

Abstract
The aim of the present study was to evaluate whether, and by means of which mechanisms, the adenosine A2A receptor antagonist SCH 58261 [5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine] exerted neuroprotective effects in a rat model of Huntington's disease. In a first set of experiments, SCH 58261 (0.01 and 1 mg/kg) was administered intraperitoneally to Wistar rats 20 min before the bilateral striatal injection of quinolinic acid (QA) (300 nmol/1 μl). SCH 58261 (0.01 but not 1 mg/kg, i.p.) did reduce significantly the effects of QA on motor activity, electroencephalographic changes, and striatal gliosis. Because QA acts by both increasing glutamate outflow and directly stimulating NMDA receptors, a second set of experiments was performed to evaluate whether SCH 58261 acted by preventing the presynaptic and/or the postsynaptic effects of QA. In microdialysis experiments in naı̈ve rats, striatal perfusion with QA (5 mm) enhanced glutamate levels by ∼500%. Such an effect of QA was completely antagonized by pretreatment with SCH 58261 (0.01 but not 1 mg/kg, i.p.). In primary striatal cultures, bath application of QA (900 μm) significantly increased intracellular calcium levels, an effect prevented by the NMDA receptor antagonist MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate]. In this model, bath application of SCH 58261 (15–200 nm) tended to potentiate QA-induced calcium increase. We conclude the following: (1) the adenosine A2A receptor antagonist SCH 58261 has neuroprotective effects, although only at low doses, in an excitotoxic rat model of HD, and (2) the inhibition of QA-evoked glutamate outflow seems to be the major mechanism underlying the neuroprotective effects of SCH 58261.
Adenosine is an endogenous neuromodulator involved in the regulation of many functions within the CNS (Phillis and Wu, 1981) and whose effects are mediated by at least four distinct receptors: A1, A2A, A2B, and A3 (Fredholm et al., 1994). Unlike A1 receptors, which are widely expressed in the CNS, and A2B and A3receptors, whose central expression is rather low, adenosine A2A receptors are mainly expressed in the striatum, although lower levels of expression do exist in other brain areas, such as the cortex and the hippocampus (for review, seeImpagnatiello et al., 2000). Besides their role in the regulation of dopamine-dependent behaviors in both normal and pathological conditions (Ferré et al., 1997), adenosine A2Areceptors seem also to be involved in excitotoxic–neurodegenerative processes: (1) selective A2A receptor ligands have been shown to regulate striatal glutamate release (Popoli et al., 1995; Corsi et al., 1999, 2000); (2) the intrastriatal injection of an adenosine A2>A1 receptor antagonist prevented the electroencephalographic (EEG) abnormalities induced by an excitotoxic striatal lesion in rats (Reggio et al., 1999); (3) A2A receptor antagonists showed neuroprotective effects in models of diseases, such as brain ischemia, in which excitotoxic mechanisms are thought to play a pathogenetic role (Gao and Phillis, 1994; Bona et al., 1997; Monopoli et al., 1998b); (4) mice lacking A2A receptors have been reported to be less vulnerable to ischemia- and MPTP-induced neuronal damage (Chen et al., 1999, 2001). Together, these observations support the view that adenosine A2A receptor antagonists may possess neuroprotective effects in neurodegenerative diseases (Ongini et al., 1997; Abbracchio and Cattabeni, 1999; Impagnatiello et al., 2000), although the mechanisms responsible for such effects are still primarily unknown. Given the anatomical distribution of A2A receptors, their blockade could be a particularly reliable approach in the treatment of neurodegenerative diseases of the striatum. In particular, because adenosine A2A receptors are selectively expressed in the striopallidal neurons (Schiffmann et al., 1991), a population of medium-sized spiny neurons that degenerate early in Huntington's disease (HD) (Glass et al., 2000), it is conceivable that such receptors may play a role in triggering neuronal death in HD. If so, blockade of striatal A2A receptors should exert neuroprotective effects in experimental models of the above disease. Besides the more recent genetic models (transgenic mice expressing the HD mutation) (Bates et al., 1997; Hodgson et al., 1999), several “pathogenetic” models of the disease have been developed. In particular, the model of excitotoxic striatal lesion by quinolinic acid (QA) in the rat has been reported to mimic both the clinical and the neuropathological features of human HD (Beal et al., 1986; Popoli et al., 1994).
The aims of the present work were as follows: (1) to study the possible neuroprotective influence of SCH 58261 [5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine], an adenosine A2A receptor antagonist showing selectivity for striatal versus cortical and hippocampal binding sites (Lindström et al., 1996; Lopez et al., 1999), on QA-induced effects in rats, and (2) to investigate the mechanisms underlying such effects.
MATERIALS AND METHODS
Surgery
Adult male Wistar rats (250–280 gm) were used. The animals were kept under standardized temperature, humidity, and lighting conditions, with access to water and food ad libitum. Animal care and use followed the directives of the Council of the European Communities (86/609/EEC). Animals were anesthetized with Equithesin (3 ml/kg, i.p.) and placed in a David Kopf Instruments (Tujunga, CA) stereotaxic apparatus. Quinolinic acid (300 nmol) or vehicle (PBS) were bilaterally injected in the striatum (coordinates: anterior, +1.7 mm; lateral, +2.7 mm; ventral, −4.8 mm from bregma and dura) by means of a Hamilton syringe (model 701); the injection volume was 1 μl. Experimental groups (n = 8–12 animals per group) were as follows: sham-lesioned animals (intrastriatal injection of 1 μl vehicle); lesioned animals (intrastriatal QA); and animals treated with SCH 58261 (0.01 and 1 mg/kg, i.p., dissolved in DMSO) 20 min before QA injection. The doses of SCH 58261 to be used were selected on the basis of preliminary experiments in which a wider range of doses of SCH 58261 had been tested in a limited number of animals. On the basis of such experiments, a low-dose range (0.01–0.05 mg/kg) and an a high-dose range (0.5–2 mg/kg) in the effects of SCH 58261 were identified, and it was established that 0.01 and 1 mg/kg SCH 58261 were the two most representative doses within the above ranges. To evaluate the possible influence of SCH 58261 alone on the motor, EEG, and cognitive parameters studied here, in separate experiments, three groups of five rats each were treated with SCH 58261 (0.01 and 1 mg/kg, i.p.) or vehicle (1 ml/kg) 20 min before being subjected to a sham lesion. The EEG tracing, spatial learning, and motor response tod-amphetamine of these animals were then evaluated 3, 4, and 5 weeks after surgery, respectively.
EEG experiments
Two weeks after surgery, the animals were newly anesthetized with Equithesin, and screw cortical electrodes were implanted at the level of the frontal cortex and fixed with dental acrylic to the skull surface. Five to 6 d thereafter, the animals were individually placed in a cylindrical Plexiglas container in a soundproof experimental room. After a 30 min habituation period, each animal was connected to an Ote (model 10b) polygraph. The EEG was then continuously recorded over 45 min. The methods used for EEG recording and analysis have been described previously (Reggio et al., 1999). Briefly, sequential power spectra of 20 sec EEG epochs (1 epoch every minute) were analyzed by fast Fourier transformation with a frequency resolution of 0.35 Hz (software by Enrico Staderini, IADA Sistemi, Rome, Italy). All of the power spectra relevant to an EEG tracing were recorded on a optical disk and then analyzed to calculate the relative power in each frequency band. Frequency bands were as follows: 1.2–4 Hz (δ), 4.35–7 Hz (θ), 7.35–9.5 Hz (α1), 9.85–12.5 Hz (α2), 12.85–16 Hz (β1), and 16.35–30 Hz (β2). One-way ANOVA and Tukey'spost hoc test were used for the statistical analysis of the results.
Motor activity recording
To measure their motor response to d-amphetamine, 5–6 weeks after surgery, the animals were placed, one per cage, in an animal activity motor (model Automex II; Columbus Instruments, Columbus, OH), in a soundproof experimental room. Experiments were always performed between 9:00 A.M. and 12:00 A.M. The motor activity of each rat, expressed as counts, was recorded on a computer and analyzed by a computer counter software (version 3.3;Columbus Instruments). After an habituation period of 30 min, each animal was injected with d-amphetamine (1 mg/kg, i.p.), and the motor activity was then recorded for an additional period of 90 min.
Evaluation of spatial learning by the Morris water maze
Experiments were performed 4–6 weeks after the lesion. A circular black pool (100 cm diameter; 48 cm height) was filled up to 30 cm with water (23–24°C). Each experiment consisted of 18 learning trials (two daily blocks of three consecutive trials each), over 3 consecutive days. Within each block, the rats were put into the pool from three different starting points (one for each trial). The rats were able to escape from the water by climbing on an invisible platform, which was submerged under water and whose location remained unchanged over the whole experiment. A trial was terminated as soon as the animal found the platform or after 70 sec of unsuccessful swimming (in this case, the animal was placed on the platform by the experimenter). Rats were allowed to stay on the platform for 10 sec before the next trial started. All trials were video recorded, and a computer-assisted analysis of escape latency (i.e., the time required to find the platform), swim distance, and swimming paths was performed (Software Delta Sistemi, Rome, Italy). One-way ANOVA and Tukey'spost hoc test were used for the statistical analysis of the results.
In vitro hippocampal electrophysiology
To assess whether the memory impairment observed in the spatial learning test correlated with alterations in hippocampal synaptic plasticity, experiments were performed in hippocampal slices obtained from sham, QA-lesioned, and SCH 58261-pretreated rats. The animals were decapitated under ether anesthesia, the brain was quickly removed, and the hippocampi were dissected free. Transverse slices (400–450 μm) were cut with a tissue chopper and maintained at room temperature (22–24°C) in oxygenated artificial CSF (ACSF) containing (in mm): 126 NaCl, 3.5 KCl, 1.2 NaH2PO4, 1.3 MgCl2, 2 CaCl2, 25 NaHCO3, and 11 glucose, pH 7.3 (saturated with 95% O2 and 5% CO2). After incubation for at least 1 hr, an individual slice was transferred to a submerged recording chamber and continuously superfused at 32–33°C with oxygenated ACSF at a rate of 3 ml/min. Field EPSPs (fEPSPs) were recorded with a glass microelectrode filled with NaCl (2 m; pipette resistance of 2–5 MΩ). A bipolar twisted NiCr-insulated electrode (50 μm outer diameter) was used to stimulate the medial perforant path and the Schaffer collaterals (test frequency of 0.05 Hz) while recording in the middle molecular layer of the dentate gyrus and the apical dendritic region of CA1, respectively. The duration of the stimulus was 100 μsec. The stimulation intensity corresponded to that necessary to obtain a response equal to 50% of the maximal fEPSP. In the CA1 region, early long-term potentiation (LTP) was produced by high-frequency stimulation (HFS) administered at 100 Hz for 1 sec using the same stimulation intensity and repeated after 15 min. In the dentate gyrus, early LTP was induced, in the presence of 10 μm bicuculline, by two trains (frequency of 100 Hz, 1 sec) delivered 10 sec apart to the medial perforant path with the stimulation intensity necessary to obtain the maximal field response. The induction of early LTP was measured 30 min after HFS as the percentage of increase of fEPSP slope with respect to pretetanus values. Signals were acquired with a DAM-80 AC differential amplifier (World Precision Instruments, Sarasota, FL) and analyzed with the LTP Software (courtesy of Dr. W.W. Anderson, University of Bristol, Bristol, UK). Mann–Whitney U test was used for the statistical analysis of the data.
Histological studies
Gliofibrillary acidic protein immunolocalization. The animals (sham, QA only, and 0.01 mg/kg SCH 58261 plus QA) were decapitated under ether anesthesia, and the brains were removed and frozen in liquid nitrogen. Ten- to 20-μm-thick coronal sections were cut on a cryostat microtome, mounted on a slide, and fixed in 4% paraformaldehyde in PBS containing 0.12 msucrose, for 15 min at room temperature. After washes in PBS, sections were incubated with 10% fetal calf serum in PBS to abolish nonspecific staining. gliofibrillary acidic protein (GFAP) was immunolabeled by monoclonal antibody (1:250; Sigma, St. Louis, MO), followed by fluoresceinated goat anti-mouse IgG (Sigma). Three sections for each brain were examined at a Nikon (Tokyo, Japan) Optiphot microscope, and three fields for each section (a total of nine fields for each brain) were chosen randomly in the lesion area (excluding areas of complete cell loss) recorded at 200× magnification. Images were captured by a color CCD camera, obtaining frames of 3 mm2 (nine frames per brain). Images were analyzed by the Optilab software (Graftek, Mirmande, France). After background subtraction, the area occupied by GFAP-positive staining was identified and measured. Positive areas were expressed as percentage of total areas. Statistical comparisons were made using the nonparametric Mann–Whitney U test.
Measurement of the lesion size. For the measurement of the striatal lesions, two groups of animals (QA only and 0.01 mg/kg SCH 58261 plus QA) were decapitated under ether anesthesia, and the brains were removed and frozen in liquid nitrogen. Serial 20 μm coronal sections were cut on a cryostat microtome, stained with cresyl violet, and examined by light microscopy. For each brain, a series of consecutive sections taken at the level of the maximal extension of the lesion was identified, and, within this series, the slide (or one of the slides) showing the largest lesion was chosen for the quantitative analysis. Images were captured by a color digital camera and analyzed by the Optilab software (Graftek). The lesion area was expressed as percentage of the total area of the dorsal striatum. Statistical comparisons were made using the nonparametric Mann–WhitneyU test.
Microdialysis experiments
Under Equithesin anesthesia, naı̈ve Wistar rats were placed in a stereotaxic frame and implanted with a concentric dialysis probe (model CMA/12; 3 mm length; CMA Microdialysis, Solma, Sweden) into the striatum. Stereotaxic coordinates in mm from bregma, sagittal suture, and dura, respectively, were as follows: anterior, +1.7; lateral, +2.7; ventral, −6.2. Twenty-four hours later, the probe was perfused at a rate of 2 μl/min with a Ringer's solution (in mm: 147 NaCl, 2.3 CaCl2, and 4.0 KCl). After a washout period of at least 90 min, samples were collected every 5 min into a refrigerated fraction collector (model CMA/170; CMA Microdialysis) and then frozen until assay. Because the intracerebral injection of QA induces tremors and convulsions in rodents, these experiments were performed under general anesthesia (3 ml/kg Equithesin). Each experimental group was made up of four to five animals. Results were expressed as percentage of changes of extracellular glutamate levels induced by probe perfusion with QA (5 mm over 30 min) with respect to basal (predrug) values (mean of three to four samples collected after the induction of general anesthesia). SCH 58261 (0.01 and 1 mg/kg) was administered intraperitoneally 20 min before starting QA perfusion. At the end of the experiments, each rat was killed with an overdose of Equithesin, the brain was fixed with 4% paraformaldehyde, and coronal sections (20-μm-thick) were cut to verify the probe location. The glutamate content of all samples was measured by reverse-phase HPLC coupled to a fluorometric detector (model LC240 at wavelength of 335 nm and emission cutoff filter of 425 nm; PerkinElmer Life Sciences, Emeryville, CA), using a 15 min gradient elution program (methanol from 20 to 80% with 50 mmNaH2PO4 and CH3COONa) and automatic precolumn derivatization with ophthalaldehyde and β-mercaptoethanol. Cysteic acid was used as internal standard. The concentration of the standard was linear (r2 = 0.99) between 0.2 and 25 ng/10 μl. Basal glutamate levels were calculated by comparison of sample peak height with external standard peak height, both corrected for the internal standard peak height and expressed as 10 μl/ng without probe recovery correction. Data were processed by two-way ANOVA, followed by post hoc Student's ttest.
Intracellular calcium measurement on primary striatal cultures
Striatal cells from 17-d-old rat embryos were mechanically dissociated and plated in Eagle's Basal Medium with a density of ∼30.000/cm2. Experiments were started 13–15 d after plating. Optical fluorimetric recordings with fura-2 AM were used to evaluate the intracellular calcium concentration ([Ca2+]i). Fura-2 AM stock solutions were obtained by adding 50 μg of fura-2 AM to 50 μl of 75% DMSO plus 25% pluronic acid. Cells were bathed for 60 min at room temperature with 5 μl of stock solution diluted in 1 ml of extracellular solution (in mm: 125 NaCl, 1 KCl, 5 CaCl2, 1 MgCl2, 8 glucose, and 20 HEPES, pH 7.35). This solution was then removed and replaced with extracellular solution, and the dishes were quickly placed on the microscope stage. To measure fluorescence changes, an Hamamatsu (Shizouka, Japan) Argus 50 computerized analysis system was used, recording every 6 sec the ratio between the values of light intensity at 340 and 380 nm stimulation. Drugs [QA, SCH 58261, CGS 21680 (carboxyethyl phenethylamino-5′-N-ethylcarboxamido adenosine HCl), and MK-801 ((+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate)] were applied by directly dropping in the bath.
RESULTS
Experiment 1: study of the influence of pretreatment with SCH 58261 on the effects induced by QA; experiments in striatally lesioned rats
As a consequence of QA lesion, three of 12 animals (25%) died within 6.5 ± 2.7 d. In animals pretreated with 0.01 mg/kg SCH 58261, the occurrence of death was significantly delayed (20.3 ± 0.88 d; p = 0.008 versus QA alone according to Student's t test), whereas the mortality rate was not reduced (18.7%). Pretreatment with 1 mg/kg SCH 58261 did not significantly affect the above parameters, although a tendency to a reduced mortality was seen (10%; NS versus QA alone according to Fisher's exact test). As reported previously (Popoli et al., 1994; Reggio et al., 1999), QA-lesioned rats show an exaggerated motor response tod-amphetamine (Fig. 1) and a marked reduction in EEG voltage amplitude (Figs.2, 3), accompanied by an altered distribution of EEG spectral power (increase in δ band and reductions in α and β bands) (Fig. 4). The exaggerated response to d-amphetamine is thought to depend on the loss of striatal GABAergic inhibitory projections (Rozas et al., 1996). QA-induced EEG changes at the level of the frontal cortex, which are very similar to those observed in HD patients (Bylsma et al., 1994), seem to depend on the impairment of the striocortical pathway, which follows the striatal lesion (Schwarz et al., 1992; Popoli et al., 1994). QA-induced motor hyperactivity (Fig. 1), EEG voltage reduction (Figs. 2, 3), and relative power distribution (Fig. 4) were fully antagonized by pretreatment with 0.01 mg/kg SCH 58261 but not 1 mg/kg. Animals treated with SCH 58261 before receiving a sham lesion did not differ from sham controls in terms of motor response tod-amphetamine (data not shown). As for EEG analysis, both the mean EEG voltage and the relative EEG power distribution of the animals pretreated with SCH 58261 were indistinguishable from those observed in sham controls (data not shown). In the Morris water maze, QA-lesioned rats showed impairment in their spatial learning performances compared with controls. As shown by their swimming paths (Fig. 5), whereas sham animals had already developed a clear spatial strategy at block 5, QA-lesioned rats did not. Such an impairment was reflected by the mean escape latencies of lesioned rats, which were significantly higher than those of control animals over the whole experiment (Fig.6A). Rats pretreated with SCH 58261 (either 0.01 or 1 mg/kg) before the lesion did not differ from QA-lesioned animals in their mean escape latencies (Fig.6B). A significant reduction in mean distance traveled (70 sec/cm) was also observed in QA-lesioned versus sham rats (1277 ± 94 and 1616 ± 93, respectively; p< 0.05 according to one-way ANOVA and Tukey's post hoctest). Such an effect, which is an expression of QA-induced motor impairment, was prevented by pretreatment with the lower dose of SCH 58261 only (0.01 SCH 58261, 1565 ± 82, p < 0.05 vs QA-lesioned; 1 SCH 58261, 1320 ± 77, NS vs QA-lesioned according to one-way ANOVA and Tukey's post hoc test). Rats treated with SCH 58261 (0.01 and 1 mg/kg) before receiving a sham lesion did not differ from sham controls in terms of mean escape latencies or mean distance traveled (data not shown). In electrophysiological studies, slices from QA-lesioned rats showed no significant alterations with respect to controls (slices from sham animals) in basal synaptic transmission. In experiments aimed at evaluating the induction of long-term synaptic plasticity, HFS applied to the input fibers elicited a robust increase in fEPSP, followed by a stable potentiation of fEPSP both in the CA1 area and in the dentate gyrus. In the CA1 area, no differences were found in the magnitude of potentiation between sham and QA-lesioned animals (+67.86 ± 14.88 and +53.05 ± 4.99% in sham and lesioned animals, respectively; data not shown). Conversely, in the dentate gyrus (Fig.7), LTP was significantly decreased in QA-lesioned animals with respect to their sham controls (+60.91 ± 10.79 and 111.18 ± 15.29%, respectively; p < 0.05). Such a decrease in dentate LTP was not prevented by pretreatment with 0.01 mg/kg SCH 58261 (+73.6 ± 17.84%).

Influence of SCH 58261 on QA-induced motor abnormalities. Rats lesioned with QA (LESQA) showed an increased motor response to d-amphetamine (1 mg/kg, i.p.) with respect to controls (SHAM). This effect was prevented by pretreatment, 20 min before the lesion, with 0.01 (SCH 0.01) but not 1 (SCH 1) mg/kg SCH 58261 intraperitoneally. Each group was composed of 8–12 animals. °p < 0.05 versus sham; *p < 0.05 versus QA-lesioned rats (one-way ANOVA and Tukey's post hoc test).

Influence of SCH 58261 on QA-induced EEG voltage reduction. The figure shows some representative EEG tracings recorded from a sham (SHAM), a QA-lesioned (QA), and two SCH 58261 (SCH 0.01 + QAand SCH 1 + QA)-pretreated rats. At the level of the frontal cortex (F-F leads), the QA-lesioned rat shows a marked reduction in the EEG voltage amplitude compared with the sham animal. Such a reduction is prevented in the animal that had been pretreated, 20 min before the lesion, with 0.01 but not 1 mg/kg SCH 58261 intraperitoneally. Leads are as follows: F-F, fronto-frontal; rF-P, right fronto-parietal;lF-P, left fronto-parietal.

Quantitative analysis of EEG voltage amplitude. The mean EEG voltage amplitude of rats lesioned with QA (LESQA) is significantly reduced with respect to controls (SHAM). This effect was prevented by pretreatment, 20 min before the lesion, with 0.01 (SCH 0.01) but not 1 (SCH 1) mg/kg SCH 58261 intraperitoneally. For EEG recording and analysis, see Materials and Methods. Each group was composed of 8–12 animals. °p < 0.05 versus sham; *p < 0.05 versus QA-lesioned rats (one-way ANOVA and Tukey's post hoc test).

Influence of SCH 58261 on QA-induced alterations in relative EEG power distribution. Bilateral intrastriatal injection of QA altered the distribution of total EEG power into the different frequency bands. In particular, relative EEG power was increased in the δ band and decreased in the α and β bands. These changes were prevented by 0.01 (SCH 0.01) but not 1 (SCH 1) mg/kg SCH 58261 intraperitoneally. For EEG recording and analysis, see Materials and Methods. Each group was composed of 8–12 animals. °p < 0.05 versus controls (SHAM); *p < 0.05 versus rats lesioned with QA (LESQA) (one-way ANOVA and Tukey's post hoc test).

Impairment of space learning in animals lesioned with QA. Rats were trained over six consecutive blocks of trials in the Morris water maze. The figure shows some representative swimming paths recorded from four sham (SHAM) and four QA-lesioned (LES) rats during block 5. Thesquare indicates the location of the platform. Whereas all sham animals were able to find the platform at this stage, most the of QA-lesioned rats were still greatly impaired.

Pretreatment with SCH 58261 does not prevent QA-induced impairment in place learning. The mean escape latencies of QA-lesioned rats (LESQA) were significantly higher than those of sham animals (SHAM) over the whole experiment (A). Animals pretreated with SCH 58261 [0.01 (SCH 0.01) and 1 (SCH 1) mg/kg, i.p.] did not differ significantly from QA-lesioned rats, although a slight reduction in their escape latencies was observed in the last two blocks of trials (B). *p < 0.05 versus sham (one-way ANOVA and Tukey's post hoctest).

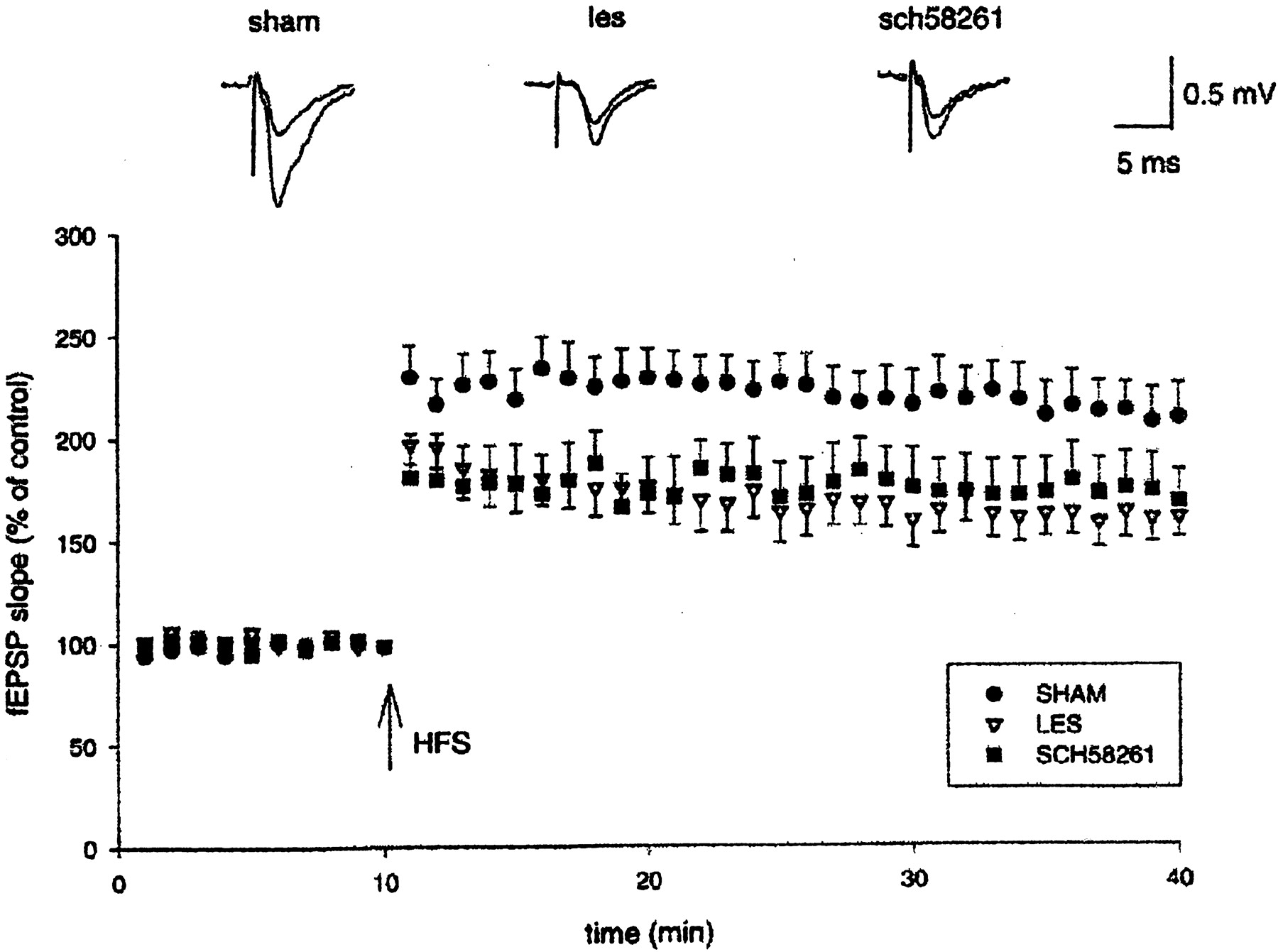
Induction of early LTP in hippocampal slices. Pooled data (mean ± SEM) of fEPSPs (percentage of control) plotted as a function of time. HFS (arrow) delivered to the medial perforant path induced LTP that was significantly decreased in QA-lesioned animals (LES; n = 8) with respect to sham controls (SHAM;n = 7). Pretreatment with 0.01 mg/kg SCH 58261 (SCH58261; n = 8) did not prevent the reduction in the magnitude of LTP. The insets show representative fEPSPs recorded before and 30 min after HFS. Eachtrace is the average of three successive fEPSPs (artifacts of stimulation have been truncated).
In histological studies, the brain tissue surrounding necrosis induced by quinolinic acid was characterized by a marked astrocytic hyperplasia. Whereas in the sham group GFAP immunolabeling identified sparse astrocytic elements, with few, thin cytoplasmic processes (Fig.8A), in sections from animals lesioned with QA, astrocytes increased in number and dimensions, showing plump cell bodies with thicker and longer processes (Fig. 8B). Quantification of hyperplasia revealed an increase of GFAP-positive area (6.2 ± 0.4%; n = 4), which significantly exceeded that of the sham group (1.8 ± 0.5%; n = 4; p = 0.02). In animals pretreated with 0.01 mg/kg SCH 58261, astrocytic hyperplasia was less evident (Fig. 8C), and GFAP-positive areas were significantly reduced (3.7 ± 0.43%; n = 5) with respect to lesioned rats (p < 0.05).

Pretreatment with SCH 58261 prevents QA-induced striatal gliosis. The figure shows representative striatal sections obtained from a sham rat (A), a QA-lesioned rat (B), and a rat pretreated with 0.01 mg/kg SCH 58261 (C). GFAP immunolabeling shows sparse astrocytic elements in A and a marked astrocytic hyperplasia in B. In C, GFAP-positive elements are markedly reduced with respect to B.
QA-lesioned animals showed marked striatal shrinkage and enlarged ventricular cavities. At the core of QA lesion, areas of complete cell loss were visible in the dorsal striatum. Such areas were surrounded by densely packed dark-stained cells and reactive gliosis. The lesion extended from −0.8 and +3.6 mm from bregma in the rostrocaudal axis and showed its maximal extension between +1.4 and +2.7 mm from bregma. At the site of its largest extension, the lesion occupied 82.9 ± 6% of the dorsal striatum (n = 6). SCH 58261-pretreated animals showed slightly enlarged ventricular cavities and small areas of marked to complete cell loss. Most of the striatal tissue surrounding such areas appeared normal at the light microscopy. The lesion was maximal between +1.4 and +2.7 mm from bregma, and its rostrocaudal extension was always included between +1.2 and +3.0 mm. At the site of its largest extension, the lesion occupied in mean 22 ± 6.6% of the dorsal striatum (n = 8;p < 0.05 vs QA alone). No histopathological damage was observed in the hippocampus of QA-lesioned or SCH 58261-pretreated rats.
Experiment 2: study of the possible mechanisms of the protective effects of SCH 58261
The excitotoxin QA is an endogenous metabolite of tryptophan, which acts by both elevating extracellular levels of glutamate and directly stimulating NMDA receptors (Connick and Stone, 1988;Stone, 1993; Mena et al., 2000). Thus, the possible influence of SCH 58261 on presynaptic and postsynaptic effects of QA was tested by microdialysis experiments in naı̈ve rats and fura-2 AM experiments on striatal neurons, respectively.
Microdialysis experiments in naı̈ve rats
In the striatum of naı̈ve rats, the perfusion of QA (5 mm) through the dialysis probe dramatically increased (approximately +500%) the extracellular levels of glutamate with respect to basal values (Fig. 9). The dose of QA was chosen on the basis of a previous report, showing that QA elevated extracellular glutamate levels in the rat cortex when injected in the low millimolar range (Connick and Stone, 1988). The effects of QA were fully prevented by intraperitoneal injection (20 min before starting the perfusion of QA) of 0.01 mg/kg SCH 58261. At the dose of 1 mg/kg, SCH 58261 did not influence QA-stimulated glutamate release (Fig. 9).

Influence of SCH 58261 on QA-evoked glutamate outflow in the rat striatum. Microdialysis probes were inserted in the striatum of naı̈ve rats. Probe perfusion with QA (5 mm over 30 min) dramatically increased extracellular glutamate levels with respect to basal values. Pretreatment with SCH 58261 (SCH) (0.01 mg/kg, i.p.) completely prevented QA-stimulated glutamate outflow. The higher dose of SCH 58261 (1 mg/kg) did not influence the effects of QA perfusion. Thebar indicates the period of QA perfusion through the probe. The time of injection of SCH 58261 is indicated by thearrow. Each experimental group was made up of four to five animals. °p < 0.01 versus QA.
Fura-2 AM experiments on striatal neurons
In striatal neurons, bath application of 900 μm QA markedly raised [Ca2+]i: the mean 340/380 ratio measured at the time of peak effect (average of 50 cells from four different experiments) was 4.01 ± 0.19. The effect of QA was potentiated by 15–200 nm SCH 58261. The mean 340/380 ratio with 30 nm SCH 58261 plus QA (n = 53/4) was 5.56 ± 0.23 (p < 0.05 vs QA alone according to Student'st test). Bath application of the selective adenosine A2A receptor agonist CGS 21680 (100 nm) reduced the effect of QA (mean 340/380 ratio, 0.96 ± 0.66; p < 0.05 vs QA alone;n = 34 cells from three experiments). The NMDA receptor antagonist MK-801 (100 μm) fully prevented QA-induced effects (n = 38 cells from three experiments). Single representative experiments performed with QA, SCH 58261 plus QA, CGS 21680 plus QA, and MK-801 plus QA are shown in Figure 10.

Measurement of intracellular calcium in striatal neurons. Optical fluorimetric recordings with fura-2 AM were performed to evaluate changes in intracellular calcium levels [Ca2+]i in rat striatal neurons. Bath application of 900 μm QA induced a sustained [Ca2+]i increase (A) that is significantly potentiated by preapplication of 30 nm SCH 58261 (B) and reduced by the selective A2A receptor agonist CGS 21680 (100 nm) (C). The effects of QA were fully prevented by bath application of the NMDA receptor antagonist MK-801 (100 μm) (D). Each panelshows a single representative experiment (average of 8–14 cells from the same dish). Ratio, Ratio between the values of light intensity at 340 and 380 nm stimulation.
DISCUSSION
Two main findings arise from this study: (1) the adenosine A2A receptor antagonist SCH 58261 shows neuroprotective effects in an excitotoxic rat model of HD; and (2) the inhibition of QA-evoked increase in extracellular glutamate seems to be the main mechanism of the effects elicited by SCH 58261.
The finding that SCH 58261 significantly prevented most of the effects induced by the intrastriatal injection of an excitotoxin is in line with some findings suggesting that activation of A2A receptors could participate in the generation of excitotoxicity. Adenosine A2A receptor agonists have been reported indeed to stimulate glutamate release in the rat striatum (Popoli et al., 1995; Corsi et al., 1999). Moreover, in previous experiments, we observed that the intrastriatal injection of the adenosine A2A receptor agonist CGS 21680, together with QA, potentiated QA-induced mortality in a dose-dependent way (50, 75, and 83% of mortality after 3, 6, and 12 nmol QA plus CGS 21680, respectively; P. Popoli, A. Pèzzola, and R. Reggio, unpublished results). Thus, although only a single compound was tested in the present investigation, on the basis of the above observations, the protective effects exerted by SCH 58261 can be actually ascribed to a blockade of adenosine A2A receptors. On the other hand, SCH 58261 is a specific and selective A2A receptor ligand, because in binding studies it showed A2A receptor affinity in the low nanomolar range (Ki of 2.3 nm), lower A1 receptor affinity (Ki of 121 nm), and no affinity for A3receptors up to micromolar concentrations (Zocchi et al., 1996; Varani et al., 1998). Moreover, in a recent study, [3H]SCH 58261 has been reported to directly and selectively label striatal A2Areceptors after peripheral administration in rodents (El Yacoubi et al., 2001).
Striatal perfusion with 5 mm QA dramatically increased extracellular glutamate levels. An abnormal glutamate outflow is thought to play a crucial role in triggering the cellular events leading to excitotoxic neuronal death (Choi, 1988; Choi and Rothman, 1990; Rossi et al., 2000). The observation that SCH 58261 fully prevented the QA-induced increase in glutamate levels when administered at the same dose (0.01 mg/kg, i.p.), which was effective in protecting QA-lesioned animals, suggests that the reduction of QA-stimulated glutamate outflow plays a major role in the effects of the drug. Although it has been observed that an increase in glutamate extracellular levels may be not a good index of excitotoxicity (Obrenovitch et al., 2000) and that reducing glutamate release does not necessarily imply neuroprotection (Calabresi et al., 2000), a contribution of increased glutamate outflow in inducing excitotoxic neuronal death cannot be ruled out. An inhibition of evoked glutamate release has been reported indeed to parallel the neuroprotective effects of some compounds (O'Neill et al., 2000; Mauler et al., 2001). Thus, the modulation of glutamate outflow can well be invoked to explain the neuroprotective effects of SCH 58261, although alternative mechanisms, such as a possible regulation of excessive microglial cell activation (Fiebich et al., 1996; Picano and Abbracchio, 2000), should also be taken into account.
According to the present findings, a possible influence of SCH 58261 on the postsynaptic, NMDA agonistic effects of QA should be ruled out. In fact, bath application of SCH 58261 did amplify the increase in intracellular Ca2+ levels induced by QA on striatal neurons. Conversely, the adenosine A2Areceptor agonist CGS 21680 attenuated QA-induced effects in the same preparation. The present results are in line with previous reports showing that CGS 21680 inhibits the conductance of NMDA receptor channels in rat neostriatal neurons (Nöremberg et al., 1997). The fact that an adenosine A2A receptor antagonist can exert protective effects while potentiating QA-stimulated intracellular calcium increase is not entirely surprising, because the NMDA agonistic effects of QA seem to contribute poorly to the toxic effect of this compound. In fact, the removal of corticostriatal projections did reduce QA-induced neuronal death in the rat striatum by ∼90% (Orlando et al., 2001). Thus, a reduction of glutamatergic input to the striatum is likely to represent the major step in protecting neurons from QA.
As for the finding that 1 mg/kg SCH 58261 was no longer able to prevent QA-induced effects, the most obvious explanation would be that adenosine receptors other than A2A (i.e., adenosine A1 receptors, whose blockade would be detrimental in models of excitotoxicity) (Phillis, 1995; Ongini and Schubert, 1998) may also be blocked by higher doses of SCH 58261. As a possible alternative explanation, the occurrence of peripheral effects after the administration of the higher dose of SCH 58261 might play a role in the inversely dose-related effects of the drug. Interestingly, after intraperitoneal administration in rats, SCH 58261 did not induce hemodynamic changes up to the dose of 0.1 mg/kg, although it increased blood pressure and heart rate starting from the dose of 1 mg/kg (Monopoli et al., 1998a). Thus, low doses of SCH 58261 might have protective effects in excitotoxic processes by the inhibition of adenosine A2A receptor-stimulated glutamate release, whereas higher doses could also block adenosine A2A receptor-mediated effects on blood pressure (Stella et al., 1996) and on platelet aggregation (Dionisotti et al., 1992), thus eventually reducing blood and nutrient supply to the compromised brain area (Jones et al., 1998) and further stimulating glutamate release.
The inability of SCH 58261 to prevent QA-induced impairment in place learning and dentate LTP deserves some consideration. Whereas the motor and EEG alterations induced by QA seem to depend directly on the striatal damage (Schwarz et al., 1992; Popoli et al., 1994; Rozas et al., 1996), the reduced ability to learn the location of an invisible platform in the water maze is rather related to an impairment of hippocampal function (Devan and White, 1999). Thus, both the impaired performance in the Morris maze and the reduced dentate LTP can be considered the expression of an “indirect” hippocampal dysfunction, which most probably depends on the QA-induced reduction in the functional link between the hippocampus and the striatum (Devan and White, 1999). If so, even a mild striatal damage may be able to perturb the functional connection between these two structures, which would explain the inability of SCH 58261 to influence “cognitive” (“hippocampal”) parameters. This view is supported by the observation that cognitive alterations, such as loss of visuospatial abilities, occur in preclinical stages of HD, when the extent of striatal damage may be still insufficient to bring about the onset of motor symptoms (Bamford et al., 1995; Lawrence et al., 1998). Interestingly, although animals pretreated with SCH 58261 were not protected in terms of place learning impairment, their distance traveled was comparable with that of control animals. Because the reduction in mean distance traveled observed in QA-lesioned rats is an expression of QA-induced motor impairment (Block et al., 1993), this finding confirms that pretreatment with SCH 58261 does prevent the motor (“striatal”) effects of QA lesion. Another implication of this finding is that QA-induced motor deficits are not responsible for the poor performances of lesioned rats in the Morris maze, because SCH 58261-treated rats still showed an impaired place learning despite their normal values in terms of distance traveled.
In conclusion, the present data show that blockade of striatal adenosine A2A receptor by SCH 58261 exerts neuroprotective effects in QA-lesioned rats. They also show, for the first time, that the neuroprotective effects of SCH 58261 are paralleled by an inhibition of QA-induced glutamate outflow. Because the bilateral striatal lesion by QA may be considered a model of HD and QA in itself may have a pathogenetic role in this disease (Stone, 2001), the finding of a beneficial effect of SCH 58261 (at least at low doses) in this model suggests that striatal adenosine A2A receptors could represent an interesting target for the development of neuroprotective strategies for HD. This hypothesis appears even more intriguing in the light of a very recent report showing that, in striatal cells expressing mutant huntingtin (a genetic cellular model of HD), adenosine A2A receptor signaling (i.e., A2A-stimulated adenylyl cyclase) is aberrantly increased (Varani et al., 2001). Interestingly, changes in A2A receptor signaling were much more evident in cells expressing truncated mutant huntingtin, which is significantly more toxic than the full-length protein. Thus, an aberrant increase in A2A receptor signaling seems to be specifically associated with the expression of mutant huntingtin cytotoxicity. This finding, together with the selective expression of adenosine A2A receptors in the population of striatal neurons that degenerate in early phases of HD (Glass et al., 2000), supports the hypothesis that A2A receptors may play a role in the pathogenesis of HD. Given the limitations of the QA lesion as a model of HD, the therapeutic potential of adenosine A7 2A receptor antagonists in such disease, for which only symptomatic treatments are available to date (Marshall and Shoulson, 1997), is worthy to be confirmed in more relevant models.
Footnotes
Correspondence should be addressed to Patrizia Popoli, Department of Pharmacology, Istituto Superiore di Sanità, Viale Regina Elena, 299 00161 Rome, Italy. E-mail: patrizia.popoli{at}iss.it.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}