by TNF-α of METH-induced DA responses. A, Under basal conditions, plasmalemmal DAT is involved in the reuptake of extracellular DA into the cytosol; subsequently the cytosolic DA is stored into synaptic vesicles via VMAT-2. B, Treatment with METH results in an increase in extracellular DA levels through the inhibition of DA reuptake, redistribution of DA from synaptic vesicles to the cytosol, and promotion of reverse transport (Sulzer et al., 1995). METH induces TNF-α expression in target neurons through the activation of DA receptors. C, TNF-α activates its receptor TNFR-1, expressed in cell bodies and processes of dopaminergic neurons (Boka et al., 1994), and potentiates plasmalemmal and vesicular DA uptake and thereby attenuates the METH-induced increase in extracellular DA levels. By way of these actions of TNF-α, TNF-α may inhibit the METH-induced acute behavioral changes and neurotoxicity. DAT, DA transporter; DA1-R, DA D1 receptors; DA2-R, DA D2 receptors; (+), activate; (-), inhibit.
The molecular mechanisms underlying TNF-α-induced neuroprotection against METH-induced neurobehavioral toxicity remain to be determined. Previous studies, which use TNF receptor knock-out mice and cell culture, have demonstrated that TNF-α upregulates manganese superoxide dismutase and calcium-binding protein calbindin expression through activation of NFκB and directly protects neurons against CNS insults (Cheng et al., 1994; Bruce et al., 1996; Mattson et al., 1997). NFκB is a ubiquitous transcription factor that is activated by a variety of cytokines, including TNF-α, and is thought to be a key regulator of genes involved in inflammation, infection, and stress. It is interesting that expression of TNF-α is dependent on activation of NFκB, as described above, and in turn this cytokine can stimulate activation of this transcription factor. Thus, it seems that TNF-α uses NFκB to amplify its own signals (Berliner et al., 1995). Although it was proposed that NFκB is involved in the cell death process (Grill et al., 1996), recent findings suggest that activation of NFκB represents neuroprotective responses after CNS insults (Sullivan et al., 1999; Yu et al., 1999). Accordingly, it is important to determine whether NFκB plays a role in the TNF-α-induced neuroprotection against METH-induced neurobehavioral toxicity.
In conclusion, the present study established a functional interaction between TNF-α and METH. The involvement of TNF-α, and perhaps other immune systems in drug-induced neural plasticity, could be important for the long-lived changes in brain function associated with drug addiction.
Footnotes
This study was supported in part by Special Coordination Funds for Promoting of Science and Technology, Target-Oriented Brain Science Research Program from the Minister of Education, Culture, Sports, Science, and Technology of Japan, a Grant-in-Aid for Health Sciences Research from the Ministry of Health, Labour and Welfare of Japan, Japan Society for the Promotion of Science Joint Research Project under the Japan-Korea Basic Scientific Cooperation Program, and NOVARTIS Foundation (Japan) for the Promotion of Science.
Correspondence should be addressed to Dr. Toshitaka Nabeshima, Department of Neuropsychopharmacology and Hospital Pharmacy, Nagoya University Graduate School of Medicine, Showa-ku, Nagoya 466-8560, Japan. E-mail: tnabeshi{at}med.nagoya-u.ac.jp.
Copyright © 2004 Society for Neuroscience 0270-6474/04/242212-14$15.00/0
↵* A.N. and K.Y. contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}