

Abstract
Collapsin response mediator proteins (CRMPs) mediate growth cone collapse during development, but their roles in adult brains are not clear. Here we report the findings that the full-length CRMP-3 (p63) is a direct target of calpain that cleaves CRMP-3 at the N terminus (+76 amino acid). Interestingly, activated calpain in response to excitotoxicity in vitro and cerebral ischemia in vivo also cleaved CRMP-3, and the cleavage product of CRMP-3 (p54) underwent nuclear translocation during neuronal death. The expression of p54 was colocalized with the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-positive nuclei in glutamate-treated cerebellar granule neurons (CGNs) and in ischemic neurons located in the infarct core after focal cerebral ischemia, suggesting that p54 might be involved in neuronal death. Overexpression studies showed that p54, but not p63, caused death of human embryonic kidney cells and CGNs, whereas knock-down CRMP-3 expression by selective small interfering RNA protected neurons against glutamate toxicity. Collectively, these results reveal a novel role of CRMP-3 in that calpain cleavage of CRMP-3 and the subsequent nuclear translocation of the truncated CRMP-3 evokes neuronal death in response to excitotoxicity and cerebral ischemia. Our findings also establish a novel route of how calpain signals neuron death.
Introduction
Collapsin response mediator proteins (CRMPs) are important brain-specific proteins with distinct functions during development and under disease conditions. In developing neurons, CRMPs mediator growth cone collapse in response to repelling guidance cues, such as semaphorin 3A or lysophosphatidic acid (Arimura et al., 2000). Specifically, CRMP-2, expressed in growth cones, modulates axonal length through modifying microtubule assembly, actin filaments, and cytoplasmic flow (Bradke and Dotti, 1999, 2000; Inagaki et al., 2001; Fukata et al., 2002; Nishimura et al., 2003; Yuasa-Kawada et al., 2003; Arimura et al., 2004; Uchida et al., 2005; Yoshimura et al., 2005). CRMP-4 has also been shown to regulate actin bundling (Rosslenbroich et al., 2005). Although how CRMPs precisely modulate the cytoskeleton to induce growth cone collapse remains unclear, it is known that CRMP-2 acts as a common intracellular target by integrating both positive and negative effects on axonal extension (Fukata et al., 2002; Yuasa-Kawada et al., 2003).
In adult brain, the functions of CRMPs are less clear. The expression levels of CRMPs are generally decreased, but CRMP-3 was selectively expressed in the spinal cord, cerebellum, and, to a lesser extent, forebrain (Wang and Strittmatter, 1996). Members of the CRMP family may have distinct functions in diseased brains (Weitzdoerfer et al., 2001; Suzuki et al., 2003). For example, CRMP-3 and CRMP-4 expression increased in Down syndrome fetal brains, whereas CRMP-2 expression was significantly decreased (Weitzdoerfer et al., 2001;). CRMPs may directly participate in axonal damage and neuronal death, as exemplified by the fact that expression of semaphorins and its downstream effector CRMP were associated with neuronal injury in epileptic brains, Alzheimer’s disease brains, and cerebral ischemia (Kee et al., 2001; Czech et al., 2004; Good et al., 2004). Antibodies and peptides against semaphorin 3A blocked CRMP-2 activities and neuronal death (Gagliardini and Fankhauser, 1999; Shirvan et al., 1999, 2002). Overexpression of CRMP-2 in mouse N2A neuroblastoma cells caused blebbing of the cytoplasm and cell death (Gu and Ihara, 2000). These findings are in sharp contrast to the fact that overexpression of CRMP-2 induced the formation of multiple axons of hippocampal neurons and was associated with neuronal survival (Inagaki et al., 2001). It is, therefore, important to investigate the role that each CRMP plays in adult disease brains.
Calpains plays a central role in ischemic neuronal death (Roberts-Lewis and Siman, 1993; Lipton, 1999) by cleaving a large number of substrates, including cytoskeletal proteins such as spectrins and important regulatory proteins such as cyclin-dependent kinase-5 (Patzke and Tsai, 2002). The activities of calpain are modulated by an endogenously expressed inhibitory protein, calpastatin, and can be inhibited by synthesized inhibitors ALLN [N-acetyl-Leu-Leu-Nle-CHO] and calpeptin. Expression of calpain inhibitor calpastatin is neuroprotective to several neurological diseases, such as Parkinson’s disease and cerebral ischemia (Lankiewicz et al., 2000; Crocker et al., 2003). Identification of targets of calpain in response to NMDA receptor activation will shed light on the molecular mechanisms of neuronal death.
In the present report, we specifically investigated the role of CRMP-3 during neuronal death caused by excitotoxicity and show that calpain directly targets CRMP-3 during neuronal death. Nuclear translocation of calpain-cleaved CRMP-3 signals neuronal death.
Materials and Methods
Materials.
All chemicals and reagents, unless stated otherwise, were purchased from Sigma (Burlington, Ontario, Canada). Antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from Advanced Immunochemicals (Long Beach, CA). Antibodies to CRMP-1, CRMP-2, CRMP-3, and CRMP-4 have been described previously (Franken et al., 2003) and were used at 1:10,000 for Western blotting.
Cell cultures and treatment.
Primary cultures of mouse (C57BL/6) cerebellar granule neurons (CGNs) were prepared from 6- to 9-d-old postnatal mice as described previously (Smith et al., 2003). Briefly, cerebella were explanted and cleaned free of meninges. Mechanical and enzymatic dissociation in a 0.025% w/v trypsin solution for 25 min followed. A trypsin inhibitor was then added to block the enzyme, and 0.05% w/v DNase was added to break DNAs from dead cells. A series of trituration and mild centrifugation steps were included to disperse the neurons before resuspension in medium and to remove undissociated debris before plating in Eagle’s minimum essential medium containing 0.8 mm glutamine, 27 mm glucose, 0.01% gentamycin, and 9% FBS and supplemented with K+ to a final concentration of 23 mm. Cells were plated onto 24-well dishes containing poly-l-lysine-coated coverslips at a density of 6 × 105 per well. After ∼18 h, cytosine α-d-arabinofuranoside was added to a final concentration of 5 μm to prevent glial cell proliferation. One hundred-millimeter culture dishes were seeded with 21 × 106 cells in 10 ml of culture medium. After 7 d in vitro (DIV), calpain inhibitors were added to CGNs at 37°C for 15–20 min before treatment with 50 μm glutamate. The plates were then incubated for 6 h at 37°C. Untreated controls were also included. At the end of the treatment period, neuronal viability was measured as described below. Human embryonic kidney 293 (HEK293) cell cultures were prepared exactly as described previously (Huang et al., 2004).
Cerebral ischemia produced by middle cerebral artery occlusion.
All procedures using animals were approved by the Institute for Biological Sciences Animal Care Committee following the guidelines established by the Canadian Council on Animal Care. C57BL/6 mice (20–23 g) were obtained from Charles River Laboratories (Wilmington, MA) and bred locally. Under temporary isofluorane anesthesia, mice were subjected to middle cerebral artery occlusion (MCAO) using an intraluminal filament as described previously (MacManus et al., 2003; Jiang et al., 2005; Weaver et al., 2005). After 1 h of MCAO, the filament was withdrawn, blood flow was restored to normal as monitored by laser Doppler flowmetry, and wounds were sutured. Animals were killed after 24 h of reperfusion. Brains were removed, and the brain infarction was measured as described previously (MacManus et al., 2003; Jiang et al., 2005). Brains were fixed and embedded in paraffin. Serial sections were cut and stained for terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) and/or with specific antibodies.
Immunostaining and TUNEL labeling.
Neuronal death was confirmed in fixed tissue by reaction for TUNEL (Roche Products, Welwyn Garden City, UK) or propidium iodide (PI) assay (Hou et al., 2002). Immunolabeling was performed using specific primary antibodies with a cyanine 3 (Cy3)-conjugated anti-mouse IgG secondary antibody (Jackson ImmuoResearch, West Grove, PA) as described previously (Tu et al., 1998; Hou et al., 2002). The primary antibodies were used at the following concentrations: mitogen-activated protein (MAP-2), 1:100; CRMP-3, 1:10,000; and neuronal-specific nuclear protein (NeuN), 1:200. The secondary antibody (conjugated with Cy3) was used at 1:200. Finally, cells were mounted in medium containing 5 μg/ml Hoechst 33258 to stain the nuclei. Slides were dried at 4°C overnight and examined under a fluorescent microscope.
Western blotting.
Protein at 10 μg was electrophoresed in a 10% SDS minigel and then electroblotted onto a nitrocellulose membrane in transfer buffer exactly as described previously (Hou et al., 2002; Smith et al., 2003). The membrane was then probed with a primary antibody at 4°C overnight. After washing with 0.01 m PBS, horseradish peroxidase-conjugated secondary antibody was applied to the membrane for 1 h at room temperature. Enhanced chemiluminescence detection of the target protein was performed using a LumiGlo substrate kit (KP Laboratories, Gaithersburg, MD) and x-ray film.
Mass spectrometry analysis of protein gel bands.
The protein gel bands of interest were excised and automatically destained and digested using Investigator Progest (Genomic Solutions, Ann Arbor, MI). Briefly, destaining was achieved using a 1:1 ratio of 30 mm potassium ferricyanide and 100 mm sodium thiosulfate (Gharahdaghi et al., 1999). The gel bands were then washed with deionized water, shrunk with acetonitrile, and reswollen with 50 mm ammonium bicarbonate containing 200 ng of modified trypsin (Promega, Madison WI). Digestion was performed overnight at 37°C, after which the supernatant was collected and the gel bands were extracted with 50% methanol and 0.5% acetic acid. The extracts were combined with the digest solutions, concentrated to ∼10 μl, and analyzed by nanoHPLC-MS/MS using a Q-TOF Ultima (Waters, Milford, MA). The entire samples were injected onto a 0.3 × 5 mm C18 micro precolumn cartridge (Dionex/LC Packings, Sunnyvale, CA), which was then brought on-line with a 75 μm × 50 mm Picofrit nanocolumn (New Objective, Woburn, MA) packed with BioBasic C18 reversed-phase media. The peptides were separated using a gradient supplied by a Waters CapLC pump (5–40% acetonitrile, 0.2% formic acid in 25 min, ∼300 nl/min flow rate). The mass spectrometer was set to automatically acquire dual mass spectrometric spectra on double-, triple-, and quadruple-charged ions. Database searching was performed in batch mode using Mascot Daemon (Matrix Science, London, UK) against the NCBInr protein sequence database. All search returns were confirmed manually.
Calpain cleavage of CRMP-3 in vivo.
The calpain inhibitors ALLN (Calbiochem, La Jolla, CA) or calpeptin (Calbiochem) was added to CGN cultures at the specified concentrations for 30 min before the addition of glutamate. After 2–6 h, cells were lysed and collected for Western blotting or fixed for immunostaining. To demonstrate that the endogenous calpain cleaves CRMP-3, normal brain extracts were mixed with 5 mm calcium in the presence or absence of calpain inhibitors. The mixture was incubated at 4°C. After 1, 4, or 18 h, reactions were terminated by the addition of protein loading buffer. Equal amount of brain extract was subjected to SDS-PAGE and Western blotting to detect CRMP-3 cleavage.
Calpain cleavage of CRMP-3 in vitro.
Constitutively active calpain I or II (Calbiochem) at the specified concentrations was mixed with equal amount of normal brain extracts or bacterially expressed full-length CRMP-3 (His-tagged CRMP-3). After incubation at 37°C for 20 min, protein loading buffer was added and samples were subjected to SDS-PAGE, followed by Western blotting using the primary antibody against CRMP-3.
Determination of calpain cleavage site on CRMP-3.
His-tagged CRMP-3 was purified using a nickel column. After digestion with active calpain I or II, duplicate samples were subjected to SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane (Amersham Biosciences, Arlington Heights, IL). One set of samples on the membrane was subjected to Western blotting, and the other was subjected to N-terminal microsequencing by cutting out the bands corresponding to p63 and p54. Protein sequencing was performed at the National Research Council Biotechnology Research Institute Protein Sequencing Facility (Montreal, Quebec, Canada). The obtained N-terminal polypeptide sequence was analyzed using Proteinprospector (http://prospector.ucsf.edu) and then aligned with the mouse CRMP-3 sequence using Clustal W to identify the N-terminal sequence of the cleaved CRMP-3.
Small interfering RNA design and transfection.
Several small interfering RNAs (siRNAs) targeted to various regions of the CRMP-3 mRNA were designed, synthesized commercially by Qiagen (Mississauga, Ontario, Canada), and used to reduce the expression of CRMP-3. The most effective siRNA that was capable of downregulating CRMP-3 was selected empirically through transfection into CGNs and Western blotting to detect the reduced expression of CRMP-3. The following duplex siRNA tagged with Alexa Fluor 546 targeted to the CRMP-3 cDNA sequence between 860 and 881 nt were selected and synthesized: 5′-r(UCUUGCUCCAGUAGUGUGAGC)d(TT)-3′-Alexa Fluor 546. A negative control siRNA to CRMP-3 tagged with Alexa Fluor 546 to the 3′ end was also synthesized: r(UUCUCCGAACGUGUCACGU)d(TT)-Alexa Fluor 546–3′. The annealed double-stranded siRNAs at 0.3–1 μg/well (Falcon 24-well plate) were transfected into 7 DIV CGNs using FuGene 6.0 (Qiagen) transfection reagent kit exactly following the instructions of the manufacturer. After 2 d of transfection, CGNs were either fixed with 4% paraformaldehyde for morphological examination or collected for protein analysis by Western blotting.
Plasmid construction, purification, and transfection.
The full-length CRMP-3 (p63) and the N-terminally truncated CRMP3 (p54) were amplified from the plasmid CRMP3-pBKCMV as template using the primer pairs rCRMP3-pr14/rCRMP3-pr15 and rCRMP3-pr16/rCRMP3-pr15, respectively. These primer pairs contain sequences encoding the restriction sites KpnI and BamHI at the end. The primer sequences are as follows: rCRMP3-pr14, 5′-ggggtaccatgtccttccaaggcaagaagag-3′; rCRMP3-pr15, 5′-gcggatccctaagaaagtgaagtgatgttg-3′; and rCRMP3-pr16, 5′-ggggtaccccagccgatgatttctgtcag-3′.
The amplified fragments were then digested with KpnI and BamHI and subcloned into the KpnI/BamHI restriction sites of the plasmid pEGFP-C1 (Clontech, Palo Alto, CA). The plasmid was purified using the plasmid midi-preparation kit purchased from Sigma (Sigma-Aldrich, Oakville, Ontario, Canada) following exactly the protocol of the manufacturer. Purified plasmid at 10 μg was used to transfect HEK293 cells using Lipofectamine 2000 transfection reagent (Invitrogen, Burlington, Ontario, Canada) as described previously (Hou et al., 2002; Huang et al., 2004).
Generation of lentiviral vectors and infection of neurons.
The full-length CRMP-3 [enhanced green fluorescent protein (EGFP)-p63] and truncated CRMP-3 (EGFP-p54) were obtained by restriction digest of the plasmids pEGFP-CRMP-3 and pEGFP-Nt-CRMP-3, respectively, using AgeI/BamHI, followed by blunt ending with treatment using the Klenow fragment. The blunt-end fragments were then subcloned into the BamHI/SalI restriction sites of the plasmid pWPT-GFP, which were first treated with the Klenow fragment and dephosphorylated using calf intestine phosphatase. The recombinant plasmids were transfected into HEK293 cells. HEK293T cells were seeded into 10-cm-diameter plastic dishes at the density of 2 × 106 cells per dish 1 d before cotransfection. Cells were then cotransfected with 10 μg of DNA constructs, 5 μg of envelop plasmid DNA pMD.G, and 5 μg of packaging plasmid DNA pCMV-dR8.74 by standard calcium phosphate method. The transfected cells were allowed to continue to grow for an additional 3 d before the culture supernatant was collected for additional purification. The supernatants were first passed through a 0.45 μm filter and then concentrated using an Amicon column (100,000 molecular weight cutoff; Millipore, Bedford, MA) by spinning at 3500 rpm for 15 min at 4°C. The supernatant was then pelleted by ultracentrifugation with the Beckman Coulter (Fullerton, CA) TLA-100.3 rotor at 50,000 rpm for 90 min at 16°C. The pellet containing the recombinant viral particles was resuspended in 0.01 m PBS. The virus was immediately used for infection of CGNs, and the rest was stored at −80°C. The expression of EGFP was monitored under a fluorescent microscope. After 4–5 d infection, CGNs were then fixed and immunostained with an antibody to NeuN to confirm that the expression of EGFP was indeed in neurons.
Subcellular fractionation.
Nuclear fractions were obtained using the following method. Cells were collected into a 15 ml tube and spun at 1500 rpm for 5 min at 4°C. Cell pellet was transferred into a 1 ml Eppendorf tube and washed briefly in buffer I (in mm: 2 KH2PO4, pH 6.55, 1 EGTA, 5 MgCl2, 0.1 DTT, and 0.5 PMSF). The pellet was finally resuspended in 3 ml of buffer I containing 10% Triton X-100 and left on ice for 10 min. After centrifugation at 3000 rpm at 4°C for 3 min, cell pellet was resuspended in 3 vol of buffer II (20 mm HEPES, pH 7.9, 420 mm NaCl, 25% glycerol, 0.2 mm EDTA, 1.5 mm MgCl2, 0.5 mm DTT, and 0.5 mm PMSF). After incubation with buffer II on ice for 30 min, the nuclear lysate was spun at 55,000 rpm for 10 min at 4°C. The supernatant contained soluble nuclear fractions. The pellet was sonicated for 15 s in buffer II and spun at 55,000 rpm for 10 min at 4°C. This supernatant contained proteins from the insoluble nuclear fraction.
Neuronal viability assays.
Neuronal viability was measured using the 5-(6)-carboxyfluorescein diacetate (CFDA) assay (for live cells) or PI (for dead cells) as described previously (Hou et al., 2002; Smith et al., 2003). The CFDA stock solution was diluted using 0.01 m PBS to a final concentration of 5 μg/ml. Cultures were incubated with 500 μl of the CFDA solution at 37°C for 30 min. The intensities of fluorescence was quantified using a Cytofluor 2350 Fluorescence Measurement System (Millipore) at λex of 485 nm and λem of 530 nm. Cellular viability was normalized against the fluorescent reading from the control cells. Neuronal death induced by glutamate was determined using a modified serial quantitative measurements of PI fluorescence at an excitation wavelength of 645 ± 40 nm and an emission wavelength of 530 ± 20 nm in a multiwell plate fluorescence scanner (Cytofluor 2350 fluorescence measurement system; Millipore) as described previously (Lin et al., 2004, 2005). Neurons were also fixed in 4% formaldehyde and mounted in Dako (High Wycombe, UK) fluorescent mounting medium containing 5 μg/μl Hoechst 33258 for examination of cell morphology with a fluorescent microscope. Duplicate assessment of each treatment was made on each plate in at least three separate experiments per treatment.
Data analysis.
Data were analyzed using Microsoft (Seattle, WA) Excel. Error bars in all graphs represent SD of at least three independent repeats. Statistical significance was determined by one-way ANOVA and an additional post hoc test for significant groups using Tukey’s test. p < 0.05 was considered statistically significant.
Results
Calpain cleaves CRMP-3
CRMP-3 protein appeared as a single band of 63 kDa (p63) on Western blot (Fig. 1A,B). However, in glutamate-treated CGNs (Fig. 1A) and focal cerebral ischemic brains (Fig. 1B), a smaller band at 54 kDa (p54) appeared below p63. Treating CGNs with the NMDA receptor antagonist MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] prevented the appearance of p54 (Fig. 1A), which had been shown to protect neurons from glutamate-induced death (Lin et al., 2005). Although treating CGNs with semaphorin 3A significantly inhibited axonal outgrowth of CGNs, this treatment did not induce the production of the p54 band (Fig. 1A, last lane). To confirm that p54 was the breakdown product of p63, the following four experiments were performed. (1) For confirmation of the specificity of the CRMP-3 antibody, Western blotting was performed using CRMP-3 antibody against overexpressed His-tag purified CRMP-1, CRMP-2, CRMP-3, and CRMP-4 proteins. Only His-CRMP-3 cross-reacted with the antibody, confirming the specificity of CRMP-3 antibody (data not shown). (2) To eliminate the possibility that the double-banding pattern was the result of CRMP-3 phosphorylation, calf intestine phosphatase was used to treat CGN neuronal protein extract. No visible changes in the levels of p63 and p54 occurred (data not shown). (3) The p54 band was excised for mass spectrometry analysis, which confirmed that p54 was CRMP-3. An example of one of the mass spectra obtained is shown in Figure 1C. (4) For determination of the N-terminal sequence of p54, N-terminal microsequencing of the p54 band determined that the cleavage site was at the N terminus of CRMP-3 starting at amino acid 76 (Fig. 1D, arrow). Interestingly, the region between CRMP-3 amino acids 7 and 83 shared high sequence homology (>50%) to the calpain cleavage site of spectrin, a known calpain target during neuronal death (Roberts-Lewis and Siman, 1993).

CRMP-3 is cleaved toward the N terminus. Protein extracts from glutamate-treated CGNs (A) and ischemic brains (B) were subjected to SDS-PAGE (10% gel) and Western blotting with an antibody against CRMP-3. The full-length CRMP-3 appeared as a 63 kDa band, whereas the breakdown product of p63 had a molecular weight of 54 kDa. The p54 band appeared in glutamate-treated CGNs (A) and ischemia in the brain (B). MK-801 at 10 μm protected CGNs against glutamate toxicity and prevented the breakdown of CRMP-3 (A). Purified recombinant semaphorin 3A was added to CGNs at 5 μg/ml for 18 h before cell extract was collected for Western blotting with CRMP-3 antibody (A, last lane). Bands corresponding to p63 and p54 were cut out on a clean surface and subjected to mass spectrometry analysis. A representative mass spectrum of CRMP-3 is shown in C. The p54 band was also subjected to N-terminal protein sequencing to confirm the identity of p54 and the cleavage site of CRMP-3 (arrow in D). The cleavage site sequence of CRMP-3 was compared with the calpain cleavage site of spectrin, which shared >50% homology (D).
To determine whether calpain directly cleaves CRMP-3 during excitotoxicity, a series of experiments were performed as shown in Figure 2A–D. First, as shown in Figure 2A, cultured CGNs were pretreated without or with calpain inhibitors ALLN (at 1 or 10 μm) or calpeptin (50 μm) before the addition of glutamate. Western blot showed that calpeptin at 50 μm (whereas ALLN was used at the concentration of 10 μm but not 1 μm) completely prevented the breakdown of CRMP-3 evoked by glutamate (Fig. 2A). Second, normal mouse brain extracts were mixed with 5 mm calcium to activate endogenous calpain (Patzke and Tsai, 2002). After coincubation with or without the presence of calpain inhibitors ALLN and calpeptin at 4°C, Western blotting was used to detect p54 (Fig. 2B). Treatment of brain samples with calpain inhibitors ALLN or calpeptin prevented the appearance of p54 (Fig. 2B, last two lanes). Third, to demonstrate that calpain directly cleaves CRMP-3, constitutively active calpain I or II was mixed with normal brain extracts (Fig. 2C) or recombinant CRMP-3 expressed in Escherichia coli (Fig. 2D). At the concentrations of 0.01 and 0.005 U of active calpain I, cleavage of CRMP-3 could be detected in both samples, whereas the lower concentrations of active calpain I (0.001 U) failed to cleave CRMP-3 (Fig. 2C,D). ALLN and calpeptin completely prevented the cleavage of CRMP-3 by added calpain I (Fig. 2C). Calpain II was also able to cleave CRMP-3, albeit at a relatively higher concentration compared with calpain I (data not shown). Fourth, for N-terminal sequencing, bacterial-expressed His-tagged CRMP-3 was purified using a nickel column, and the purified protein was cleaved with calpain I. The cleavage product was transferred onto a PVDF membrane, and the p54 band was excised for N-terminal microsequencing, which confirmed the cleavage site on CRMP-3 as shown in Figure 1D. Collectively, these data demonstrate that CRMP-3 is a direct target of calpain and that p54 is a cleavage product of p63.

CRMP-3 is a direct target of calpain. A, Proteins extracted from glutamate-treated CGNs either with or without preincubation with calpain inhibitors, ALLN or calpeptin, were subjected to Western blotting to detect changes in CRMP-3 breakdown. Calpain inhibitors ALLN at 10 μm and calpeptin at 50 μm protected CGNs and prevented the breakdown of CRMP-3 (A). B, Normal mouse brain protein extracts were stimulated with 5 mm CaCl2 to activate endogenous calpain, which led to the cleavage of the endogenous CRMP-3, as shown by Western blotting. The addition of calpain inhibitors ALLN (10 μm) and calpeptin (50 μm) before calcium stimulation prevented calpain activation and the breakdown of CRMP-3. Western blotting for GAPDH was used to show equal protein loading (A, B). C, Active calpain I at the concentrations indicated were added into normal brain protein extracts without calcium stimulation. At the concentrations of 0.01 and 0.005 U, CRMP-3 was cleaved after 20 min incubation with calpain I, whereas lower concentrations of calpain were not sufficient to cleave CRMP-3. Calpain inhibitors ALLN and calpeptin effectively prevented the breakdown of CRMP-3 (last two lanes). D, Active calpain I at the concentrations indicated was mixed with 10 μg of recombinant CRMP-3 expressed in E. coli. At the concentrations of 0.01 and 0.005 U of calpain I used, CRMP-3 was cleaved into a p54 fragment.
Nuclear localization of CRMP-3 after excitotoxicity and MCAO
Glutamate-induced death of CGNs was preceded by the appearance of neurite shortening, followed by the condensation of the nucleus and neuronal death. We examined CRMP-3 expression during this death process using immunostaining. As shown in Figure 3A, CRMP-3 was exclusively expressed in the cytoplasm of untreated control CGNs. After 4 h exposure to glutamate, most of the CRMP-3 staining appeared either around or in the nucleus (Fig. 3B, arrows). Pretreatment of CGNs with calpain inhibitors ALLN or calpeptin prevented the nuclear accumulation of CRMP-3 (Fig. 3C) and protected CGNs against glutamate toxicity (Fig. 3D).

Altered expression of CRMP-3 during neuronal death. A–C, CGNs were treated with glutamate (Glut) (50 μm) in either the absence (B) or presence (C) of calpain inhibitor ALLN (10 μm). Untreated CGNs were used as a negative control (Ctl) (A). ALLN protected CGNs against glutamate toxicity (C). After 4–6 h of treatment with 50 μm glutamate, cells were either fixed and subjected to immunostaining for CRMP-3 (A–C) or double stained with TUNEL (E, F). Note that glutamate-treated CGNs had shorter neurites and higher levels of CRMP-3 immunostaining in areas in and around the nucleus (B) compared with those in the untreated and ALLN-protected neurons. Blue in A–C indicates Hoechst 32558-positive nuclei. Neuroprotection by calpain inhibitors was also quantified using PI staining as described in Materials and Methods (D). G–I, Nuclear fractions from glutamate-treated CGNs (G) and ischemic brains (H) were prepared as described in Materials and Methods and were probed with antibodies to CRMP-3. Histone H1 and GAPDH were also probed as loading controls for nuclear and cytosolic fractions, respectively. Total protein extracts were also used as positive controls. A clear p54 band appeared in the nuclear fraction of glutamate-treated CGNs (G) and ischemic brains (H). The intensity of each band was quantified using densitometry and plotted in I. Data represented the average of at least three repeats, with error bars representing the SD. Statistical analysis in D and I was performed using Student’s t test, and significant groups were identified using Tukey’s post hoc test. *p < 0.05; **p < 0.01. J–U, Mice were subjected to 1 h MCAO, followed by reperfusion. Brains were fixed with 4% paraformaldehyde and embedded in paraffin. Sections at 10 μm thickness were cut and subjected to immunostaining to detect CRMP-3. N and R show MCAO-induced nuclear localization of CRMP-3 (red) in ischemic neurons located in the ischemic side of the brain but not in the contralateral side (J). TUNEL staining was also performed on the same section (K, O, S), and images for CRMP-3 and TUNEL were merged (yellow in L, P, T). All sections were counterstained with Hoechst 32558 to show the nuclei (blue in M, Q, L). Scale bars, 100 μm.
To begin to understand the significance of CRMP-3 nuclear translocation, double labeling for CRMP-3 and TUNEL was performed on CGNs treated with glutamate. Interestingly, nuclear CRMP-3 staining colocalized with those nuclei positive for TUNEL staining, suggesting that the nuclear translocation of the CRMP-3 is associated with neuronal death (Fig. 3E,F). To further characterize nuclear CRMP-3, subcellular fractionation was performed, and the fractions were analyzed by Western blotting (Fig. 3G,H). Surprisingly, only p54, but not the full-length CRMP-3 (p63), was detected in the nuclear fraction derived from CGNs treated with glutamate (Fig. 3G) and ischemic brains (Fig. 3H). The nuclear fractions were free from cytosolic protein contaminations as confirmed by the lack of GAPDH on Western blot. Histone H1 was used as nuclear protein loading control in Figure 3, G and H. To quantitatively characterize CRMP-3 nuclear translocation, the relative intensities of nuclear CRMP-3 band were measured against histone H1 band using densitometry measurement. Nuclear p54 level was almost fivefold higher in glutamate-treated CGNs and ischemic brains compared with the normal nontreated controls (Fig. 3I). These data indicate that CRMP-3 undergoes nuclear translocation during neuronal death.
Because glutamate-mediated excitotoxicity is a major cause of neuronal death after cerebral ischemia (Hou and MacManus, 2002), we examined changes in CRMP-3 expression in mouse brains subjected to MCAO. As shown in Figure 3J–M, CRMP-3 was expressed only in the cytoplasm of normal neurons located in the non-ischemic (contralateral) side of the brain (Fig. 3J), and no TUNEL-positive nuclei were detected (Fig. 3K–M). In contrast, on the ischemic side of the brain, CRMP-3 expression was mostly associated with the nuclei having condensed morphology and, at the same time, being TUNEL positive after reperfusion for 20 h (Fig. 3N–Q) or 24 h (Fig. 3R–U). These data demonstrate that nuclear translocation of CRMP-3 is associated with ischemia-induced neuronal death.
Overexpression of p54 causes neuronal death
To investigate the function of p54, plasmid vectors expressing EGFP-tagged p63 (EGFP-p63) and p54 (EGFP-p54) were constructed (Fig. 4A). The EGFP vector plasmid was used as a control. These constructs were transfected into HEK293 or PC12 cells because these cells lack endogenous CRMP-3 (Fig. 4C). Fusion proteins were confirmed by Western blotting for CRMP (Fig. 4B). After 2 d transfection, pEGFP vector expressed EGFP in both the nucleus and cytoplasm (Fig. 4D), whereas pEGFP-p63 was exclusively detected in the cytoplasm of HEK293 cells (Fig. 4E). In contrast, pEGFP-p54 was only detected in the nucleus (Fig. 4F). Importantly, significant amount of cell death (TUNEL positive) occurred in the pEGFP-p54-transfected HEK293 cells (Fig. 4F′,F″) but not in cells transfected with pEGFP (Fig. 4D′,D″) or pEGFP-p63 (Fig. 4 E′,E″), indicating that p54 nuclear expression induces cell death. Cell death was quantified by counting TUNEL-positive nuclei (Fig. 4G, **p < 0.01).

Overexpression of p54 induces cell death. A, Plasmid constructs of EGFP-tagged full-length CRMP-3 (pEGFP-p63), p54 (pEGFP-p54), and EGFP vector control (pEGFP) were transfected into HEK293 cells. The overexpressed proteins were detected by Western blotting (B). EGFP fusion protein with p63 and p54 had a molecular weight of 98 and 89 kDa, respectively (B). Proteins from HEK293 and PC12 cells were subjected to Western blotting to confirm that they did not express endogenous CRMP-3 (C). Cultured HEK293 cells were transfected with 10 μg of plasmids. After 2 d, cells were fixed and viewed under a fluorescent microscope. EGFP vector construct expressed EGFP in both the nucleus and the cytoplasm. In contrast, EGFP-p63 only expressed in the cytoplasm (E), whereas EGFP-p54 expressed mostly in the nucleus (F). Overexpression of p54 (pEGFP-p54) caused nuclear condensation (F), which overlapped with TUNEL positivity (F′ and F″). The number of TUNEL-positive cells were counted and plotted in G. Scale bar, 50 μm. **p < 0.01 by one-way ANOVA and post hoc Tukey’s test. H–M are CGNs infected with lentiviral constructs. Lentiviral vector expressing EGFP, EGFP-p63, and EGFP-p54 were made as described in Materials and Methods and were used to infect 7-d-old mature postmitotic CGNs for 72 h. Cell were examined live under a fluorescent microscope, followed by fixation with fresh Formalin and TUNEL staining (I, K, M). The number of TUNEL-positive cells were counted and plotted in N. Statistical analysis was performed using one-way ANOVA, and significant groups were identified by Tukey’s post hoc analysis. **p < 0.01.
These plasmids were then packaged into a lentiviral vector. Equal amount of the viral construct expressing EGFP (Lenti-EGFP), p63 (Lenti-EGFP-p63), and p54 (Lenti-EGFP-p54) was used to infect CGNs in culture. As shown in Figure 4H–M, Lenti-EGFP expression was in both the cytoplasm and nucleus (Fig. 4H), whereas Lenti-EGFP-p63 was mostly expressed in the cytoplasm (Fig. 4J). These two constructs did not cause neuronal death (Fig. 4I,K, respectively, TUNEL negative). However, Lenti-EGFP-p54 was mostly expressed in the nucleus (Fig. 4L), and EGFP was associated with TUNEL-positive nuclei (Fig. 4M). The number of TUNEL-positive nuclei was counted and plotted in Figure 4N. Together, these studies demonstrate that p54 nuclear expression is sufficient to cause neuronal death.
Downregulation of CRMP-3 protects CGNs from glutamate toxicity
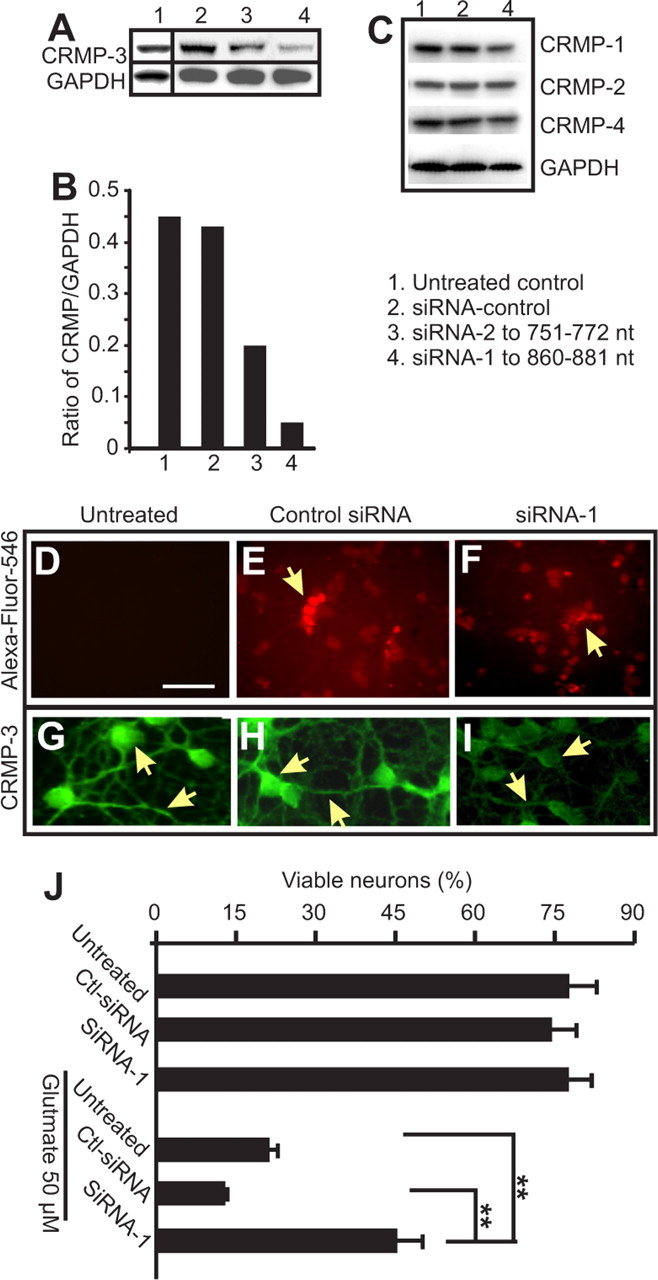
To further investigate the role of p54 in neuronal death, specific siRNA to CRMP-3 was designed and used to downregulate CRMP-3 expression to see whether the reduction in CRMP-3 provides neuronal protection. As described in Materials and Methods, control siRNA and siRNA specific to CRMP-3 (siRNA-1 and siRNA-2) were commercially tagged with fluorescent Alexa Fluor 546 and transfected into cultured CGNs (Fig. 5D–F). CRMP-3 expression was downregulated more than fivefold by siRNA-1 compared with the control siRNA-treated CGNs and nontransfected CGNs (Fig. 5A,B). A second siRNA sequence to CRMP-3, siRNA-2, was less effective compared with siRNA-1 (Fig. 5A, lane 3). The effect of siRNA-1 on the reduction of CRMP-3 expression was selective because the expression levels of other CRMPs (i.e., 1, 2, and 4) were not affected (Fig. 5C). The reduced expression of CRMP-3 in the cytoplasm of neurons were confirmed by immunostaining for CRMP-3 (Fig. 5G–I). After 2 d transfection with siRNA, neurons were treated with glutamate (50 μm). Viable neurons were detected using CFDA and quantified using a plate reader (Fig. 5J). siRNA alone did not induce neuronal death (Fig. 5J). After 4 h treatment with 50 μm glutamate, CGNs transfected with siRNA-1 had significantly more viable neurons compared with glutamate-treated CGNs either transfected with the control siRNA or without transfection (Fig. 5J). Together, these experiments demonstrate that CRMP-3 plays a role in glutamate-induced neuronal death.

Downregulation of CRMP-3 expression protects neurons against glutamate toxicity. CGNs were cultured in a 24-well plate for 7 d, followed by transfection for 2 d with 1 μg of CRMP-3-specific siRNA (1 or 2) or a control siRNA (Ctl-siRNA) using FuGene 6. CGNs were subsequently collected for Western blotting to show the selective downregulation of CRMP-3 (A, B) but not CRMP-1, CRMP-2, or CRMP-4 (C) with GAPDH as a loading control. Densitometry measurement of the CRMP-3 band intensity in A showed a clear downregulation of CRMP-3 by siRNA-1 (B; values represent the average of two independent repeats). Transfection of neurons by siRNA was confirmed by examining the fluorescence of Alexa Fluor 546 (D–F). Immunostaining of these CGNs showed that siRNA-1 (I), but not the control siRNA (Ht), reduced expression of CRMP-3 in neurons. The siRNA-1 was subsequently used to transfect 7-d-old CGNs for 2 d. CGNs were treated with 50 μm glutamate and followed by CFDA staining to detect live cells in culture using a plate reader. Data in J represent at least four independent repeats. Statistical analysis was performed using one-way ANOVA, and significant groups were determined by Tukey’s post hoc analysis. **p < 0.01.
Collectively, these data led us to propose a working model in that Ca2+-activated calpain cleaves CRMP-3. Truncated CRMP-3 (p54) translocates into the nucleus to cause neuronal death (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). In addition, evidence based on the series of loss-and-gain of function studies described here also suggests that the full-length CRMP-3 plays a distinct role from other family members such as CRMP-2, in that, when overexpressed in neurons, unlike CRMP-2, CRMP-3 does not cause the growth of multiple axons.
Discussion
The present study focused on the role of CRMP-3 and demonstrated for the first time that CRMP-3 is a direct target of calpain and that the cleavage product of CRMP-3 functions as a positive injury signal to cause neuronal death after cerebral ischemia and excitotoxicity. To the best of our knowledge, this is the first report describing such a novel property of CRMP-3.
Excitotoxicity through NMDA receptor-mediated intracellular calcium influx has been regarded as the major cause of toxicity to neurons after cerebral ischemia (Hou and MacManus, 2002; Bano et al., 2005). Early response to excitotoxicity of cultured neurons is axonal retraction, followed by, over time, neuronal death. Accumulating evidence suggests that intracellular signaling pathways are important contributing factors to transmit injury signals from distal axons to the cell body to cause neuronal degeneration (Raff et al., 2002; Aarts and Tymianski, 2004; Perlson et al., 2004). In the present study, we clearly demonstrate that, in response to glutamate toxicity and cerebral ischemia, the expression pattern of CRMP-3, normally in the cytoplasm of neurons, was altered in dying neurons and CRMP-3 cleavage is correlated with neuronal death. Calpain-cleaved CRMP-3 was translocated into the nucleus (Fig. 3). Neuroprotection by NMDA receptor antagonist MK-801 and calpain inhibitors prevented CRMP-3 cleavage, nuclear translocation, and neuronal death. Collectively, these studies show that CRMP-3 has a unique role in functioning as a retrograde-transported positive injury signal to induce neuronal death in adult neurons in response to excitotoxicity.
The mechanism of CRMP-3-induced neuronal death is still not clear but is obviously involved in the nuclear translocation of calpain-cleaved CRMP-3. Inhibitors to caspases failed to prevent the breakdown of CRMP-3, suggesting that CRMP-3 is specifically cleaved by calpain. Calpain cleaves many important signal transduction molecules (Tompa et al., 2004). It is perhaps not surprising that calpain targets CRMP-3 during neuronal death. However, it is important to note that the cleaved product of CRMP-3 propagates a death signal by spatial translocation into the nucleus. We also attempted to examine p54 nuclear translocation by constructing a C-terminally tagged CRMP-3. However, such attempts have failed because tagging CRMP-3 at the C terminus with either six histidines or EGFP rendered the fusion protein unstable (data not shown), confirming published findings (Yuasa-Kawada et al., 2003). Although the present study did not examine how p54 was translocated into the nucleus, searches for a typical nuclear localization signal amino acid sequence did not yield any positive hits. It has been shown recently in the literature that activated MAP kinase in injured nerves can be translocated into the nucleus in a nuclear localization signal-independent, but vimentin-dependent, manner (Perlson et al., 2004, 2005). Whether truncated CRMP-3 can also be translocated into the nucleus using such a mechanism requires additional investigation.
The tissue-selective expression of CRMP family members in the adult nervous system argues for possible distinct functions of CRMPs that may not only be limited solely to growth cone collapse (Luo et al., 1993; Wang and Strittmatter, 1996). Indeed, the present study, for the first time, demonstrated that CRMP-3 had a completely different role compared with CRMP-2. In contrast to CRMP-2, one of the most well studied members of CRMPs, neither the overexpression of CRMP-3 nor the downregulation of CRMP-3 by siRNA affected neuronal survival and the morphology of the dendrites of mature neurons. However, downregulation of the full-length CRMP-3 expression rendered neurons more resistant to excitotoxicity-induced death, suggesting that CRMP-3 plays an important role in neuronal susceptibility to excitotoxicity. These observations are in sharp contrast to the role of CRMP-2 in that its overexpression promotes neurite elongation and axon induction by modifying actin filaments, microtubules, and cytoplasmic flow (Inagaki et al., 2001; Fukata et al., 2002; Nishimura et al., 2003; Arimura et al., 2004; Yoshimura et al., 2005). CRMP-2 overexpression transformed the established dendrites to become an axon without affecting the total number of processes emerging from the cell body (Arimura et al., 2004). Overexpression of CRMP-2 also accelerates nerve regeneration (Pasterkamp et al., 1998a; Suzuki et al., 2003), suggesting that CRMP-2 is associated with neuronal survival.
Under the normal physiological conditions, CRMPs are downstream of semaphorin 3A receptors, which encompass neuropilin, plexin, and L1 subunits and associated tyrosine kinases and act as cytosolic mediators for axonal guidance by collapsing growth cones. Importantly, under the conditions of neuronal injury and recovery, inhibitory factors secreted from the scar tissues, such as semaphorins, Nogo, MAG, oligodendrocyte myelin glycoproteins, and myelin-associated chondroitin sulfate proteoglycans, form major impediments to the successful repair of CNS connections (Pasterkamp et al., 1998a, b; Sandvig et al., 2004; Domeniconi and Filbin, 2005) by either inducing neuronal death or preventing the formation of new axonal connections, possibly through CRMP-mediated axonal retraction and death. Evidence from the present study showing that CRMP-3 was indeed part of the death response to excitotoxicity, together with recent demonstrations that semaphorin and neuropilin expression are associated with neuronal death, including cerebral ischemia (Shirvan et al., 1999, 2002; Fujita et al., 2001; Kee et al., 2001; Zhang et al., 2001; Beck et al., 2002; Good et al., 2004), lend additional support to the suggestion that CRMPs may be potential targets for therapeutic modulation to facilitate neuroprotection and regeneration.
Footnotes
-
This work was supported by Heart and Stroke Foundation of Ontario Grant-in-Aid NA5393 (S.T.H.). We thank the Institute for Biological Sciences animal facility for the timely supply of animals.
- Correspondence should be addressed to Dr. Sheng T. Hou, Institute for Biological Sciences, National Research Council of Canada, 1200 Montreal Road, Building M54, Ottawa, Ontario, Canada K1A 0R6. Email: sheng.hou{at}nrc-cnrc.gc.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}