

Abstract
Cav1.3 (L-type) voltage-gated Ca2+ channels have emerged as key players controlling Ca2+ signals at excitatory synapses. Compared with the more widely expressed Cav1.2 L-type channel, relatively little is known about the mechanisms that regulate Cav1.3 channels. Here, we describe a new role for the PSD-95 (postsynaptic density-95)/Discs large/ZO-1 (zona occludens-1) (PDZ) domain-containing protein, erbin, in directly potentiating Cav1.3. Erbin specifically forms a complex with Cav1.3, but not Cav1.2, in transfected cells. The significance of erbin/Cav1.3 interactions is supported by colocalization in somatodendritic domains of cortical neurons in culture and coimmunoprecipitation from rat brain lysates. In electrophysiological recordings, erbin augments facilitation of Cav1.3 currents by a conditioning prepulse, a process known as voltage-dependent facilitation (VDF). This effect requires a direct interaction of the erbin PDZ domain with a PDZ recognition site in the C-terminal domain (CT) of the long variant of the Cav1.3 α1 subunit (α11.3). Compared with Cav1.3, the Cav1.3b splice variant, which lacks a large fraction of the α11.3 CT, shows robust VDF that is not further affected by erbin. When coexpressed as an independent entity with Cav1.3b or Cav1.3 plus erbin, the α11.3 CT strongly suppresses VDF, signifying an autoinhibitory function of this part of the channel. These modulatory effects of erbin, but not α11.3 CT, depend on the identity of the auxiliary Ca2+ channel β subunit. Our findings reveal a novel mechanism by which PDZ interactions and alternative splicing of α11.3 may influence activity-dependent regulation of Cav1.3 channels at the synapse.
Introduction
In the nervous system, L-type voltage-gated Ca2+ channels play essential roles in controlling membrane excitability (Marrion and Tavalin, 1998), gene expression (Bading et al., 1993; Weick et al., 2003), and synaptic plasticity (Johnston et al., 1992). Of multiple L-type Ca2+ channels that have been identified (Cav1.1–Cav1.4), Cav1.2 and Cav1.3 are widely expressed in the brain and in many of the same neuronal cell groups (Hell et al., 1993b; Ludwig et al., 1997). However, a number of experimental mouse models indicate neural functions for Cav1.3 that differ from those of Cav1.2. Cav1.2 is important for processes underlying spatial memory (Moosmang et al., 2005), whereas Cav1.3 activation selectively causes depression-like behavior (Sinnegger-Brauns et al., 2004), promotes synaptic changes associated with Parkinson's disease (Day et al., 2006), and is essential for auditory transduction (Platzer et al., 2000; Dou et al., 2004). In addition, Ca2+ influx through Cav1.2 and Cav1.3 causes transcriptional activation in different brain regions (Hetzenauer et al., 2006). These findings suggest that Cav1.3 participates in distinct cellular and subcellular tasks compared with Cav1.2 and therefore may be subject to unique modes of regulation.
In support of this hypothesis, Cav1.3 but not Cav1.2 channels interact with Shank, a postsynaptic protein associated with excitatory synapses (Zhang et al., 2005). This interaction targets Cav1.3 to postsynaptic sites in neurons (Zhang et al., 2005, 2006) and permits Cav1.3 inhibition by G-protein-coupled receptors (Olson et al., 2005). Shank contains a single PSD-95 (postsynaptic density-95)/Discs large/ZO-1 (zona occludens-1) (PDZ) domain that binds to a C-terminal sequence in the α1 subunit of Cav1.3 (α11.3) (Zhang et al., 2005). Although PDZ domains are generally known to localize and scaffold membrane proteins with intracellular signaling partners, some PDZ interactions directly regulate the gating properties of ion channels (Ou et al., 2003). Given the wealth and diversity of PDZ proteins in the brain (Kim and Sheng, 2004), PDZ partners other than Shank could differentially modulate Cav1.3 and the role of these channels in various neurobiological contexts.
Here, we identify a novel regulatory mechanism for Cav1.3 channels involving erbin, a multifunctional PDZ protein concentrated at the neuromuscular junction and at excitatory synapses in the CNS (Huang et al., 2001; Kolch, 2003). Erbin binding to the C-terminal domain (CT) of α11.3 augments voltage-dependent facilitation (VDF) of Cav1.3 channels containing the auxiliary β1b but not the β4 subunit by relieving an inhibitory influence of the CT on this process. Cav1.3b channels in which the CT is removed by alternative splicing (Safa et al., 2001; Xu and Lipscombe, 2001) show robust VDF that is not further influenced by erbin. Erbin coimmunoprecipitates with and colocalizes with Cav1.3 in neurons, in which it may regulate postsynaptic Cav1.3 signaling. Our results define a new role for PDZ proteins in directly modulating Cav1.3 channels through intermolecular suppression of an autoinhibitory domain in the α11.3 CT.
Materials and Methods
Constructs and molecular biology.
The α11.3b and α11.3 variants of the rat brain Cav1.3 α1 subunit (GenBank accession numbers AF370009 and AF370010; provided by Dr. D. Lipscombe, Brown University, Providence, RI) and auxiliary channel subunits β1b (GenBank accession number NM017346) or β4 (GenBank accession number L02315; provided by Dr. E. Perez-Reyes, University of Virginia, Charlottesville, VA) and α2δ (GenBank accession number M21948) were used in this study. FLAG-α11.3 was generated by PCR amplification of a FLAG-tagged fragment (nucleotides 1–660 of α11.3) and cloned into NheI and AleI sites of rat α11.3-pcDNA6/V5-His (Xu and Lipscombe, 2001). FLAG-α11.2 was described previously (Zhou et al., 2004). For pull-down assays, glutathione S-transferase (GST)-tagged constructs containing the cytoplasmic C-terminal domain of rat α11.3 [GST-α11.3 CT, nucleotides 5886–6465 (see Fig. 1) and 6360–6465 (see Fig. 8B)] or rat α11.3b [GST-α11.3b CT, nucleotides 4822–4930 (see Fig. 8B)] were subcloned into BamHI/NotI sites of pGEX4T.1 (GE Healthcare, Piscataway, NJ). Green fluorescent protein-tagged protein containing the C-terminal 500 aa of α11.3 not present in α11.3b (GFP-CT500) corresponds to nucleotides 4903–6465 of rat α11.3 subcloned into HindIII/SacII of pEGFPN1 (Clontech, Mountain View, CA). Hexahistidine- and S-tagged constructs (his-S-erbinPDZ, amino acids 1275–1371; his-S-MAGI1PDZ1, amino acids 316–456) were described previously (Fam et al., 2005). Myc-erbin, myc-erbinΔPDZ, GFP-erbin965, and GFP-erbin965ΔPDZ were also described previously (Huang et al., 2001).

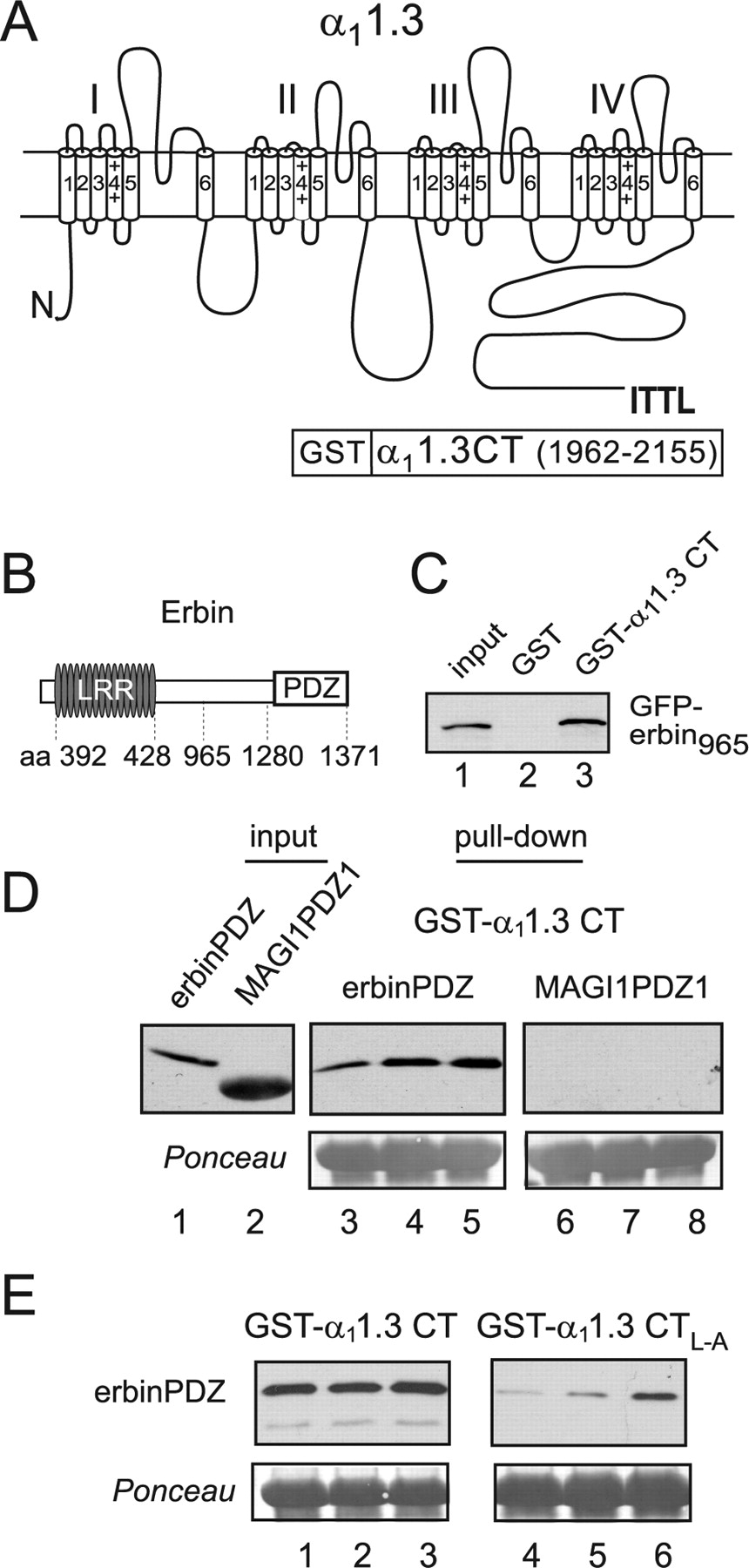
The PDZ domain of erbin binds to the C-terminal domain of α11.3. A, Schematic of rat α11.3 showing class I PDZ-binding consensus sequence (ITTL) in the cytoplasmic C-terminal domain and the region used for GST-α11.3 CT in pull-down assays (amino acids 1962–2155). B, Schematic of mouse erbin indicating position of LRR (amino acids 392–428) and the PDZ domain (amino acids 1280–1371). C, Binding of erbin to α11.3 CT. GST-α11.3 CT (lane 3) or GST alone (lane 2) was immobilized on glutathione-agarose beads and incubated with lysates from cells transfected with GFP-erbin965 (amino acids 965–1371). Bound GFP-erbin965 was detected by Western blot with anti-GFP antibodies. Lane 1 shows ∼15% of GFP-erbin965 input used in the pull-down assay. D, Binding of erbinPDZ but not MAGI1PDZ1 to α11.3 CT. GST-α11.3 CT was incubated with purified his-S-erbinPDZ (lanes 3–5) or his-S-MAGI1PDZ1 (lanes 6–8). Input his-S-tagged protein was the following (in μg): 1 (lanes 3, 6), 2 (lanes 4, 7), and 4 (lanes 5, 8). Input lanes 1 and 2 show his-S-tagged proteins used in the assay (1 μg). E, Impaired binding of erbinPDZ to α11.3 CTL–A. His-S-erbinPDZ protein (lanes 1, 4, 1.5 μg; lanes 2, 5, 3 μg; lanes 3, 6, 6 μg) was incubated with GST-α11.3 CT (lanes 1–3) or GST-α11.3 CTL–A (lanes 4–6). In D and E, bound proteins were detected by Western blot (top) with anti-S-protein antibodies, and Ponceau staining (bottom) shows equal levels of GST-α11.3 CT or GST-α11.3 CTL–A in each group.
Antibodies.
For the generation of rabbit polyclonal α11.3 antibodies, a GST fusion protein containing amino acids 1–41 (MQHQRQQQEDHANEANYARGTRLPISGEGPTSQPNSSKQTV) of rat α11.3 (GenBank accession number AF370009), GST-α11.3 NT1–41, was used as an immunogen, and the resulting antiserum was generated by a commercial source (Covance, Denver, PA). For goat polyclonal α11.3 antibodies, two peptides corresponding to an N-terminal sequence (amino acids 24–37, PISGEGPTSQPNSS) and a sequence in the cytoplasmic loop linking domains II and III (amino acids 810–827, DNKVTIDDYQEEAEDKD) were used as dual immunogens for antisera generated by a commercial source (ProSci, Poway, CA). The N-terminal sequence was chosen because it shows relatively low sequence homology with the corresponding regions of other L-type channel α1 subunits (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). The loop II–III sequence was selected because it has been used previously to generate α11.3-selective antibodies (Hell et al., 1993b). α11.3 antibodies were purified on affinity columns (AminoLink Coupling Gel; Pierce Biotechnology, Rockford, IL) coupled with the immunogens according to manufacturer's protocol. Rabbit α11.3 antibodies were purified first by passing antiserum through the GST column to remove anti-GST antibodies, and the flow-through was loaded onto the GST-α11.3 NT1–41 column overnight. Goat α11.3 antibodies were purified by passing antiserum over a column coupled with both immunogenic peptides. Antibodies were eluted with 5 m MgCl2, dialyzed against PBS, and stored at 4°C in PBS containing 0.02% sodium azide.
The specificity of α11.3 antibodies was characterized by the following criteria. First, the antibodies recognized α11.3, but not other L-type channel α1 subunits, in transfected cell lysates by immunoprecipitation (see Fig. 2B), Western blot (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), and immunocytochemistry (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material). Second, these antibodies immunoprecipitated and detected a protein consistent in size with α11.3 in brain lysates of wild-type mice but not mice with targeted disruption of the α11.3 gene (Platzer et al., 2000) (see Fig. 2D). Third, immunofluorescent staining of primary neurons with both rabbit and goat antibodies was abolished by preadsorption of the antibodies with the immunogen (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). Together, these results validated the specificity and sensitivity of the antibodies for detection of transfected and native α11.3 in our experiments.

Coimmunoprecipitation of erbin with Cav1.3 from transfected cells and brain. A, Schematic of α11.3 and α11.2 showing C-terminal PDZ-binding motifs. B, Coimmunoprecipitation of erbin and Cav1.3 from transfected cells. HEK293T cells were cotransfected with myc-erbin and Cav1.3 (FLAG-α11.3, β1b, and α2δ; lanes 1, 4, 6) or Cav1.2 (FLAG-α11.2, β1b, and α2δ; lanes 2, 3, 5) and subjected to lysis and immunoprecipitation using rabbit antibodies against α11.3 (lanes 1, 2) or α11.2 (lanes 3, 4). Immunoprecipitated proteins (I.P.; lanes 1–4) were detected by Western blotting (WB) with antibodies recognizing FLAG (top) or myc (bottom) epitopes. Lanes 5 and 6 represent 5% of the cell lysate input used for coimmunoprecipitation. C, Coimmunoprecipitation of erbin and Cav1.3 from the brain. Rat brain lysates were incubated with goat α11.3 antibodies or control goat IgG for immunoprecipitation, and α11.3 and erbin were detected by Western blotting with rabbit α11.3 and erbin antibodies, respectively. D, Lack of α11.3 immunoprecipitation in Cav1.3−/− mouse brain. α11.3 immunoprecipitation protocol used in C was applied to brain lysates from Cav1.3+/+ or Cav1.3−/− mice. An ∼200 kDa protein corresponding to α11.3 was detected in the brain lysate, and samples were immunoprecipitated with α11.3 antibodies from Cav1.3+/+ but not Cav1.3−/− mice. Lysate lanes represent 3% of the input used for coimmunoprecipitation.
Rabbit polyclonal α11.2 antibodies were generated against a peptide corresponding to a sequence in rat α11.2 (KYTTKINMDDLQPSENEDKS) used previously for the generation of α11.2-specific antibodies (Hell et al., 1993b). The criteria for determining specificity of these antibodies for α11.2 were similar to those for the α11.3 antibodies and will be described in a later report. Anti-erbin polyclonal antibodies were produced and characterized previously (Huang et al., 2001).
Other antibodies used were mouse monoclonal antibodies against FLAG M2 and myc (Sigma-Aldrich, St. Louis, MO), GFP (Santa Cruz Biotechnology, Santa Cruz, CA), and HRP-conjugated antibodies against S-protein (Novagen, La Jolla, CA).
Cell culture, ransfection, and lysate preparation.
A human embryonic kidney cell line transformed with SV40 T antigen (HEK293T) was maintained in DMEM with 10% fetal bovine serum (Invitrogen, Gaithersburg, MD) at 37°C in a humidified atmosphere under 5% CO2. Cells were grown to 70–80% confluence and transfected using Gene Porter reagent (Gene Therapy Systems, San Diego, CA) according to the manufacturer's protocols. For immunoprecipitation, HEK293T cells were transfected with cDNAs encoding Cav1.2 (4 μg of FLAG-α11.2, 2 μg of β1b, and 2 μg of α2δ) or Cav1.3 (6 μg of FLAG-α11.3, 2 μg of β1b, and 2 μg of α2δ) and myc-erbin (4 μg). For electrophysiological experiments, HEK293T cells plated on 35 mm dishes were transfected with ∼5 μg of total DNA (1.5 μg of FLAG-α11.3 or FLAG-α11.3b, 0.5 μg of β1b or β4, and 0.5 μg of α2δ with or without 1 μg of erbin) and GFP expression plasmid (0.01 μg) for fluorescent detection of transfected cells.
Pull-down binding assays.
GST- or his-S-tagged fusion proteins were prepared and purified on glutathione-agarose beads or Ni2+-nitrilotriacetic acid-agarose beads, respectively, as described previously (Zhou et al., 2004). GST-α11.3 CT or -α11.3b CT proteins immobilized on beads were incubated with purified his-S-erbinPDZ or his-S-MAGI1PDZ1 proteins or GFP-erbin965-transfected cell lysate and brought to a total volume of 400 μl with binding buffer [50 mm Tris-buffered saline (TBS; 50 mm Tris-HCl, pH 7.5, and 150 mm NaCl)/0.1% Triton X-100/protease inhibitors (1 μg/ml each of PMSF, pepstatin, aprotinin, and leupeptin)]. Binding reactions were incubated, rotating at 4°C for 1–2 h. Beads were washed three times with ice-cold binding buffer (1 ml), and bound proteins were eluted, resolved by SDS-PAGE, and transferred to nitrocellulose. Western blotting was performed with appropriate antibodies followed by HRP-conjugated secondary antibodies and enhanced chemiluminescent detection reagents (GE Healthcare). Ponceau staining was used to verify that equal levels of immobilized GST-α11.3 CT proteins were used in each experimental group. Interpretations of results from pull-down assays were based on at least three independent experiments.
Coimmunoprecipitation assays.
Two days after transfection, lysates from transfected HEK293 cells were harvested and solubilized in 1 ml/100 mm cell culture plate of radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mm EDTA, and protease inhibitors) at 4°C for 30 min and subjected to centrifugation at 100,000 × g (30 min) to remove insoluble material. Lysates were incubated with α11.3 or α11.2 antibodies (5 μg) and 50 μl of protein A-Sepharose (50% slurry) for 4 h, rotating at 4°C. After three washes with RIPA buffer (1 ml), proteins were eluted with SDS-containing sample buffer and subjected to SDS-PAGE. Coimmunoprecipitated proteins were detected by Western blotting with specific antibodies as described for pull-down assays. Conclusions were drawn from results of three independent experiments.
For coimmunoprecipitation assays from rat brain, crude membrane fractions were generated as follows: three frozen rat brains were homogenized in buffer [(in mm) 320 sucrose, 4 HEPES, 5 EDTA, 5 EGTA, pH 7.4 with NaOH, and protease inhibitors]. After homogenization, large debris was removed by spinning samples at 1000 × g for 6 min at 4°C. The supernatant was then subjected to ultracentrifugation at 100,000 × g (35 min), and the resulting pellet was solubilized for 3 h in 9 ml of buffer containing 1.2% digitonin, 300 mm KCl, 150 mm NaCl, 10 mm sodium phosphate, pH 7.2, and protease inhibitors at 4°C. A final centrifugation step (100,000 × g for 30 min) was performed to remove insoluble material. The resulting supernatant was precleared for 1 h with protein G agarose (200 μl; 50% slurry) and goat IgG (∼100 μg). The samples (∼9 ml) were then incubated with goat anti-α11.3 antibodies (∼120 μg) or an equivalent amount of goat IgG (final antibody concentration, ∼13 μg/ml) and protein G agarose (200 μl; 50% slurry) for 4 h, rotating at 4°C. After three washes with 0.1% digitonin/PBS buffer (1 ml), proteins were eluted with SDS-containing sample buffer and subjected to SDS-PAGE. Coimmunoprecipitated proteins were detected by Western blotting with erbin or rabbit α11.3-specific antibodies. For immunoprecipitation experiments from brain lysates of Cav1.3+/+ or Cav1.3−/− mice, an identical protocol was used, except that one-half of a mouse brain was used per immunoprecipitation with 30 μg of antibodies. Coimmunoprecipitation experiments from brain tissue were repeated at least twice with consistent results.
Immunocytochemistry.
For double-label immunofluorescence, primary cultures of neurons were prepared as follows. Neocortical tissue was dissected from rat embryos (embryonic day 19). The tissue was then digested with papain for 1 h at 37°C and triturated in inactivation solution [minimum essential medium (MEM; Invitrogen) and 10% FBS]. The resulting cell suspension was plated at a density of ∼300,000 neurons per 60 mm plate containing glass coverslips precoated with poly-d-lysine. Neurons were maintained in medium containing MEM, 1 mm pyruvate, 0.6% dextrose, 5% FBS, 1× B-27 (Invitrogen), 0.5 mm penicillin/streptomycin/glutamine, and 0.001% MITO+ serum extender (BD Biosciences, San Jose, CA). For immunostaining, coverslips (8–14 d in culture) were fixed in 4% paraformaldehyde/4% sucrose/PBS and incubated in blocking buffer [10% normal goat serum (NGS) or 10% donkey serum (DS), 0.1% Triton X-100, in TBS]. All antibodies were diluted in TBS containing 2.5% NGS or DS and 0.1% Triton X-100, and samples were rinsed three times for 5–10 min between each step. After blocking, samples were incubated with rabbit anti-α11.3 antibodies (1:50) 4 h to overnight at 4°C and, after rinsing in TBS, with donkey anti-rabbit rhodamine-conjugated Fab fragments (1:300; Jackson ImmunoResearch, West Grove, PA) for 60 min. Sections were rinsed and blocked with goat anti-rabbit Fab fragments (1:50; Jackson ImmunoResearch) for ∼2.5 h to block any rabbit IgG sites before the second labeling step. Sections were then incubated overnight at 4°C with anti-erbin antibodies (1:100–1:400), rinsed, and incubated with donkey FITC–anti-rabbit IgG (1:300; Jackson ImmunoResearch). The double-labeled sections were mounted and coverslipped with Vectashield (Vector Laboratories, Burlingame, CA) for viewing on a Zeiss (Oberkochen, Germany) LSM510 Meta confocal microscope. Image processing was withZeiss LSM Image Browser and Adobe Photoshop (Adobe Systems, San Jose, CA) software.
Because the double-labeling procedure involved two rabbit antibodies, a number of control experiments were performed to confirm the pattern of erbin and α11.3 colocalization. In particular, to ensure that the incubation with goat anti-rabbit Fab fragments was sufficient to prevent recognition of the first primary antibody (α11.3) by the second secondary antibody (FITC–anti-rabbit IgG), anti-erbin antibodies were omitted (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Loss of FITC fluorescence in this control experiment (supplemental Fig. 2D, available at www.jneurosci.org as supplemental material) argued against the possibility that overlap in α11.3 and erbin immunofluorescence resulted from insufficient blocking and therefore reactivity of both secondary antibodies with α11.3-labeled sites. Interpretations from immunocytochemical experiments were based on consistent results obtained from more than five independent experiments.
Electrophysiological recordings.
At least 48 h after transfection, whole-cell patch-clamp recordings of transfected cells were acquired with a HEKA Elektronik (Lambrecht/Pfalz, Germany) EPC-9 patch-clamp amplifier. Data acquisition and leak subtraction using a P/−4 protocol were done with Pulse software (HEKA Elektronik). Extracellular recording solutions contained the following (in mm): 150 Tris, 1 MgCl2, and 10 BaCl2 or 10 CaCl2. Intracellular solutions consisted of the following (in mm): 140 N-methyl-d-glucamine, 10 HEPES, 2 MgCl2, 2 Mg-ATP, and 5 EGTA. The pH of intracellular and extracellular recording solutions was adjusted to 7.3 with methanesulfonic acid. Electrode resistances were typically 1–2 MΩ in the bath solution, and series resistance was ∼2–4 MΩ, compensated up to 80%. Normalized tail current–voltage and prepulse voltage-dependence data were fit with a single Boltzmann function: A/{1 + exp[(V − V1/2)/k] + b}, where V is the test or prepulse voltage, V1/2 is the half-maximal voltage, k is a slope factor, A is the amplitude, and b is the baseline. Curve fits and data analyses were done with Igor Pro software (Wavemetrics, Lake Oswego, OR). All averaged data are presented as the mean ± SEM. Statistical significance of differences between two groups was determined by Student's t test as indicated (SigmaPlot; Systat Software, San Jose, CA). For statistical analyses of prepulse voltage and I–V curves, data were compared by two-way repeated-measures ANOVA (SigmaStat; Systat Software).
Results
Erbin binds to the α11.3 C-terminal domain
The extreme C-terminal residues of α11.3 (ITTL) (Fig. 1A) are consistent with a signature sequence favored by class I PDZ domains, (S/T)-X-φ-COOH, where φ is a hydrophobic amino acid, and X is any amino acid (Songyang et al., 1997). To identify PDZ proteins interacting with this sequence in α11.3, we used a GST fusion protein containing the last 200 aa of α11.3 CT (Fig. 1A) to screen a proteomic array of 96 putative class I PDZ domains, as described previously (Fam et al., 2005). Several PDZ domains on the array interacted with α11.3 (data not shown), including one belonging to erbin, a member of the LAP (leucine-rich region and PDZ) family (Borg et al., 2000; Huang et al., 2001) Fig. 1B). The interaction between erbin and α11.3 was confirmed in a pull-down assay with immobilized GST-α11.3 CT and a truncated version of erbin containing the PDZ domain (GFP-erbin965) (Fig. 1C). GST-α11.3 CT, but not the GST control, precipitated GFP-erbin965 from transfected cell lysates (Fig. 1C). To discount the possibility that GFP-erbin965 could have associated indirectly with α11.3 CT via a second protein in the transfected cell lysate, we performed pull-down assays with purified protein corresponding to the PDZ domain of erbin (erbinPDZ) (Fig. 1D). Although GST-α11.3 CT showed dose-dependent binding to erbinPDZ, the class I PDZ of a different protein [membrane-associated guanylate kinase inverted 1 (MAGI1)] did not bind to GST-α11.3 CT (Fig. 1D). These results confirm that α11.3 CT interacts directly and specifically with the PDZ domain of erbin.
Although most PDZ domains interact with a canonical sequence at the extreme CT of their ligands, some also recognize internal motifs not at the CT (Penkert et al., 2004). Because the GST-α11.3 CT fusion protein encompassed a relatively large region (∼200 aa) (Fig. 1A), binding to erbin could have been mediated by residues other than the C-terminal PDZ recognition site. To confirm the importance of the PDZ signature sequence (ITTL) in α11.3 CT for binding to erbin, we tested how substitution of alanine for the final leucine in this sequence affected the interaction (α11.3 CTL–A) (Fig. 1E). This substitution eliminates the hydrophobic nature of the final residue, which is critical for maintaining PDZ–ligand interactions (Songyang et al., 1997). If erbin binds to α11.3 CT via a conventional class I PDZ interaction, the L–A substitution should significantly weaken the interaction with erbin. Consistent with this prediction, binding of erbinPDZ to GST-α11.3 CT L–A was greatly reduced relative to GST-α11.3 in pull-down experiments Fig. 1E). Therefore, erbin binding to α11.3 CT depends on the C-terminal PDZ recognition site.
To confirm that erbin interacts not only with the isolated CT fragment of α11.3 but also with the intact channel in mammalian cells, coimmunoprecipitation experiments were performed with lysates of HEK293T cells cotransfected with Cav1.3 subunits (FLAG-α11.3, β1b, and α2δ) and myc-tagged erbin. Because the α1 subunit of Cav1.2 (α11.2) also contains a class I PDZ-binding element (VSNL) (Fig. 2A), we determined whether myc-erbin might selectively interact with either channel. Using α11.3 antibodies, we found that myc-erbin coimmunoprecipitated with α11.3 (Fig. 2B, lane 1). Myc-erbin was not detected when α11.3 antibodies were applied to lysates from cells cotransfected with α11.2 and erbin, arguing against nonspecific immunoprecipitation of erbin by α11.3 antibodies (Fig. 2B, lane 2). This result also confirmed that the antibodies specifically recognized α11.3 and not α11.2. However, the amount of myc-erbin that coimmunoprecipitated with Cav1.3 was relatively small (lane 1) compared with the total amount of myc-erbin that was detected in the cell lysates (lane 6). This could be explained by the presence of erbin endogenously expressed in HEK293 cells (Huang et al., 2001), which might have competed with transfected myc-erbin for interacting with Cav1.3. In addition, previous analyses indicate that the erbin PDZ domain has a strong preference for ligands with the CT sequence (E/D)-(S/T/V)-X-V and binds relatively weakly to sequences more similar to the ITTL α11.3 motif (Jaulin-Bastard et al., 2001). Such low-affinity interactions might be difficult to maintain during immunoprecipitation procedures that required strong detergent solubilization conditions.
With α11.2 antibodies, myc-erbin could not be coimmunoprecipitated with α11.2, despite the fact that α11.2 expression and immunoprecipitation was consistently greater than that observed for α11.3 (Fig. 2B, lanes 1, 3, 5, 6). The α11.2 antibodies did not immunoprecipitate α11.3, confirming the specificity of these antibodies for α11.2 (Fig. 2B, lane 2). Inability of myc-erbin to coimmunoprecipitate with α11.2 was not related to insufficient levels of myc-erbin, because Western blots of the transfected cell lysates indicated that equal levels of myc-erbin were coexpressed with Cav1.3 and Cav1.2 (Fig. 2B, lanes 5, 6). These results confirm that erbin selectively forms a complex with Cav1.3 channels and support previous observations that Cav1.2 and Cav1.3 are distinguished by the types of PDZ proteins with which they interact (Weick et al., 2003; Zhang et al., 2005).
Cav1.3 and erbin coimmunoprecipitate and colocalize in neurons
To address the neurobiological significance of erbin/Cav1.3 interactions, we determined whether erbin and Cav1.3 formed a complex in rat brain in coimmunoprecipitation experiments. Cav1.3 channels were immunoprecipitated from solubilized rat brain membrane fractions with goat polyclonal α11.3 antibodies. Cav1.3 and associated erbin were detected by Western blot with rabbit polyclonal α11.3 and erbin antibodies, respectively. In these experiments, erbin was coimmunoprecipitated with α11.3 using α11.3 antibodies (Fig. 2C). The coimmunoprecipitation was specific in that a similar result was not obtained with an equivalent concentration of control IgG (Fig. 2C). The ∼200 kDa protein immunoprecipitated and immunoblotted with our α11.3 antibodies corresponded to α11.3, because it was observed in brain lysates from wild-type mice (Cav1.3+/+) but not mice lacking α11.3 (Cav1.3−/−) (Fig. 2D), which were characterized previously (Platzer et al., 2000). These results confirm the existence of erbin and Cav1.3 interactions in the brain.
If erbin is a significant partner of neuronal Cav1.3 channels, these proteins should show similar cellular and subcellular localization in the brain. To test this, we used confocal microscopy and double-label immunofluorescence of cortical neurons in culture. We observed erbin and Cav1.3 immunofluorescence in neuronal cell bodies (Fig. 3A–C,G–I). In addition, punctate erbin and Cav1.3 immunofluorescence was observed in proximal dendrites (Fig. 3D–F). In developing neurons, erbin and Cav1.3 immunofluorescence coincided strongly at the distal ends of dendritic processes (Fig. 3G–I). Colocalization of erbin and Cav1.3 in the soma and proximal dendrites was extensive in virtually all neurons examined (Fig. 3C,F,I). However, colocalization of erbin and Cav1.3 was not complete, and there were many puncta that showed only Cav1.3 or erbin labeling (Fig. 3D–F). In addition to control experiments (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), these results discounted the possibility that overlapping fluorescence was artifactual as a result of cross-reactivity of the secondary reagents in the double-labeling protocol. The nucleus of most neurons was labeled with erbin but not Cav1.3 antibodies (Fig. 3A–C,G–I), which is not unexpected, because erbin is involved in multiple cellular signaling functions (Kolch, 2003). Nevertheless, the strong colocalization of erbin and Cav1.3 in neurons and coimmunoprecipitation from brain lysates support the potential for erbin to physiologically interact with neuronal Cav1.3 channels.
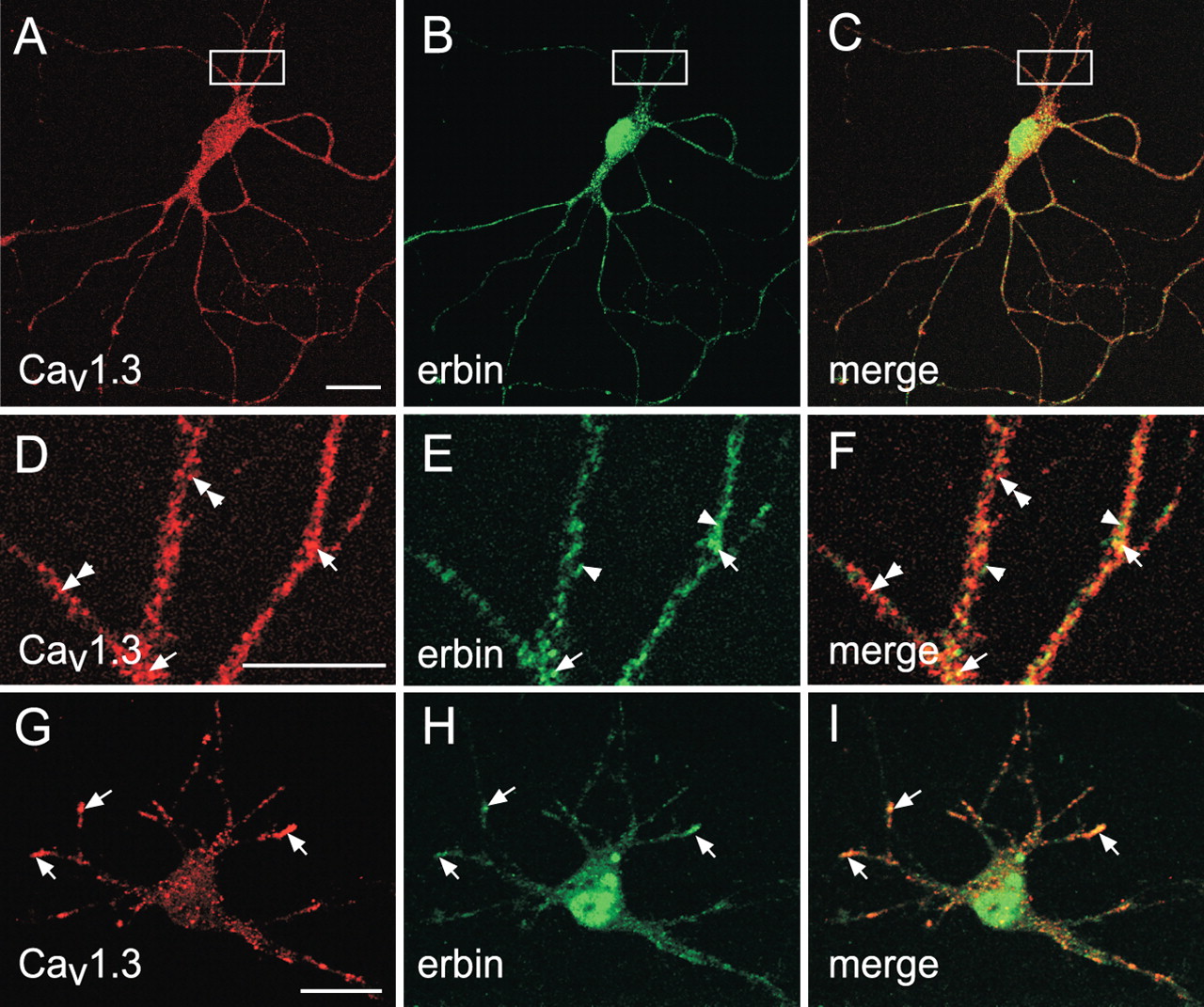
Cav1.3 and erbin colocalize in somatodendritic domains of cortical neurons in culture. A–I, Confocal images of primary cortical neuron cultures (A–F, 14 d in culture; G–I, 8 d in culture) sequentially double labeled with antibodies against α11.3 (to detect Cav1.3) and erbin are shown. Immunofluorescence was viewed under optics for rhodamine for Cav1.3 (A, D, G) or fluorescein for erbin (B, E, H). Regions of colocalization appear yellow in the merged images (C, F, I). Extensive colocalization of erbin and Cav1.3 was detected in cell bodies (A–C, G–I) and dendrites (D–F; higher-magnification view of boxed region in panels A–C). In D–I, elements immunoreactive for Cav1.3 alone (double arrowheads), erbin alone (arrowheads), or Cav1.3 plus erbin (arrows) are indicated. Scale bars: A (for A–C), G (for G–I), 20 μm; (in D) D–F, 10 μm.
Erbin augments voltage-dependent facilitation of Cav1.3
The cytoplasmic C-terminal domain of the Cav1.2 α1 subunit interacts with a variety of regulatory proteins that directly modulate channel activation or inactivation (Catterall, 2000). To test the hypothesis that erbin binding to α11.3 CT might similarly influence the function of Cav1.3, we screened for modulatory effects of erbin in electrophysiological recordings of HEK293T cells cotransfected with Cav1.3 subunits (α11.3, β1b, and α2δ). This system allows transfection and electrophysiological analysis of recombinant channels in the relative absence of competing influences. Similar experiments in primary neurons are complicated by the existence of multiple classes of voltage-gated Ca2+ channels and subsequent difficulty of isolating modulatory effects specific for Cav1.3 channels. Despite the low levels of endogenous erbin in HEK293 cells (Huang et al., 2001), functional effects of erbin on signaling have been studied in this cell line through transfection of exogenous erbin cDNAs (Dai et al., 2006), which we found to increase erbin expression significantly over endogenous levels (data not shown). Therefore, modulatory effects of erbin could be resolved by comparing Cav1.3 properties in cells cotransfected with Cav1.3 and erbin and in cells transfected with Cav1.3 alone.
Consistent with previous reports (Koschak et al., 2001; Safa et al., 2001; Scholze et al., 2001; Xu and Lipscombe, 2001), Cav1.3 showed a relatively hyperpolarized range of activation (Fig. 4). However, parameters for tail current activation curves were not significantly different in cells transfected with Cav1.3 alone (V1/2=−21.7 ± 1.6 mV; k=−5.8 ± 0.5; n=9) and those cotransfected with erbin (V1/2=−22.7 ± 1.9 mV; k=−6.8 ± 0.7; n=6; p=0.70 for V1/2; p=0.25 for k) (Fig. 4A,B). In addition, erbin did not influence the average amplitude of the Cav1.3 peak current evoked by various test voltages (Fig. 4C,D). There was no significant difference in the peak current amplitudes in current–voltage (I–V) relationships in cells transfected with Cav1.3 alone and those cotransfected with erbin (F(1,12)=0.0005; p=0.983) (Fig. 4C,D). These results show that erbin does not regulate the expression level or activation properties of Cav1.3 channels in mammalian cells.

Erbin does not affect voltage-dependent activation or mamplitude of Cav1.3 currents. A, B, Normalized tail current (Norm. Itail) activation curves for HEK293T cells transfected with Cav1.3 alone (n=9) (A) or cotransfected with Cav1.3 plus erbin (n=6) (B). Test pulses (10 ms) were applied from a holding voltage of −90 mV to various voltages, and peak tail currents were measured after repolarization to −70 mV for 2 ms. Tail currents were normalized to the largest in the series and plotted against test voltage. Representative current traces are shown at top. C, D, Current–voltage relationships for Cav1.3 alone (C) and Cav1.3 plus erbin (D). Data were from same voltage protocol as in A and B, except that peak current amplitudes during the test pulse were plotted against test voltage. Error bars represent SEM.
Cav1.3 channels undergo prominent VDF that, in recombinant systems, is independent of heterotrimeric G-proteins or phosphorylation (Safa et al., 2001). To determine whether erbin influenced this aspect of Cav1.3 function, we used a paired-pulse protocol in which the amplitude of two test currents (P1 and P2) evoked by the same voltage step was compared before and after a conditioning (facilitating) prepulse Fig. 5A). Consistent with previous studies (Safa et al., 2001), a short (20 ms) conditioning prepulse from −90 to +60 mV caused a significant increase (30.3 ± 4.1%; n=12; p < 0.01) in Cav1.3 currents evoked by a test pulse from −90 to −20 mV Fig. 5A). With the same voltage protocol, the amount of facilitation in cells cotransfected with erbin was dramatically increased (47.8 ± 6.6%; n=10) and was significantly greater than in cells transfected with Cav1.3 alone (p < 0.01) Fig. 5A). The enhanced facilitation attributable to erbin could have resulted from a negative shift or steeper dependence of facilitation on prepulse voltage. To test this, we compared the relationship between prepulse voltage and percentage of maximal facilitation in cells transfected with Cav1.3 alone or those cotransfected with erbin Fig. 5B). However, Boltzmann fits of prepulse voltage curves revealed no significant effect of erbin on either V1/2 (−12.2 ± 3.4 for Cav1.3 alone vs −8.3 ± 3.4 for Cav1.3 plus erbin; p=0.62) or slope (−19.0 ± 2.7 for Cav1.3 alone vs −17.6 ± 1.7 for Cav1.3 plus erbin; p=0.71). A second mechanism by which erbin could augment VDF is by increasing the number of channels undergoing facilitation as a result of a given prepulse. This was tested by plotting net facilitation, which was the amplitude of the P2 relative to the P1 current [Fratio (P2/P1)] Fig. 5C) against prepulse voltage. With prepulse voltages ranging from −20 to +60 mV, net facilitation was significantly greater (F(1,20)=5.25; p < 0.05) in cells cotransfected with erbin than in cells with Cav1.3 alone Fig. 5C). These results confirm that erbin promotes VDF by increasing the pool of channels undergoing prepulse-induced facilitation, rather than enhancing voltage dependence of entry into the facilitated state.

Erbin augments VDF of Cav1.3 Ba2+ currents. A, Representative Ba2+ current traces and double-pulse voltage protocol for measuring VDF in cells transfected with Cav1.3 alone or cotransfected with erbin. Currents were evoked by 10 ms test pulses from −90 to −20 mV before (P1) and after (P2) a conditioning 20 ms prepulse (Pre) to +60 mV. B, No effect of erbin on prepulse voltage dependence of facilitation. Percentage of facilitation was calculated as [(IP2/IP1) − 1] × 100 for cells for different prepulse voltages, normalized to that for a +60 mV prepulse, and plotted as percentage of maximum (Max.) facilitation against prepulse voltage for cells transfected with Cav1.3 alone (n=11) or cotransfected with erbin (n=10). C, Increased net facilitation attributable to erbin. Fratio represents the ratio of the P2 and P1 test currents and is plotted against prepulse voltage for Cav1.3 alone (n = 12) or Cav1.3 plus erbin (n = 10). *p < 0.05. Error bars represent SEM.
In these experiments, Ba2+ rather than Ca2+ was used as the charge carrier to resolve VDF without the competing effects of Ca2+-dependent inactivation, which would significantly diminish the amplitude of the P2 test current in double-pulse protocols. However, because Ca2+ is the physiologically relevant charge carrier, we also tested whether erbin influenced facilitation of Cav1.3 Ca2+ currents (Fig. 6). With a modified double-pulse protocol, Ca2+-dependent inactivation of Cav1.3 currents was evident as a decrease in the P2 relative to the P1 current (Fratio < 1) with prepulse voltages ranging from −20 to +30 mV (Fig. 6B). As would be expected of a Ca2+-dependent process, inactivation paralleled the amount of Ca2+ influx during the prepulse such that Fratio was smallest with a prepulse to 0 mV (Fig. 6B), which evoked the maximal inward Ca2+ current (data not shown). Although erbin did not affect the extent to which Cav1.3 Ca2+ currents inactivated (p > 0.18, for prepulses from −40 mV to +30 mV), prepulse steps from +50 to +80 mV caused facilitation (Fratio > 1) of Cav1.3 Ca2+ currents that was significantly greater (p < 0.05) in cells cotransfected with erbin than in cells with Cav1.3 alone (Fig. 6A,B). That erbin increased facilitation across this voltage range for Cav1.3 when either Ca2+ (Fig. 6) or Ba2+ Fig. 5) was used as the permeant ion supports modulation of voltage- rather than Ca2+-dependent facilitation. As for Cav1.3 Ba2+ currents, the effect of erbin on facilitation of Ca2+ currents was not accompanied by effects on current amplitude, because I–V relationships for Ca2+ currents in cells with Cav1.3 alone and Cav1.3 plus erbin were not significantly different (p=0.16; data not shown). Because the effects of erbin on facilitation of Cav1.3 Ca2+ currents were smaller and occurred across a narrower set of prepulse voltages than for Ba2+ currents, the molecular mechanism underlying erbin-dependent increases in VDF was analyzed further with Ba2+ as the charge carrier.

Erbin increases VDF of Cav1.3 Ca2+ currents. A, Representative Ca2+ current traces and double-pulse voltage protocol for measuring VDF in cells transfected with Cav1.3 alone (n=6) or cotransfected with erbin (n=7). Currents were evoked by 5 ms test pulses from −90 to −10 mV before (P1) and after (P2) a conditioning 10 ms prepulse (Pre) to varying voltages. B, Increased net facilitation for prepulse voltages greater than +40 mV in cells cotransfected with erbin. Fratio was determined and plotted against prepulse voltage as in Figure 5C. The dotted line indicates Fratio=1. Points falling below the line result from Ca2+-dependent inactivation with prepulse voltages evoking significant Ca2+ entry. *p < 0.05 by two-factor ANOVA and Tukey's post-test. Error bars represent SEM.
Erbin binding to α11.3 supports VDF of Cav1.3
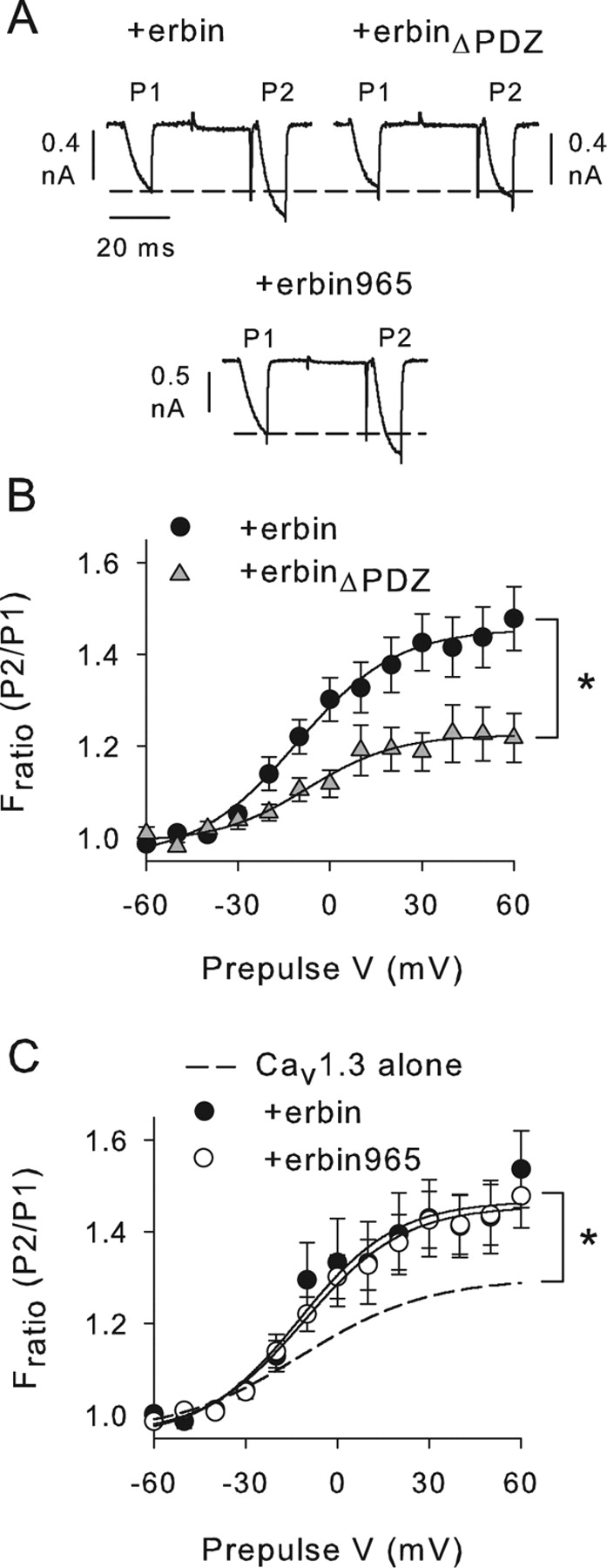
Based on our biochemical evidence supporting a direct interaction between erbin and α11.3 (Figs. 1, 2), we hypothesized that this interaction is required for the modulation of Cav1.3 VDF by erbin. If so, then preventing erbin from binding to α11.3 should abolish the increased VDF in cells with Cav1.3 plus erbin. This prediction was first tested with truncated erbin constructs that either lacked or were generally restricted to the PDZ domain (erbinΔPDZ and erbin965, respectively) (Fig. 7). Compared with cells cotransfected with full-length erbin, VDF was significantly reduced when Cav1.3 was cotransfected with erbinΔPDZ (F(1,16)=6.46; p < 0.05) (Fig. 7A,B). This result confirms the importance of the erbin PDZ domain for VDF. However, erbin contains other functional domains, including a leucine-rich repeat region (LRR) Fig. 1B) implicated in subcellular trafficking of membrane proteins (Legouis et al., 2003). Thus, it was possible that the PDZ domain acted in concert with the LRR of erbin to promote VDF of Cav1.3 through a mechanism independent of binding to α11.3. This was not the case, because erbin965, which did not contain the LRR, caused increased VDF compared with cells with Cav1.3 alone (F(1,15)=5.44; p < 0.05) (Fig. 7A,C). Moreover, the effect of erbin965 on VDF was not significantly different from full-length erbin (F(1,13)=0.05; p=0.83) (Fig. 7C). These results show that the PDZ domain of erbin is necessary and sufficient for modulation of Cav1.3 VDF.

Increased VDF depends on PDZ domain of erbin. A, Representative current traces obtained with the same voltage protocol as in Figure 5A for cells cotransfected with Cav1.3 plus erbin, erbinΔPDZ, or erbin965 (amino acids 965–1371). B, Deletion of erbinPDZ prevents effects on VDF. Fratio is plotted against prepulse V for cells cotransfected with Cav1.3 plus erbin (n=10) or erbinΔPDZ (n=8). *p < 0.05. C, Deletion of LRR of erbin does not prevent effects of erbin on VDF. Fratio was plotted against prepulse voltage for cells cotransfected with Cav1.3 plus erbin (n=10) or erbin965 (n=5). The dashed line represents data replotted from Figure 5C for cells transfected with Cav1.3 alone. *p < 0.05 for Cav1.3 plus erbin965 compared with Cav1.3 alone. Error bars represent SEM.
To characterize the importance of the PDZ-binding site in the α11.3 CT for erbin modulation, we took advantage of a naturally occurring α11.3 variant (α11.3b) in which inclusion of an alternatively spliced exon leads to truncation of the C-terminal domain and subsequent elimination of the PDZ-binding sequence (Safa et al., 2001; Xu and Lipscombe, 2001) (Fig. 8A). If erbin binding specifically to α11.3 CT was required for increasing VDF, channels containing α11.3b (Cav1.3b) should be insensitive to the effects of cotransfected erbin. The potential of erbin to interact with α11.3b was first tested in pull-down assays with his-tagged erbinPDZ and GST-tagged α11.3 CT or α11.3b CT (Fig. 8A). Relatively weak binding of erbinPDZ was observed with α11.3b CT compared with α11.3 CT (Fig. 8B). Consistent with these results, VDF of Cav1.3b channels was not significantly affected by cotransfected erbin (F(1,20)=0.06; p=0.81) (Fig. 8C). This was not attributable to the inability of Cav1.3b channels to undergo VDF, because VDF was quite robust in cells transfected with Cav1.3b alone (Fratio=1.42 ± 0.09 with a +60 mV prepulse; n=14). Together, our results support a mechanism in which the PDZ domain of erbin binding to the C-terminal domain α11.3 mediates enhanced VDF that is specific for Cav1.3 L-type channels.

Erbin does not cause increased VDF of Cav1.3b channels. A, Schematic showing differences in C-terminal domain of α11.3 and α11.3b. Parentheses indicate unique sequence including the PDZ-binding site (ITTL) in α11.3 but not α11.3b. Splicing of this region in α11.3b results in a truncated C-terminal domain with a distinct C-terminal sequence (ERML). B, ErbinPDZ interacts weakly with α11.3b CT. GST-α11.3 CT or GST-α11.3b CT was used in pull-down assay with his-S-erbinPDZ (1.5 or 3.0 μg). Top, Western blot detection of erbinPDZ. Bottom, Ponceau staining showing equal levels of immobilized GST-α11.3 CT (left) and GST-α11.3b CT (right) used in assay. C, No effect of erbin on VDF of Cav1.3b. Fratio was plotted against prepulse voltage for cells transfected with Cav1.3b alone (n = 14) or cotransfected with erbin (n=8). The dashed line represents data replotted from Figure 5C for cells transfected with Cav1.3 alone. Representative current traces obtained with a +60 mV prepulse are shown above. Error bars represent SEM.
Erbin relieves the inhibitory effect of α11.3 CT on VDF
In recordings of the short Cav1.3b variant, we found that VDF was on average greater than for Cav1.3 and was not different from that caused by erbin in cells cotransfected with the long Cav1.3 variant (F(1,22)=0.04; p=0.85) (Figs. 5C, 8C). This result suggested an intriguing mechanism in which the α11.3 CT, which is missing in Cav1.3b, may act as an inhibitory module for VDF. The absence of the inhibitory CT domain in α11.3b might therefore permit enhanced facilitation of Cav1.3b, which cannot be influenced further by erbin (Fig. 8C). If so, then α11.3 CT when expressed as a separate entity should suppress VDF of Cav1.3b. We tested this prediction with GFP-CT500 (Fig. 9). Compared with cells with Cav1.3b alone, VDF was significantly impaired by cotransfection with CT500 (F(1,16)=14.32; p < 0.01) (Fig. 9A). In contrast, CT500 did not significantly affect VDF for Cav1.3 (F(1,6)=0.49; p=0.51) (Fig. 9B). These results confirm an inhibitory role for the CT of α11.3 in VDF.

Autoinhibition of VDF by the CT of α11.3. A–C, Cotransfection of α11.3 CT fragment (CT500; amino acids 1655–2155) suppresses VDF. Effect of CT500 on Fratio is plotted against prepulse V in cells with Cav1.3b (n=6) (A), Cav1.3 (n=4; B), or Cav1.3 plus erbin (n=10; C). Representative current traces obtained with a +60 mV prepulse are shown above. Vertical scale bars, 0.8 nA; horizontal scale bars, 40 ms. *p < 0.01. D, Voltage-dependent inhibition of VDF by CT500. Percentage inhibition was measured as [1 − (Fratio+CT500/Fratio)] × 100, where Fratio is for Cav1.3b, Cav1.3, or Cav1.3 plus erbin, and Fratio+CT500 represents the mean Fratio for these groups cotransfected with CT500. Stronger prepulse voltage dependence of CT500 inhibition is observed for Cav1.3b and Cav1.3 plus erbin compared with Cav1.3. *p < 0.001. Error bars represent SEM.
Erbin interactions might facilitate voltage-dependent removal of the α11.3 CT, thus disinhibiting VDF as in Cav1.3b channels. If so, then CT500 should similarly “reinhibit” VDF of Cav1.3 channels modulated by erbin. As predicted, coexpression of CT500 strongly suppressed the increase in VDF caused by erbin (F(1,19)=23.18; p < 0.001) (Fig. 9C). However, in this experiment, VDF inhibition could have resulted simply from CT500 competitively binding to erbin and displacing it from the channel. This would then allow the endogenous CT of the channel to inhibit VDF, as in cells transfected with Cav1.3 alone. If this were the case, inhibition by CT500 in cells cotransfected with Cav1.3 plus erbin should be relatively invariant with prepulse voltage and similar to that seen in cells transfected with Cav1.3 alone. Alternatively, if CT500 inhibition involved interaction with an inhibitory site in Cav1.3 that was increasingly accessible in facilitated channels, CT500 inhibition in cells cotransfected with Cav1.3 plus erbin should increase with prepulse voltage, as in cells transfected with Cav1.3b alone. To resolve the underlying mechanism, we compared the prepulse voltage dependence of VDF inhibition by CT500 for Cav1.3, Cav1.3b, and Cav1.3 plus erbin (Fig. 9D). In cells transfected with Cav1.3 plus erbin, VDF inhibition by CT500 increased steeply with prepulse voltage and was not significantly different from that in cells transfected with Cav1.3b alone (F(1,15)=0.29; p=0.60) (Fig. 9D). In contrast, percentage of inhibition of VDF by CT500 in cells with Cav1.3 alone did not vary greatly with prepulse voltage and was significantly different from that in cells with either Cav1.3b (F(1,8)=31.0; p < 0.001) or with Cav1.3 plus erbin (F(1,13)=25.0; p < 0.001) (Fig. 9D). These results show that CT500 does not reverse the effects of erbin by displacing it from the channel. Rather, our data suggest that CT500 suppresses VDF in cells with Cav1.3b or Cav1.3 plus erbin through the same voltage-dependent mechanism.
Because VDF of L-type channels can be affected by the auxiliary Ca2+ channel β subunit (Cens et al., 1998), we determined whether substituting the β1b subunit used in our experiments with the β4 subunit influenced modulation of Cav1.3 VDF by erbin and α11.3 CT. Like β1b, β4 is highly expressed in the brain (Castellano et al., 1993; Ludwig et al., 1997; Vendel et al., 2006) and has been shown to associate with a large fraction of brain L-type channels (Pichler et al., 1997), although the identity of the β subunits interacting specifically with Cav1.3 in the brain has yet to be characterized. Although Cav1.3 channels containing β4 (Cav1.3-β4) exhibited VDF similar to that with β1b, there was no significant difference in VDF in cells with Cav1.3-β4 alone and those cotransfected with erbin (F(1,13)=0.05; p=0.83) Fig. 10A). However, VDF of the Cav1.3b variant with β4 (Cav1.3b-β4) was significantly increased compared with Cav1.3-β4 (F(1,11)=13.8; p < 0.01) Fig. 10A,B). Moreover, coexpression of CT500 with Cav1.3b-β4 significantly blunted VDF (F(1,11)=44.5; p < 0.001) Fig. 10B), similar to that observed for Cav1.3b channels containing β1b (Fig. 9A). We conclude that VDF modulation by erbin depends on which β subunit is associated with Cav1.3, and the inhibition of VDF imposed by the α11.3 CT is a fundamental regulatory feature of α11.3, which is removed by alternative splicing of the CT in Cav1.3b.

Effect of erbin but not α11.3 CT depends on auxiliary Ca2+ channel β subunit. A, Erbin does not increase VDF of Cav1.3 channels containing the β4 subunit (Cav1.3-β4). Fratio was determined from double-pulse protocol as described in Figure 5, except that P1 and P2 currents were evoked by 7 ms test pulses from −90 to −20 mV in cells transfected with Cav1.3-β4 (n=7) or cotransfected with erbin (n=8). B, CT500 inhibits VDF of β4-containing Cav1.3b channels (Cav1.3b-β4). Fratio was determined and plotted as in A for cells transfected with Cav1.3b-β4 (n=6) or cotransfected with CT500 (n=7). *p < 0.001. For A and B, representative current traces obtained with +80 mV prepulse are shown above. Error bars represent SEM.
Discussion
Our results reveal a novel mechanism controlling VDF of Cav1.3 Ca2+ channels. Erbin binding to the CT of the Cav1.3 α1 subunit alleviates an inhibitory effect of the CT, causing increased facilitation of Cav1.3 currents after a conditioning prepulse. This effect is seen for Cav1.3 channels containing the auxiliary β1b but not β4 subunit (Figs. 5C, 10A). The Cav1.3b variant lacks the PDZ-binding site with which erbin interacts and, more importantly, the autoinhibitory CT domain. As a consequence, Cav1.3b channels are “disinhibited,” exhibiting more robust VDF than Cav1.3, regardless of the identity of the auxiliary β subunit (Figs. 8⇑–10). Thus, the extent to which neuronal Cav1.3 channels undergo facilitation in response to membrane depolarization may depend on α11.3 splice variation and cell-type specific interactions with different β subunits and PDZ proteins.
Mechanisms of L-type channel facilitation
Facilitation of voltage-gated Ca2+ channels in response to a hormonal or depolarizing stimulus is an adaptive mechanism that can boost Ca2+ entry in excitable cells (Pietrobon and Hess, 1990; Dolphin, 1996). For Cav1.2 L-type channels, various mechanisms for facilitation have been characterized, including those involving phosphorylation of the channel by cAMP-dependent protein kinase (PKA) (Catterall, 2000) and association with calmodulin-dependent protein kinase II (CaMKII) (Dzhura et al., 2000; Hudmon et al., 2005; Grueter et al., 2006; Lee et al., 2006). Although PKA and CaMKII also cause facilitation of Cav1.3 channels (Qu et al., 2005; Gao et al., 2006), the enhanced facilitation caused by erbin and splice variation of the α11.3 CT in our study differs from these mechanisms in a number of ways. First, although PKA and CaMKII cause an increase in Cav1.3 peak current amplitude (Qu et al., 2005) and negative shift in voltage-dependent activation (Gao et al., 2006), respectively, erbin had neither effect on Cav1.3 activation in our study (Fig. 4). Second, CaMKII activation by insulin-like growth factor causes facilitation of both Cav1.3 and Cav1.3b (Gao et al., 2006), whereas we find that Cav1.3 but not Cav1.3b channels are subject to erbin-dependent facilitation (Figs. 5, 8). Third, neither PKA nor CaMKII-dependent facilitation of Cav1.3 requires conditioning depolarization (Qu et al., 2005; Gao et al., 2006), unlike the clearly voltage-dependent effects of erbin on Cav1.3 facilitation Fig. 5). Therefore, we conclude that erbin promotes facilitation of Cav1.3 channels via a different molecular pathway than that described previously for these channels by PKA or CaMKII. Although we cannot rule out the possibility that other second messengers are involved, our data implicate the α11.3 CT as a key regulatory element in this process.
Autoinhibitory function of the α11.3 CT and modulation by PDZ proteins
An autoinhibitory role for the CT of the Cav1.1 and Cav1.2 α1 subunits is well established (Wei et al., 1994; Morrill and Cannon, 2000; Gao et al., 2001). Deletion of the distal C-terminal domain of the α1 subunit enhances activation of Cav1.1 (Morrill and Cannon, 2000) and Cav1.2 (Wei et al., 1994) when these channels are expressed in recombinant systems. This upregulation of channel activity is reversed by reintroduction of the distal CT fragment as a separate entity (Gao et al., 2001; Hulme et al., 2005, 2006), much like the inhibitory effects on VDF that we observed when the α11.3 CT (CT500) was coexpressed with Cav1.3b (Figs. 9A,D, 10B) or Cav1.3 plus erbin C,D). How the α11.3 CT inhibits VDF is not clear but may involve intramolecular interactions with other domains in α11.3. Previous studies have shown that the CT of α11.2 and α11.1 is subject to proteolytic processing in vivo (De Jongh et al., 1991; Hell et al., 1993a) and that the cleaved distal CT product remains associated with the α1 subunit (Gerhardstein et al., 2000; Hulme et al., 2005, 2006). For Cav1.1 and Cav1.2, the distal CT interacts directly with the proximal CT of the α1 subunit at sites that are conserved in the corresponding regions of α11.3 (Hulme et al., 2005, 2006). The putative proximal CT interaction site is included in the CT500 fragment that inhibited VDF in our study (Figs. 9, 10B), whereas the distal CT interaction site is retained in the truncated CT of the α11.3b (Fig. 8A). Thus, it is possible that for Cav1.3, the interaction of distal and proximal CT sites forms an autoinhibitory module for VDF, which is disrupted when erbin binds to the distal CT of β1b-containing channels or when the distal CT is removed by alternative splicing in Cav1.3b. Coexpression of the CT500 fragment would inhibit VDF of Cav1.3b (Figs. 9A,D, 10B) or Cav1.3 plus erbin (Fig. 9C,D) through reassociation with the proximal CT, therefore restoring the autoinhibition. Although additional experiments are required to confirm this mechanism, our results are consistent with previous observations that interdomain interactions involving the α11.2 CT regulate Cav1.2 activation and inactivation (Ivanina et al., 2000; Kobrinsky et al., 2005), which might parallel the role of the CT in modulating Cav1.3 VDF.
Our findings that the Cav1.3 channels containing the β4 subunit were insensitive to modulation by erbin further underscore the importance of auxiliary β subunits in differentially regulating not only gating but also modulation of voltage-gated Ca2+ channels (Richards et al., 2004). Although additional investigation is necessary, β4 may impair physical association of erbin with Cav1.3 or channel transitions to facilitated states that are favored by erbin. Alternatively, given that β4 variants interact with cellular signaling molecules other than voltage-gated Ca2+ channels (Hibino et al., 2003; Vendel et al., 2006), β4 could recruit other modulators to the Cav1.3 channel complex that oppose the effects of erbin. Because both β1b and β4 are highly expressed in the brain (Ludwig et al., 1997; Pichler et al., 1997), whether neuronal Cav1.3 channels are modulated by erbin could vary according to which β subunit is associated with the channel complex.
PDZ proteins as modulators of voltage-gated Ca2+ channels
PDZ domain-containing proteins have emerged as multifaceted regulators of voltage-gated Ca2+ channel targeting and localization (Maximov et al., 1999; Zhang et al., 2005, 2006), modulation by G-protein-coupled receptors (Olson et al., 2005), and signal transduction (Weick et al., 2003). By selectively targeting and assembling Ca2+ channels with various signaling molecules, PDZ proteins can confer specific forms of Ca2+ channel regulation in different subcellular contexts. For example, the PDZ protein Shank clusters Cav1.3 but not Cav1.2 at postsynaptic sites with D1 dopamine receptors, enabling dopaminergic suppression of Cav1.3 L-type currents (Olson et al., 2005; Zhang et al., 2005). Our study is the first to show a more direct role for a PDZ protein in regulating Ca2+ channel gating. We have not tested whether related PDZ proteins, such as Shank, that interact with the α11.3 CT have an effect similar to erbin in increasing VDF of Cav1.3-β1b. Our findings that a truncated version of erbin containing primarily the PDZ domain (erbin965) (Fig. 7A,C) was sufficient for increasing VDF suggest that a PDZ/α11.3 CT interaction is the essential determinant for the modulation. However, PDZ proteins, including erbin and Shank, differ significantly in their overall structure and can vary in terms of the number of PDZ domains and the existence of other protein interaction motifs (Kim and Sheng, 2004). Such structural variations may alter the modulatory potential of PDZ proteins, perhaps by allowing them to scaffold multiple distinct signaling molecules. This raises the possibility that a wide range of PDZ interactions may influence other aspects of Cav1.3 function, thus enhancing the signaling capabilities of these channels in neurons. In this context, it is important to note that peptide mimetics of the class I PDZ-binding site in α11.3 Fig. 1A) may competitively disrupt interactions with multiple PDZ proteins and thus may not reflect the functional consequences of disturbing a single PDZ interaction.
Physiological significance of Cav1.3 channel facilitation and modulation by erbin/α11.3 splicing
Compared with previous descriptions of native L-type channel VDF (Pietrobon and Hess, 1990; Forti and Pietrobon, 1993; Kavalali and Plummer, 1996), VDF of Cav1.3 channels in our experiments occurred with relatively moderate depolarizing prepulses (−20 to +60 mV) (Figs. 5⇑⇑⇑⇑–10). This feature of VDF is consistent with previous reports of Cav1.3 splice variants cloned from a pituitary cell line (Safa et al., 2001) and L-type channels in neostriatal medium spiny neurons (Song and Surmeier, 1996) and hippocampal pyramidal neurons (Kavalali and Plummer, 1996). In cortical neurons, L-type channels influence neuronal activity via Ca2+-dependent plateau potentials (Nasif et al., 2005) and regulation of interspike intervals during repetitive firing (Pineda et al., 1998). Increased VDF of Cav1.3-β1b caused by erbin or expression of the Cav1.3b variant could amplify such activity-dependent Ca2+ signals, which normally control cortical excitability. Given that Cav1.3-β1b but not Cav1.3-β4 channels are subject to modulation by erbin (5,10 it is interesting to consider the lethargic mutation in the Ca2+ channel β4 subunit, which causes seizures and ataxia (Burgess et al., 1997). This mutation prevents interactions of β4 with the Ca2+ channel α1 subunit and causes compensatory increases in the association of α1 with other β subunits, including β1b (Burgess et al., 1999). Our results suggest a mechanism in which such pathological increases in Cav1.3-β1b channels might cause neurological defects through aberrant upregulation of Ca2+ signaling and neuronal excitability.
Footnotes
-
This work was supported by National Institutes of Health Grants R01 NS044922 (A.L.), T32 DA015040 (I.C.-J.), and R01 NS044521 (L.M.), the W. M. Keck Foundation (R.A.H.), and the Whitehall Foundation (A.L.). We thank Drs. D. Lipscombe and E. Perez-Reyes for Cav1.3 and β4 cDNAs, respectively; Dr. Jörg Striessnig for permission to use Cav1.3−/− mice; Drs. Geoff Murphy and Anjali Rajadhyaksha for providing Cav1.3−/− mouse brains; Amanda Castleberry for preparing the PDZ domain proteomic array used in the initial screen; and Dr. Frank Gordon for advice on statistical analyses.
- Correspondence should be addressed to Amy Lee, Department of Pharmacology, Emory University School of Medicine, 5123 Rollins Research Building, 1510 Clifton Road, Atlanta, GA 30322. alee{at}pharm.emory.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}