

Abstract
The specification of the intricate neuronal assemblies that characterize the forebrain is not well understood. The ventral spinal cord is specified through a concentration gradient of Sonic hedgehog (Shh) protein secreted by the notochord. Shh is expressed also in the forebrain neuroepithelium (neural Shh) and the underlying notochord and prechordal plate. Neural Shh is essential for the development of the prethalamus (ventral thalamus), but its effects on the thalamus (dorsal thalamus) are still unclear. We hypothesized that neural Shh would act on a previously regionalized dorsal diencephalic region to promote the emergence of specific thalamic nuclear and histological traits. To find out, we generated a conditional mouse mutant line specifically lacking Shh expression in the diencephalic neuroepithelium. We show that the transcription factor Gbx2, required for thalamic development downstream Shh, is expressed in our mutant in a restricted thalamic region and is necessary and sufficient for the differentiation of the medial and intralaminar thalamic nuclei. In the rest of the thalamus, neural Shh is required to promote neuronal aggregation into nuclei as well as axonal extension. In this way, the individual thalamic nuclei show differential dependence on Shh, Gbx2, or both for their differentiation. Additionally, Gbx2 is required for the survival of thalamic neurons.
Introduction
Brain regions differentiate stepwise, receiving first general regional specification during gastrulation and progressing down a genetic regulatory hierarchy. This cascade ends in the effector genes that direct formation of neuronal nuclei and confer mature functional attributes to neurons. Transient embryonic structures called organizers and their secreted products are key in this process. The best known model is the specification of ventral spinal cord neurons through Sonic hedgehog (Shh) protein secreted by the notochord (Dessaud et al., 2008). Shh is expressed in the forebrain neuroepithelium (neural Shh) and the underlying notochord and prechordal plate. If and how the mechanisms found in the ventral spinal cord have become modified to specify the convoluted three-dimensional neuronal assemblies found in the forebrain is not well understood. The thalamus (dorsal thalamus) is a forebrain region constituted mostly by neurons projecting to cortex and striatum, and, in rodents, it can be subdivided into several nuclear groups harboring altogether >30 nuclei and subnuclei (Swanson, 1992). These differ from each other specifically in position, morphology, marker expression, membrane properties, and cortical targets (Jones, 2007a). Thalamic subdivisions originate as five undifferentiated cell masses or pronuclei that gradually resolve into specific nuclear groups: central pronucleus (anterior, ventral, and intralaminar groups), dorsal pronucleus (lateral and posterior groups), medial pronucleus (medial group), lateral geniculate pronucleus, and medial geniculate pronucleus (Rose, 1942; Jones, 2007b).
Neural Shh [from the zona limitans interthalamica (ZLI) and diencephalic tegmentum] is essential for prethalamus (ventral thalamus) specification (Kiecker and Lumsden, 2004; Vieira et al., 2005; Zeltser, 2005; Scholpp et al., 2006; Vieira and Martinez, 2006), but its role in (dorsal) thalamic development is still unclear. Shh controls thalamic expression of transcription factor gene Gbx2 (Hashimoto-Torii et al., 2003), required for thalamic development (Miyashita-Lin et al., 1999; Hevner et al., 2002). We hypothesized that the role of neural Shh would be to take over the last steps of the thalamic specification cascade, which lead to the emergence of specific thalamic histogenesis and nucleogenesis, and that this would be done in part through activation of Gbx2.
To address this question, we analyzed thalamic differentiation in the Gbx2 mutant and in a conditional mutant lacking diencephalic Shh expression. We found that, in the neural Shh-deficient diencephalon, Gbx2 is expressed by default in a restricted area and is necessary and sufficient for the development of the medial and intralaminar thalamic nuclei. The rest of the thalamus requires neural Shh to promote proper migration and neuronal aggregation into nuclei as well as axonal extension. Therefore, the individual thalamic nuclei form three groups according to their dependence on neural Shh, Gbx2, or both for their differentiation. Additionally, we show that Gbx2 is essential for neuronal survival in the prenatal thalamus.
Materials and Methods
Mouse lines
Experiments with animals were performed in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC) and under authorization of Az 32.22/Vo (“Ordnungsamt der Stadt Göttingen”). To obtain embryos and fetuses for the analysis, timed-pregnant females of the appropriate crossings were killed by cervical dislocation.
Foxb1–Cre mouse line (Foxb1tm1(cre)Gabo).
Homologous recombination was used to replace the Foxb1 coding sequence by the Cre recombinase cDNA, and a mouse line was created (kept in the C57BL/6 background) (Zhao et al., 2007). This line expresses Cre under the control of the regulatory sequences of Foxb1. All mutant mice used for our study were heterozygous for Foxb1Cre, and therefore they were heterozygous for Foxb1 and showed normal phenotype as expected (Alvarez-Bolado et al., 2000; Kloetzli et al., 2001). No homozygotes were used in this study.
Floxed Shh mouse line (Shhtm2Amc).
This line was generated by Dr. MacMahon (Harvard University, Cambridge, MA) (Dassule et al., 2000) and obtained through The Jackson Laboratory. In these mice, exon 2 of the Shh locus [which encodes approximately half of the active N-terminal Shh signal that is essential for Shh function (Fan et al., 1995; Hynes et al., 1995; López-Martínez et al., 1995; Martí et al., 1995; Roelink et al., 1995)] is flanked with loxP sites. The same floxed line has been used for the study of the developing limb (Lewis et al., 2001).
Foxb1tm1(cre)Gabo × Shhtm2Amc (Shh-c).
After crossing the floxed Shh line with a Cre line, a mutant mouse line is generated (Shh-c) carrying a recombined (i.e., inactive) Shh locus in Cre-expressing cells. Because the caudal diencephalon (including the thalamus) belongs entirely to the Foxb1 lineage, i.e., expresses Cre in the Foxb1–Cre line (Zhao et al., 2008), this line lacks functional Shh in the diencephalic neuroepithelium (see Results). When Shh expression is activated, the recombined Shh locus produces a truncated mRNA lacking exon 2. In situ hybridization (ISH) probes can be designed against the 3′ untranslated region (UTR), labeling all Shh mRNA forms, either full length (functional) or truncated (nonfunctional). Probes against exon 2, however, label only the full-length form.
Gbx2 mutant mouse line.
In this line, provided by Drs. Gail Martin and Alex Joyner, all coding sequences of exon 2, including the homeobox, have been deleted (Wassarman et al., 1997). A portion of the 3′ UTR of Gbx2 remains unaltered in the targeted Gbx2 locus (Wassarman et al., 1997), and in situ hybridization probes against this sequence can label the expression of Gbx2 mRNA (although lacking the homeodomain and therefore nonfunctional) in the Gbx2 mutant (see Results).
Reporter lines.
To map the embryonic brain region in which Cre-mediated recombination takes place, we crossed our mice with the ROSA26R reporter mouse line (Soriano, 1999). In ROSA26R animals, the reporter gene β-galactosidase is inserted in the constitutively active ROSA locus downstream a floxed stop codon. During Cre-mediated recombination, the stop codon is deleted and β-galactosidase is constitutively produced. This reporter is a lineage marker because, in mice carrying both the Foxb1Cre and the ROSA26R alleles, cells expressing Foxb1 and any cell derived from them will permanently express β-galactosidase. To label the axons of neurons of the Foxb1 lineage, we crossed our Foxb1Cre mice with the Z/AP (Lobe et al., 1999) reporter mouse line (C57BL/6) (see below, Axonal labeling by alkaline phosphatase activity). In Z/AP animals, Cre-mediated recombination activates constitutive expression of human placental alkaline phosphatase (hPLAP). This reporter is also a lineage marker.
Axonal labeling by alkaline phosphatase activity
In Foxb1Cre Z/AP animals, Foxb1 lineage neurons will express hPLAP. Because hPLAP attaches to axonal membranes, it is a marker of axons of lineage-labeled neurons (Fields-Berry et al., 1992; Gustincich et al., 1997; Leighton et al., 2001).
Alkaline phosphatase activity detection.
This protocol has been described previously (Lobe et al., 1999). Briefly, brains of the appropriate genotypes were collected, fixed (4% paraformaldehyde) on ice for 60 min, agarose embedded, cut into 150-μm-thick sections, fixed again (4% paraformaldehyde/0.2% glutaraldehyde) on ice for 60 min, incubated for 30 min at 72°C to inhibit endogenous phosphatase, rinsed in alkaline phosphatase buffer (in mm: 100 Tris-Cl, pH 9.5, 100 NaCl, and 10 MgCl2), incubated with staining solution (250 μl of nitroblue-tetrazolium-chloride plus 187.5 μl of 5-bromo-4-chlor-indolyl-phosphate per 50 ml of alkaline phosphatase buffer) overnight (4°C), and fixed (4% paraformaldehyde) for 60 min (4°C).
In situ hybridization in whole mount
ISH was performed as described previously (Wilkinson, 1992). The probes were cloned by PCR (primers available on request). Briefly, the embryos were fixed in paraformaldehyde 4% in PBS overnight (4°C), washed in PBS plus 0.1% Tween 20 (PBT), and stored at −20°C in methanol. For ISH, the embryos were rehydrated, bleached (6% H2O2), digested [10 μg/ml proteinase K in PBT at room temperature (RT)], washed (2 mg/ml glycine/PBT), postfixed (4% paraformaldehyde/0.2% glutaraldehyde/PBT), prehybridized for 1–2 h (70°C), and hybridized overnight (70°C). They were then washed (50% formamide, 5× SSC, pH 4.5, and 1% SDS at 70°C), rinsed [100 mm maleic acid, 150 mm NaCl, 2 mm levamisole, and 0.1% Tween 20 (MAB)], and incubated in 10% sheep serum in MAB/2% blocking reagent (Roche Diagnostics) for 2–3 h (RT) and then in anti-digoxigenin alkaline phosphatase antibody (Roche Diagnostics) overnight (4°C). The embryos were rinsed and then left in MAB overnight (4°C). The embryos were then incubated in BM-Purple (Roche Diagnostics) with levamisole (RT) and, after color developed, washed in PBT, pH 4.5, fixed in 4% formaldehyde/0.1% glutaraldehyde overnight (4°C), and transferred into 80% glycerol/PBT.
ISH on sections
Nonradioactive ISH was performed on cryosections (15 μm thick) that were fixed in 4% paraformaldehyde and acetylated after sectioning. Prehybridization, hybridization, and washing steps were performed with the help of an automatic liquid-handling unit (Genesis RSP 200; Tecan), and the digoxigenin-labeled probe was detected by a dual-amplification procedure (Yaylaoglu et al., 2005; Visel et al., 2006).
Bromodeoxyuridine labeling
Pregnant mice from appropriate crossings were intraperitoneally injected with bromodeoxyuridine (BrdU) (RPN201; GE Healthcare) (50 μg/g body weight). For short survival data, we injected at approximately embryonic day 12.75 (E12.75) (at 6:00 P.M. on day 12 postcoitum) and collected the embryos either 1.5 or 3 h later. According to thalamic birthdating data (Altman and Bayer, 1988; Clancy et al., 2001, 2007), at this stage, neurons are being produced in all areas of the thalamic neuroepithelium. For long-term survival, we injected BrdU on day E12.5 at 12:00 P.M., 3:00 P.M., and 6:00 P.M. (Takahashi et al., 1993) and collected the fetuses at E18.5.
We detected cell proliferation on cryosections (10 μm) by means of anti-BrdU antibody M0744 (1:100) (Dako), after epitope retrieval in 2 m HCl for 30 min at 37°C. We used the nuclear marker 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Invitrogen) as a counterstain. In the short-survival animals (E12.75), we examined the sections under confocal microscopy and counted BrdU-labeled and unlabeled cells in 100-μm-wide bins encompassing the width of the neuroepithelium (apical to basal side) at three rostrocaudal levels and calculated the labeling index (BrdU-labeled cells as percentage of total cells) (Takahashi et al., 1993; Warren et al., 1999; Ishibashi and McMahon, 2002).
Apoptosis detection
We selected cryostat sections of E12.5 and E18.5 brains at three thalamic rostrocaudal levels in three individuals per genotype (wild type, Shh-c, Gbx2−/−). We pretreated the sections with 4% paraformaldehyde (20 min) and proteinase K (1.5 μg/ml, 5 min) at room temperature and then labeled the apoptotic cells with the ApopTag TUNEL (terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling) kit (Millipore Bioscience Research Reagents) according to the instructions of the manufacturer. We used DAPI as counterstain and counted absolute number of apoptotic cells per histological section of the thalamus (both sides) under high magnification in three individuals per genotype.
Statistical analysis
ANOVA of the cell counting results (BrdU and apoptosis) was performed with Prism software (GraphPad Software).
Results
Abolition of functional Shh expression and of Shh signaling in the caudal diencephalon of Shh-c mutants
At E10.0, Foxb1 is expressed in the prethalamus and thalamus (Fig. 1A), overlapping the domain of the incipient ZLI (Fig. 1B) (Zhao et al., 2008). In Shh-c mutants at this age, Shh expression has been abolished in the ZLI as well as in domains of the rostral diencephalon (Fig. 1C). As expected (Ishibashi and McMahon, 2002), the caudal diencephalon is smaller in the mutant (Fig. 1B,C, dotted line). An in situ probe specifically labeling full-length Shh mRNA (Shh–exon 2 probe; see Materials and Methods) failed to label the mutant diencephalon (Fig. 1D), demonstrating that the Shh-c neuroepithelium does not produce functional Shh mRNA. The telencephalic Shh expression domains were intact (Fig. 1D, MGE and sub), because they do not overlap with Foxb1 expression (Fig. 1A). At E12.5, all major regions of the brain can be recognized by specific marker expression (Shimamura et al., 1995). At this age, Foxb1 drives Cre recombination activity in the caudal diencephalon, dorsal and ventral (as reflected by lineage labeling in ROSA26R crossings; Fig. 1E) (Zhao et al., 2008). In wild-type embryos, Shh showed a very pronounced and characteristic expression pattern in the caudal diencephalon (Fig. 1F, ZLI and TG) as well as the rostral diencephalon (hypothalamus) (Fig. 1F, LH). In the Shh-c mutant, expression of Shh was absent from the diencephalon (Fig. 1G), and this was confirmed by an exon 2 probe (Fig. 1H). Expression of Shh receptor Ptch1 is diagnostic of a functioning Shh signaling pathway (for review, see Lewis et al., 2001). Ptch1 labeled the wild-type thalamus at this age (Fig. 2A) but was absent in the Shh-c mutant caudal diencephalon (Fig. 2B), indicating Shh pathway abolition in this region.

Abolition of functional Shh expression in the Shh-c caudal diencephalon. A–D, F–H, Whole-mount in situ hybridization for the genes and genotypes indicated. E, Foxb1 lineage mapping by β-galactosidase detection in Foxb1Cre/ROSA26R heterozygotes. A, B, At E10.0, Foxb1 expression (A) overlaps with the incipient ZLI as labeled by Shh (B). C, D, In the Shh-c mutant, Shh transcription is not active in the ZLI but in the diencephalic tegmentum (C). An exon 2 probe shows that all diencephalic Shh mRNA at this age is nonfunctional (D). The dotted lines in B and C show that the mutant thalamus is smaller. E, F, At E12.5, Foxb1 lineage labeling is present in most of the thalamic region (E), overlapping thalamus and ZLI as labeled by Shh (F). The Shh-c mutant shows almost no Shh transcriptional activation in the caudal diencephalon (G) and no functional Shh at all (H). For the abbreviations used in the figures, see Table 1.

Thalamic regionalization in Shh-c mutants. Whole-mount in situ hybridization of hemisected E12.5 mouse brains, probes, and genotypes as indicated. A, B, Lack of Ptch1 expression indicates that the Shh signaling pathway is abolished in the Shh-c mutant diencephalon. C–F, The ZLI has disappeared in the Shh-c mutant, as determined by expression of Nkx2-2 and Lhx1. G, H, Thalamic expression of Dbx1 is preserved in the mutant. I, J, Gbx2 is expressed in a small restricted thalamic domain at E12.5 in the Shh-c mutant (arrow in J).
Abolition of the ZLI in the Shh-c mutant mouse
Other ZLI markers confirmed lack of this domain in the mutant. Expression of ZLI marker gene Nkx2-2, which is downstream Shh (Kitamura et al., 1997; Kiecker and Lumsden, 2004), was absent from the Shh-c mutant (Fig. 2C,D). Expression of transcription factor gene Lhx1, a marker of the zona limitans, prethalamus, and thalamic eminence (Bachy et al., 2001), was also absent from the Shh-c ZLI (Fig. 2E,F). To evaluate the thalamus of the Shh-c mutant, we used expression of specific markers Dbx1 (Shoji et al., 1996; Ishibashi and McMahon, 2002) and Gbx2 (Miyashita-Lin et al., 1999; Hevner et al., 2002). In the Shh-c thalamus, Dbx1 was still expressed (Fig. 2G,H), indicating that initial regional specification of the thalamus has taken place in the mutant. Gbx2 was reduced to a very small domain (Fig. 2I,J).
Specific markers in the wild-type thalamus
To explore thalamic differentiation, we chose five marker genes whose thalamic expression has been established: Calb2–Calretinin (Arai et al., 1991, 1994; Winsky et al., 1992; Frassoni et al., 1998), Gbx2 (Jones and Rubenstein, 2004), Lhx2 (Nakagawa and O'Leary, 2001), Neurog2 (Nakagawa and O'Leary, 2001), and Cdh6 (Jones and Rubenstein, 2004). Because the Shh-c mutants survive up to E18.5, we performed our analysis of the wild type also at this age. We first determined marker expression on transverse sections of E18.5 wild-type thalamus, at four rostrocaudal thalamic levels (Fig. 3, Tables 1, 2).
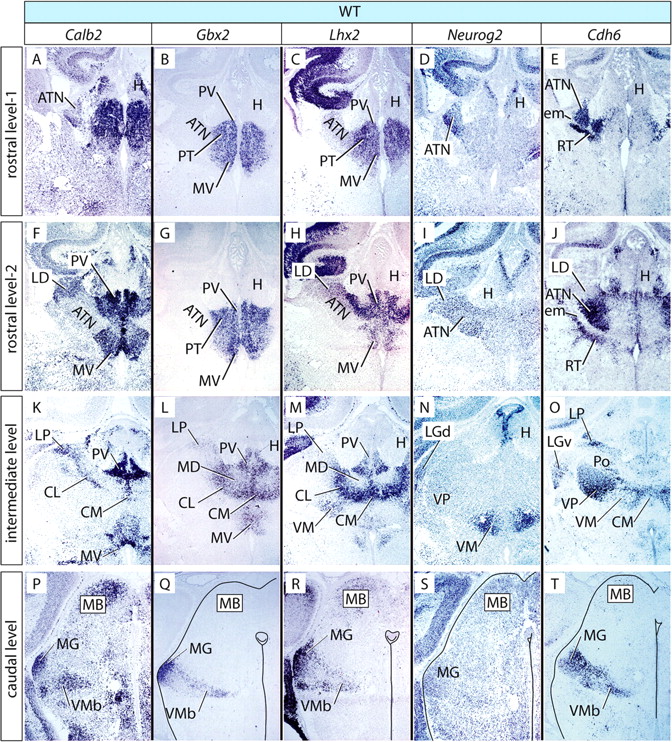
Marker expression in the E18.5 wild-type thalamus. A–T, In situ hybridization on E18.5 mouse brain sections for five marker genes as indicated on top of each column. Rows show sections at given rostrocaudal levels, as indicated on the left side, labeled for each of the five markers. Columns correspond to four rostrocaudal levels labeled with the same probe. Some structures have been outlined for clarity. For details, see Results.
Abbreviations used for thalamic and other structures
Pronuclear markers in the wild-type thalamus
Central pronucleus (anterior and ventral nuclear groups)
The anterior group was labeled by Lhx2 (weakly), Neurog2, and Cdh6 (Fig. 3C–E,H–J, ATN). The ventral group was labeled by Lhx2, Neurog2, and Cdh6 (Fig. 3M–O, VP and VM). The “tail” of the central pronucleus, represented by the ventromedial basal, was labeled by Calb2, Gbx2, Lhx2, and Cdh6 (Fig. 3P–R,T, VMb).
Medial pronucleus (medial and intralaminar nuclear groups)
We include the intralaminar nuclei in the medial pronucleus (not in the central, as did Rose, 1942) because of marker and phenotype similarity (see below and Discussion). All derivatives of this pronucleus expressed Gbx2 (Fig. 3B,G, L, CL, CM, MD, MV, PT, PV). Additionally, they (except the mediodorsal nucleus) expressed Calb2 and Lhx2 (Fig. 3A–C, F–H, K–M).
Dorsal pronucleus (lateral and posterior nuclear groups)
The laterodorsal nucleus expressed Calb2, Lhx2, and Neurog2 (Fig. 3F,H,I, LD). The lateral posterior nucleus was labeled by Calb2, Gbx2, and Cdh6 (Fig. 3K,L,O, LP). The posterior nucleus expressed Cdh6 (Fig. 3O, Po).
Lateral geniculate pronucleus
The lateral geniculate pronucleus was labeled by Neurog2 and Cdh6 (Fig. 3N,O, LG).
Medial geniculate pronucleus
The medial geniculate pronucleus was labeled by Calb2, Gbx2, Lhx2, and Cdh6 (Fig. 3P–R,T, MG).
General appearance of the Shh-c thalamus
In the Shh-c brain, the prethalamus is absent so that the thalamus “hangs” over the third ventricle, particularly on rostral sections (Fig. 4A–J). At caudal levels, the Shh-c thalamus was embedded into the midbrain (Fig. 4K–T). By counting the number of histological sections, we estimated that the Shh-c thalamus was smaller than the wild type, on the rostrocaudal axis, by ∼50%, as could be expected (Ishibashi and McMahon, 2002).

Marker expression in the E18.5 Shh-c thalamus. A–T, In situ hybridization on E18.5 mouse brain sections for five marker genes as indicated on top of each column. Rows show sections at a given rostrocaudal level, as indicated on the left side, labeled for each of the five markers. Columns correspond to four rostrocaudal levels labeled with the same probe. The arrows in J and O mark continuous rostrocaudal expression of Cdh6 in the central pronucleus. Some structures have been outlined for clarity. For details, see Results.
Despite gross morphological alteration, the general position of the nuclear groups in the rostrocaudal and dorsoventral axes was correct. Comparison of Tables 2 and 3 shows that, in the Shh-c thalamus, the medial pronucleus was well preserved, whereas the rest of them, particularly the central and dorsal pronuclei, were morphologically very altered but mostly preserved specific marker expression (only Lhx2 and Neurog2 were abolished in some nuclei).
Markers and nuclei in the Shh-c thalamus
The medial pronucleus is preserved in the Shh-c thalamus
Central pronucleus (anterior and ventral nuclear groups)
The mutant anterior thalamic nuclei could be recognized by their preserved Cdh6 expression and their relation to the external medullary lamina. The anterior thalamic nuclei lost, however, Lhx2 and Neurog2 expression (Fig. 4C,D). The ventral group lost the expression of Neurog2 (Fig. 4I) and preserved only the most reduced expression of Cdh6 (Fig. 4J,O). The entire central pronucleus (anterior plus ventral groups) seemed reduced to a rostrocaudal row of Cdh6-expressing cells (Fig. 4J,O, arrows). The tail of the central pronucleus (the ventromedial basal) was an exception, because it lost Calb2 and Cdh6 but still expressed Gbx2 and Lhx2 in the mutant (Fig. 4Q,R).
Medial pronucleus (medial and intralaminar nuclear groups)
This pronucleus was well preserved in morphology and marker expression, including Calb2 (Fig. 4A,F,K), Lhx2 (Fig. 4C,H,M), and Gbx2 (Fig. 4B,G,L). Expression of Gbx2 labeled the entire pronucleus and is an argument toward including the intralaminar nuclei in this medial pronucleus (not the central) (Rose, 1942).
Dorsal pronucleus (lateral and posterior nuclear groups)
The laterodorsal nucleus, expressing Calb2, was present in the mutant (Fig. 4F) but lacked expression of markers Lhx2 and Neurog2 (Fig. 4H,I). The lateral posterior nucleus preserved Calb2, Gbx2, and Cdh6 expression (Fig. 4K,L,O) but lost Lhx2 (Fig. 4M). The posterior nucleus appeared lost, to judge by Cdh6 expression (Fig. 4J).
Lateral geniculate pronucleus
We could not identify a lateral geniculate nucleus in the Shh-c thalamus.
Medial geniculate pronucleus
The medial geniculate pronucleus preserved expression of Calb2 and Gbx2 (Fig. 4P,Q) but lost Lhx2 and Cdh6 in the mutant (Fig. 4R,T).
In summary, the medial pronucleus (in which we include the intralaminar group) was comparatively little affected by lack of neural Shh, whereas the other four pronuclei showed major alteration.
No thalamocortical axons in the Shh-c mutant
Extension of axons projecting to the cortex is an important differentiation trait of thalamic neurons (for review, see López-Bendito and Molnár, 2003). To explore this capability in the Shh-c mutant thalamus, we crossed our Shh-c line with the reporter mouse line Z/AP (Lobe et al., 1999), which results in labeling of the axons of Foxb1 lineage cells (see Materials and Methods). Because the entire thalamus belongs to the Foxb1 lineage (Fig. 1E) (Zhao et al., 2008), Foxb1–Cre Z/AP mice showed staining of thalamocortical axons as expected (Fig. 5A). The Shh-c mutant, however, showed a completely blank cortex (Fig. 5B). The second mutant used in this study, the Gbx2-deficient mouse (see below), also lacked thalamocortical axons (Fig. 5C) as reported previously (Miyashita-Lin et al., 1999; Hevner et al., 2002).

No thalamic axons reach the cortex in the Shh-c brain. Detection of alkaline phosphatase activity in transverse sections of wild-type (A), Shh-c (B), and Gbx2−/− (C) brains at E18.5. The wild-type cortex (A) shows abundant labeled thalamic axons in the cortex, whereas the Shh-c (B) and the Gbx2−/− (C) cortices are completely unlabeled.
No regionalization defect in the Gbx2 mutant
Gbx2 is a transcription factor specifically expressed in the entire mouse thalamic primordium starting at E11.5 (Bulfone et al., 1993a,b; Hashimoto-Torii et al., 2003) and is essential for thalamic development (Miyashita-Lin et al., 1999; Hevner et al., 2002). Because thalamic expression of Gbx2 is under the control of Shh (Hashimoto-Torii et al., 2003), we were surprised to find a small domain of Gbx2 expression in the Shh-c thalamus at E12.5 (Fig. 2J, arrow), as well as essentially normal thalamic expression at E18.5 (Fig. 4B,G,L,Q). We asked whether this late, default expression of Gbx2 in the absence of neural Shh was perhaps “rescuing” some traits of the wild-type phenotype in the Shh-c mutant, making its phenotype milder. Therefore, we wanted to compare the Gbx2−/− thalamus with the Shh-c thalamus. We used a Gbx2 mutant in which most of exon 2, containing the homeobox-coding sequence, has been deleted (Wassarman et al., 1997) (see Materials and Methods). First, we detected regional marker genes at E12.5 to assess regionalization in this mutant. Probes against Ptch1, Gli1, Nkx2-2, Lhx1, Emx2, Pax6, Dlx2, Dbx1, and Irx3 detected no changes in the regionalization of the caudal diencephalon, except for the smaller size of the thalamus (data not shown). We then proceeded to analyze thalamic differentiation in the Gbx2 mutant at E18.5.
Abolition of the medial pronucleus in the Gbx2 mutant
As was the case for the Shh-c mutant, the Gbx2-deficient thalamus was smaller than normal but showed correct dorsoventral and anteroposterior axes, as judged from the presence of a morphologically recognizable habenula (epithalamus) dorsally and the medial geniculate nucleus caudally (see below) (Fig. 6, Table 4). Comparison of Tables 2⇑–4 shows that, in this mutant, the medial pronucleus is specifically altered in morphology, although it maintains marker expression. The central and dorsal pronuclei were very affected (like in the Shh-c), but the lateral and medial geniculate pronuclei seemed relatively intact.

Marker expression in the E18.5 Gbx2 thalamus. A–T, In situ hybridization on E18.5 mouse brain sections for five marker genes as indicated on top of each column. Rows show sections at a given rostrocaudal level, as indicated on the left side, labeled for each of the five markers. Columns correspond to four rostrocaudal levels labeled with the same probe. The arrows in B, C, G, H, L, and M indicate marker expression in morphologically abolished medial pronucleus. Some structures have been outlined for clarity. For details, see Results.
Markers and nuclei in the Gbx2−/− thalamus
Central pronucleus (anterior and ventral nuclear groups)
The anterior group, although very much altered morphologically, showed ectopic Calb2 expression (Fig. 6A), lost Lhx2 (Fig. 6C,H) and Neurog2 (Fig. 6D,I), and preserved Cdh6 expression (Fig. 6E,J). The ventral group seemed reduced to a single rostrocaudal region preserving, however, all its markers (Lhx2, Neurog2, and Cdh6) (Fig. 6H–J,M–O, VEN). The ventromedial basal preserved three of its four markers (Calb2, Gbx2, and Cdh6) (Fig. 6P,Q,T) and lost Lhx2 (Fig. 6R). The smaller size of this nucleus in the Gbx2 mutant brought to the fore a structure labeled by Calb2 and Lhx2 and perhaps corresponding to the subparafascicular nucleus (Fig. 6P,R, arrowheads).
Medial pronucleus (medial and intralaminar nuclear groups)
Truncated, nonfunctional Gbx2 mRNA can still be detected in the Gbx2 mutant (see Materials and Methods), allowing for recognition of the medial pronucleus. This marker as well as Lhx2 showed complete morphological abolition of the medial and intralaminar groups in the Gbx2 mutant (Fig. 6B,C,G,H,L,M, arrows).
Dorsal pronucleus (lateral and posterior nuclear groups)
The laterodorsal nucleus had disappeared, whereas the lateral posterior preserved Cdh6 expression (Fig. 6O) but lost Gbx2, Calb2, and Lhx2 expression. The posterior nucleus preserved Cdh6 (Fig. 6O).
Lateral geniculate pronucleus
The lateral geniculate pronucleus preserved its two markers, Neurog2 and Cdh6 (Fig. 6N,O).
Medial geniculate pronucleus
The medial geniculate pronucleus preserved all its markers, Calb2, Gbx2, Lhx2, and Cdh6 (Fig. 6P–R,T).
Basic helix–loop–helix gene expression in the mutant thalamic neuroepithelium
The phenotypes observed could be the result of transdifferentiation or lack of differentiation attributable to incorrect activation of proneural genes, a family of basic helix–loop–helix (bHLH) transcription factors governing fate acquisition (Guillemot, 2007a,b). Neurog1, Neurog2, and Olig3 are bHLH transcription factors with major roles in telencephalic fate acquisition (Fode et al., 2000) and also expressed in the thalamic neuroepithelium (González et al., 2002; Vue et al., 2007). Mining the expression pattern database GenePaint (www.genepaint.org) (Alvarez-Bolado and Eichele, 2006) for bHLH genes with developmental thalamic expression yielded two additional ones (Neurod1 and Neurod4). In situ hybridization labeling of the thalamic neuroepithelium at E12.5 did not show loss of expression of any of these five genes in the Shh-c and Gbx2 mutants compared with the wild type (Fig. 7).

Differentiation markers in the thalamus. In situ detection of mRNA on transverse sections of E12.5 (A–O) or E18.5 (P–U) mouse brains for the genes and genotypes indicated. At E12.5, expression of Olig3 (A–C), Neurog1 (D–F), Neurog2 (G–I), Neurod1 (J–L), and Neurod4 (M–O) is maintained in the mutants. At E18.5, expression of pan-neuronal marker gene Tubb3 (P–R) and astroglial marker gene Gfap (S–U) does not show changes in the mutant brains. In P, Q, S, and T, the thalamus has been outlined for clarity.
We then used in situ hybridization to label mRNA for the genes Tubb3 (β tubulin 3; a pan-neuronal marker) and Gfap (glial fibrillary acidic protein; an astroglial marker) to assess possible changes in cell type in the E18.5 mutant thalamus. Our results (Fig. 7P–U) showed no change in the neuron versus glia composition of the mutant thalamus.
Cell proliferation in the mutant thalamic neuroepithelium
Next we asked whether alterations in proliferation could be involved in the phenotypes observed. It is known that Shh deficiency results in decreased proliferation at approximately E9.0, i.e., during the period of expansion of neural precursors (Ishibashi and McMahon, 2002), as witnessed by the very small size of the early thalamic primordium in our Shh-c mutants. We wanted to investigate the proliferative capabilities of the Shh-c neuroepithelium during the period of neuron production (i.e., after E11.0) (Altman and Bayer, 1988; Clancy et al., 2007). To this purpose, we labeled the thalamic neuroepithelium at E12.75 with BrdU (Fig. 8A–C). At this age, all rostrocaudal levels of the thalamic region should be actively proliferating (for details, see Material and Methods). We were not able to find statistically significant differences in absolute numbers of labeled cells or in labeling index between the three genotypes investigated, after either 1.5 h (data not shown) or 3 h survival (Fig. 8D,E).

BrdU labeling in the mutant thalamus. A–C, BrdU-labeled sections of E12.75 thalamus of the three genotypes as indicated. White arrows indicate the labeled thalamic neuroepithelium. D, E, Results from counting absolute numbers of labeled cells (D) and labeling index (E) in the E12.75 thalamic neuroepithelium (±SD) after 3 h. survival. No statistically significant differences were detected. F–I, BrdU-labeled cells (injection at E12.5, collection at E18.5) in the wild-type (F, H) and Shh-c (G, I) thalamus. The thalamus has been outlined in white. Rostrally (F, G), the wild type showed labeled cells at every mediolateral level, whereas in the Shh-c (G), they were confined to a band surrounding the ventricle (dotted line), and the mantle layer (arrow) showed sparse labeling. More caudally (H, I), most labeled cells in the Shh-c (I) did not progress beyond intermediate mediolateral levels (dotted line).
Distribution of BrdU labeling in the E18.5 Shh-c thalamus
Migration alterations could contribute to the mutant thalamic phenotypes. To test this possibility, we wanted to examine the overall distribution in the thalamus of neurons born on a specific day. We labeled brains of the three genotypes with BrdU at E12.5 and collected them at E18.5, when most thalamic neurons have already been produced (Fig. 8F–I). In the wild-type thalamus (Fig. 8F,H) as well as the Gbx2 mutant thalamus (data not shown), we could find labeled neurons at every mediolateral level of the thalamus, reaching the periphery of this region. However, in the Shh-c thalamus at the rostral level almost all labeled cells were confined to a band around the ventricle (Fig. 8G). This phenotype was less striking at more caudal levels, in which most labeled cells had reached intermediate mediolateral levels (Fig. 8I, dotted line), although only few were located in the periphery of the primordium.
Increase in neuronal apoptosis in the Gbx2 mutant thalamus
Increase in neuroepithelial (or “proliferative”) apoptosis or in neuronal apoptosis could be at the root of the mutant phenotypes observed. Deficiency in Shh increases apoptosis in the diencephalic neuroepithelium during the phase of precursor expansion (Ishibashi and McMahon, 2002). To know whether Shh is necessary to prevent neuroepithelial cell death also during the phase of neuronal production, we labeled E12.5 thalamic sections by means of the TUNEL reaction. We found only very few apoptotic cells in the neuroepithelium of the three genotypes investigated and no evidence of an increase in apoptosis at this age (Fig. 9A–F).

Apoptosis in the thalamus of the Gbx2 mutant. A–F, TUNEL of apoptotic cells in the E12.5 thalamus of the three genotypes as indicated. B, D, and F show magnified views of the frames in A, C, and E. Arrowheads indicate apoptotic cells. G–I, Sections through intermediate rostrocaudal levels of the E18.5 thalamus of the three genotypes showing apoptosis detection. The frame in dotted line is shown magnified as inset in each picture. Some apoptotic cells are indicated by arrowheads. J, Mean ± SD of apoptotic cells per histological section of the E18.5 thalamus at rostral (R), intermediate (I), and caudal (C) levels in the three genotypes. The increase at every level in the Gbx2 mutant is significant with respect to the other two genotypes (p < 0.005).
Next we asked whether neuronal cell death would be increased in our mutants. TUNEL sections of E18.5 thalamus at three rostrocaudal levels in the three genotypes (Fig. 9G–I) showed no change in neuronal cell death in Shh-c versus wild type (Fig. 9J). However, the Gbx2 mutant thalamus showed a very significant increase (p < 0.005) in neuronal cell death (Fig. 9J).
Discussion
Assessing thalamic differentiation with marker genes
Our identification of thalamic nuclear markers (Table 2) agrees with the literature (Arai et al., 1991, 1992, 1994; Frassoni et al., 1991; Winsky et al., 1992; Nakagawa and O'Leary, 2001; Jones and Rubenstein, 2004) except for minor discrepancies probably attributable to species (mouse vs rat) or age [E18.5 vs postnatal day 0 (P0), P2, or adult) differences. We have not found Gbx2 expression in the anterior thalamus, but Jones and Rubenstein (2004) described it in the anterodorsal nucleus at P0. These authors do not mention lateral posterior nucleus expression of Cdh6, as we do. Lhx2 expression in the anterior, ventral, and lateral groups, as well as Neurog2 in the laterodorsal nucleus (Table 2), are not mentioned by Nakagawa and O'Leary (2001), working on P2. Finally, we do not find Calb2 in the mediodorsal nucleus, contrary to Arai et al. (1994), working on adult rat.
These discrepancies do not bear on the reliability of our conclusions because, in every case, we have other markers plus clear ISH evidence obtained through a particularly sensitive dual-amplification protocol.
Neural Shh in the cascade of thalamic differentiation
The development of a brain region proceeds by steps regulated by a hierarchical cascade of gene activation. Because the Shh-c mutant expresses thalamic marker Dbx1 (Fig. 2G,H), which is missing in the full Shh mutant (Ishibashi and McMahon, 2002), we can assume that the first steps of the thalamic cascade (regional specification) are not regulated by neural Shh. In any case, Shh is one among several factors in thalamic regionalization, because transcription factor gene Pax6, required for thalamic development (Warren and Price, 1997; Pratt et al., 2000, 2002), is expressed in the full Shh mutant (Ishibashi and McMahon, 2002). It is only after gastrulation (and general regional specification) that neural Shh signaling gets underway, activating Gbx2, Nkx2-2, and Sox14 and repressing Pax6 (Ishibashi and McMahon, 2002; Hashimoto-Torii et al., 2003; Kiecker and Lumsden, 2004) in a thalamus already specified regionally.
We hypothesized that neural Shh would promote the last steps of the thalamic specification cascade (partially through Gbx2 activation), leading to specific thalamic histogenesis and nucleogenesis. Our analysis of the mutant phenotypes confirms this hypothesis by showing that neural Shh has at least two roles: (1) promotion of specific neuronal aggregation, leading to the formation of recognizable thalamic nuclei; (2) promotion of axonal extension, leading [through a tightly regulated process (López-Bendito and Molnár, 2003; Price et al., 2006)] to appropriate thalamocortical connectivity.
Neural Shh and neuronal fate acquisition in the thalamus
However, earlier events such as incorrect neuronal fate acquisition could contribute to the mutant phenotypes reported here. The acquisition of general neuronal fate as well as some neuronal subtype traits are regulated by proneural genes, encoding bHLH transcription factors whose role in telencephalic development is well known (Guillemot, 2007a,b). Although there is evidence of a link between Shh and proneural and other bHLH genes (Blader et al., 1997; Lu et al., 2000; Ota and Ito, 2003; Andersson et al., 2006), the thalamic bHLH genes examined and the cell-type markers Tubb3 and Gfap were not changed in the mutant. This agrees with our hypothesis that neural Shh influences later stages of differentiation.
Neurogenesis and proliferative cell death are not affected by neural Shh
Shh promotes precursor proliferation in the thalamus, hypothalamus, and cortex (Dahmane et al., 2001; Ishibashi and McMahon, 2002; Manning et al., 2006). The very small size of the early Shh-c thalamic primordium confirms this role. A role for Shh in neuron generation has not yet been documented (except for an indirect function in cortical interneuron generation) (Gulacsi and Lillien, 2003). In agreement, we show that deficiency in neuroepithelial Shh does not impair proliferation during the phase of neurogenesis.
Normally occurring cell death is a key developmental process that can affect the proliferative neuroepithelium (so-called proliferative cell death) (Thomaidou et al., 1997) as well as postmitotic neurons (see below). Although Shh is a neuroepithelial survival factor during precursor proliferation (Ishibashi and McMahon, 2002), we show that later deficiency in Shh does not increase neuroepithelial cell death. This suggests that Shh (of any origin) acts specifically on the neuronal progenitors during the phase of expansion.
Migration in the Shh-c mutant thalamus
In the cortex, it is possible to follow neuronal migration to different layers by using precisely timed BrdU injections (Edgar and Price, 2001). Thalamic neuronal migration does not result in layer formation, but the principle of labeling a certain population with BrdU and recording its position at a later stage holds also for the thalamus. In this way, we have detected alteration in neuronal distribution in the E18.5 Shh-c thalamus, compatible with slow or failed migration. Inability of neurons to reach their settling places would alter specific neuronal aggregation (i.e., nucleogenesis and histogenesis) and would help explain the Shh-c mutant phenotype. Hedgehog signaling can influence the migration of glia and neurons in the developing fruit fly (Rangarajan et al., 2001). Shh has an indirect influence on the migration of newly born neurons in the adult mouse (Balordi and Fishell, 2007). Even if a comprehensive analysis of the role of Shh in migration is not intended here, our results indicate a possible function of neural Shh in the radial migration of newly born thalamic neurons.
Gbx2 is necessary for neuronal survival
Normally occurring developmental neuronal death (Oppenheim, 1991) appears in the rodent thalamus during the first postnatal week (Ashwell and Waite, 1991; Spreafico et al., 1995; Alcántara et al., 1997; Lotto et al., 2001; Luczyńska et al., 2003). Accordingly, we found few apoptotic figures in the wild-type and the Shh-c thalamus at E18.5. Thalamic neuronal survival during development depends on endogenous and target-derived factors (Lotto et al., 1997, 2001), a mechanism not altered by neural Shh deficiency.
We show, however, very increased neuronal cell death in the Gbx2 mutant thalamus, indicating that Gbx2 has a hitherto unknown role in the survival of specific neuronal populations. Obviously, a major cause of the Gbx2 thalamic phenotype is massive cell death.
Gbx2 is necessary and sufficient to specify the medial pronucleus
Gbx2 is initially expressed at E11.5 in the entire thalamic primordium downstream Shh (Hashimoto-Torii et al., 2003). However, we show delayed and restricted expression of Gbx2 independent of Shh. Because in the Shh-c mutant the medial pronucleus preserved general shape and gene expression whereas in the Gbx2 mutant it showed complete and specific morphological abolition, it follows that Gbx2 is necessary and sufficient to specify cell fate and position of the medial and intralaminar groups (in a thalamic primordium already specified as a region).
The lateral and medial geniculate pronuclei, however, depend on Shh, not Gbx2, for differentiation. Finally, the central and dorsal pronuclei depend on both neural Shh and Gbx2. The resultant Shh–Gbx2-thalamic differentiation cascade misses (at least) two components (Fig. 10A, X and Y) that would be responsible for the specific effects of Shh. Component X could simply be the early expression (E11.5) of Gbx2 in the entire primordium.

Summary of results. A, Involvement of Shh, Gbx2 and unknown factors (X, Y) in the differentiation of thalamic nuclear groups inferred from the mutant phenotypes. The Gbx2 label in a closed box symbolizes restricted default expression of Gbx2. We include the intralaminar nuclei in the medial pronucleus (see Results). Color code as in B. B, Color code for this figure. C, Diagram showing the thalamic pronuclei in four rostrocaudal levels labeled according to the severity of their phenotype in the Shh-c and Gbx2 mutants. D, Thalamic nuclear groups mapped on a flat map (Swanson, 1992) and labeled according to dependence on Shh and/or Gbx2. Each of the three groups encompasses contiguous nuclei. E, Localization of the thalamic neuroepithelium (top) and possible arrangements of the three presumptive regions that will give rise to the different nuclear groups (bottom). Dotted line, Anteroposterior axis of thalamic primordium. C, Central; D, dorsal; M, medial; ETh, epithalamus.
Prepatterning the thalamic neuroepithelium
The existence of an early, Shh-independent pattern (“prepattern”) in the spinal cord has been proposed to explain why the phenotype of Gli compound mutants is less severe than expected (Ruiz i Altaba et al., 2003). Thalamic prepatterning depends on Wnt activity upstream Irx3 (Braun et al., 2003), which makes the thalamic neuroepithelium competent to respond to Shh (Kiecker and Lumsden, 2004). We show an additional thalamic prepatterning phenomenon resulting in a late phase of neural Shh-independent Gbx2 expression. The pronuclei, labeled according to their differential Shh or Gbx2 dependence (Fig. 10A,B), have a complex tridimensional relation to each other (Fig. 10C). However, when they are depicted on a “flat map” (Fig. 10D) (after Swanson, 1992), it becomes evident that each of the three groups encompasses adjacent nuclei that could originate in contiguous neuroepithelial areas. The thalamic neuroepithelium would then be patterned into at least three areas whose spatial arrangement is not yet clear (Fig. 10E, bottom).
Finally, our results bear on a current question about the basic developmental mechanisms patterning the thalamus. Does neural Shh specify thalamic neurons through a concentration gradient? This mechanism is used by the notochord to specify the ventral spinal cord (Jessell, 2000), and it would be an elegant way to specify the intricate thalamic structure. However, the existence of a (neural) Shh-independent area in the thalamic neuroepithelium implies that a concentration gradient would have to work around a nonresponsive patch of tissue (Fig. 10E). This suggests that any concentration gradient mechanism in the thalamus would have to adopt a more complex form than in the ventral spinal cord.
Footnotes
-
This work was supported by the Max Planck Society. Drs. Gail Martin (University of California, San Francisco) and Alex Joyner (Sloan-Kettering Cancer Center, New York) provided the Gbx2 mutant mouse line. Dr. Ulrike Teichmann and her team took expert care of the mutant colonies.
- Correspondence should be addressed to Gonzalo Alvarez-Bolado, Department of Genes and Behavior, Max Planck Institute of Biophysical Chemistry, Am Fassberg 11, D-37077 Göttingen, Germany. gonzalo.alvarez-bolado{at}mpibpc.mpg.de
This article is freely available online through the J Neurosci Open Choice option.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}