

Abstract
How glutamate regulates dopamine (DA) release in striatum has been a controversial issue. Here, we resolve this by showing that glutamate, acting at AMPA receptors, inhibits DA release by a nonclassic mechanism mediated by hydrogen peroxide (H2O2). Moreover, we show that GABAA-receptor activation opposes this process, thereby enhancing DA release. The influence of glutamate and GABA on DA release was assessed in striatal slices using carbon-fiber microelectrodes and fast-scan cyclic voltammetry. Modulation by both transmitters was prevented by H2O2-metabolizing enzymes. In addition, the influence of GABAA-receptor activation was lost when AMPA receptors were blocked with GYKI-52466. Together, these data show that modulation of DA release by glutamate and GABA depends on H2O2 generated downstream from AMPA receptors. This is the first evidence that endogenous glutamate can lead to the generation of reactive oxygen species under physiological conditions. We also show that inhibition of DA release by H2O2 is mediated by sulfonylurea-sensitive K+ channels: tolbutamide blocked DA modulation by glutamate and by GABA. The absence of ionotropic glutamate or GABA receptors on DA terminals indicates that modulatory H2O2 is generated in non-DA cells. Thus, in addition to its known excitatory actions in striatum, glutamate mediates inhibition by generating H2O2 that must diffuse from postsynaptic sites to inhibit presynaptic DA release via K+-channel opening. These findings have significant implications not only for normal striatal function but also for understanding disease states that involve DA and oxidative stress, including disorders as diverse as Parkinson's disease and schizophrenia.
Introduction
The overall circuitry of the basal ganglia is well known (Kemp and Powell, 1971; Albin et al., 1989; Smith and Bolam, 1990). However, the microchemical circuitry within individual structures is only beginning to be elucidated. This issue is particularly important for the striatum, which receives excitatory input from motor cortices and thalamus and supplies the major inhibitory output of the basal ganglia to subcortical structures (Albin et al., 1989; Smith and Bolam, 1990). The principal striatal efferent cells are GABAergic medium spiny neurons (Kemp and Powell, 1971), which receive synaptic glutamate input to their dendrites (Smith and Bolam, 1990; Bernard and Bolam, 1998; Chen et al., 1998). These neurons also receive synaptic dopamine (DA) input from midbrain DA cells (Albin et al., 1989; Smith and Bolam, 1990). Dopaminergic input is critical for the control of movement by the basal ganglia; its loss leads to the motor deficits of Parkinson's disease (Albin et al., 1989; Olanow and Tatton, 1999).
The issue of how, and even whether, glutamate regulates striatal DA release has been a long-standing source of controversy (Cheramy et al., 1986; Leviel et al., 1990; Moghaddam et al., 1990; Moghaddam and Gruen, 1991; Westerink et al., 1992; Keefe et al., 1993; Wu et al., 2000). The absence of ionotropic glutamate receptors on DA terminals (Bernard and Bolam, 1998; Chen et al., 1998) suggests that any influence is indirect. Proof of this has been elusive, however. Previous studies to address glutamate–DA interactions in striatum have been complicated by two factors. First, local application of glutamate agonists at levels sufficient to elicit DA release can trigger spreading depression (Moghaddam et al., 1990; Westerink et al., 1992). This massive depolarization causes a profound increase in extracellular DA concentration ([DA]o) but indicates little about normal glutamate–DA interactions. Second, most studies have been conducted in vivo, such that interactions among basal ganglia structures could overshadow local effects. For example, an increase in striatal [DA]o with local glutamate-antagonist application could reflect the consequences of decreased activation of the inhibitory striatonigral pathway, which could increase DA-cell activity in midbrain and consequent release in striatum. The change in striatal [DA]o would be indistinguishable from one mediated locally. Moreover, in vivo experiments to distinguish this would be not only technically challenging but also difficult to interpret.
To avoid these complications, we assessed the influence of glutamate on DA release using specific receptor antagonists in slices of guinea pig striatum in vitro. Release of DA was elicited using local electrical stimulation and monitored with subsecond resolution using carbon-fiber microelectrodes and fast-scan cyclic voltammetry (Bull et al., 1990; Wu et al., 2000; Chen et al., 2001; Chen and Rice, 2002). Initial studies revealed a paradoxical increase in evoked [DA]o when ionotropic AMPA receptors were blocked, suggesting a normally inhibitory action of glutamate on DA release. This proved to be independent of conventional inhibitory circuitry involving GABA. Because we had discovered previously that hydrogen peroxide (H2O2) could inhibit DA release in striatum (Chen et al., 2001), we proposed and tested the involvement of glutamate-dependent H2O2 generation as a novel inhibitory pathway. We report here that AMPA-receptor activation generates diffusible H2O2, which opens hyperpolarizing K+ channels and thereby inhibits synaptic DA release.
Materials and Methods
DA recording in striatal slices. All animal handing procedures were in accordance with National Institutes of Health guidelines and were approved by the New York University School of Medicine Animal Care and Use Committee. Young adult guinea pigs (male, Hartley, 150–250 gm) or, in some experiments, young adult rats (male, Long–Evans, 160–210 gm) were deeply anesthetized with 40 mg/kg pentobarbital (intraperitoneal) and decapitated. Coronal slices of striatum (400 μm thick) were prepared as described previously and then kept for at least 1 hr in HEPES-buffered artificial CSF (ACSF) before experimentation (Chen and Rice, 2001; Chen et al., 2001). After transfer to a submersion recording chamber at 32°C, slices were allowed another 30 min equilibration before stimulation; the superfusing ACSF contained the following (in mm): 124 NaCl, 3.7 KCl, 26 NaHCO3, 2.4 CaCl2, 1.3 MgSO4, 1.3 KH2PO4, and 10 glucose (equilibrated with 95% O2–5% CO2).
The voltammetric method used for all experiments was fast-scan cyclic voltammetry using a Millar voltammeter (PD Systems International, West Molesey, UK). Carbon-fiber microelectrodes were made from single, 8 μm carbon fibers, etched to a 2–4 μm tip (MPB Electrodes; Queen Mary and Westfield College, London, UK). Electrodes were postcalibrated in the recording chamber at 32°C in all media used in a given experiment [e.g., ACSF with mercaptosuccinate (MCS) and MCS plus catalase]. Release of DA was elicited using a bipolar stimulating electrode placed on a slice surface, with the carbon-fiber microelectrode positioned between the electrical poles and inserted 50–100 μm into the tissue (Chen and Rice, 2001; Chen et al., 2001). The usual stimulus was a 10 Hz train of 30–50 pulses delivered at 10 min intervals. In some experiments, train stimulation was alternated with single-pulse stimulation at 7 min intervals. Pulse duration for all stimulations was 100 μsec, and pulse amplitude was 0.4–0.6 mA. Striatal DA release evoked under these conditions is sensitive to inhibition by tetrodotoxin or removal of extracellular calcium (Chen and Rice, 2001).
Data are given as mean ± SEM (n indicates number of slices) and illustrated as percentage of same-site control. Only slices with at least three consistent control responses at a given site were tested further; these were averaged, and the mean peak [DA]o was taken as 100%. Differences in peak evoked [DA]o between conditions were assessed using unpaired Student's t test.
Drug and enzyme application. For studies with glutamate- and GABA-receptor antagonists or an inhibitor of glutathione peroxidase (GSHPx), MCS, control records were obtained, and then the agent was applied; maximal changes in evoked [DA]o were typically seen within 30 min of drug application. Antagonist concentrations used were those found previously to be effective in the nigrostriatal DA system (Chen and Rice, 2002), as follows: GYKI-52466 [1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine hydrochloride] (50 μm);d(−)-2-amino-5-phosphonopentanoic acid (AP-5) (50–100 μm); picrotoxin (100 μm); and saclofen [3-amino-2-(4-chlorophenyl)-2-hydroxypro-panesulfonic acid] (50 μm). GYKI-52466, AP-5, and picrotoxin were from Sigma-RBI (St. Louis, MO); saclofen was from Tocris Cookson (Ellisville, MO). MCS (1 mm) (Chen et al., 2002) was from Sigma-RBI. For studies with catalase (500 IU/ml) (Desagher et al., 1997; Chen et al., 2001) or GSHPx (3 IU/ml) (Saito et al., 1997), slices were preincubated in the enzyme in ACSF for 30 min at room temperature, followed by enzyme superfusion in the recording chamber at 32°C for an additional 30 min to ensure complete tissue infiltration. Catalase (bovine liver) was from Calbiochem (San Diego, CA), and GSHPx (bovine erythrocyte) was from Sigma-RBI. Control slices in these enzyme studies were exposed to heat-inactivated enzyme for the same periods. Enzyme solutions were inactivated by incubation at 75°C for 15 min; inactivation was confirmed by the lack of bubbling when 1.5 mmH2O2 was added, which was always seen with active enzyme solutions. The effect of DA transport inhibition was examined in slices preincubated for least 1 hr at room temperature in GBR-12909 (1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine) (2 μm) (Chen and Rice, 2001) before transfer to the recording chamber, with continued exposure to GBR-12909 during the experiment. GBR-12909 was from Research Biochemicals (Natick, MA). To examine the role of sulfonylurea-sensitive K+ channels, we first tested 100–300 μm tolbutamide (Stanford and Lacey, 1995) in pilot studies with MCS; 200 μm was the lowest effective concentration and was used subsequently. For these studies, control records were obtained, and then tolbutamide was superfused for 30–40 min, followed by application of MCS, GYKI-52466, or picrotoxin in the continued presence of tolbutamide. Tolbutamide was from Sigma-RBI.
Results
Do glutamate and GABA modulate striatal DA release?
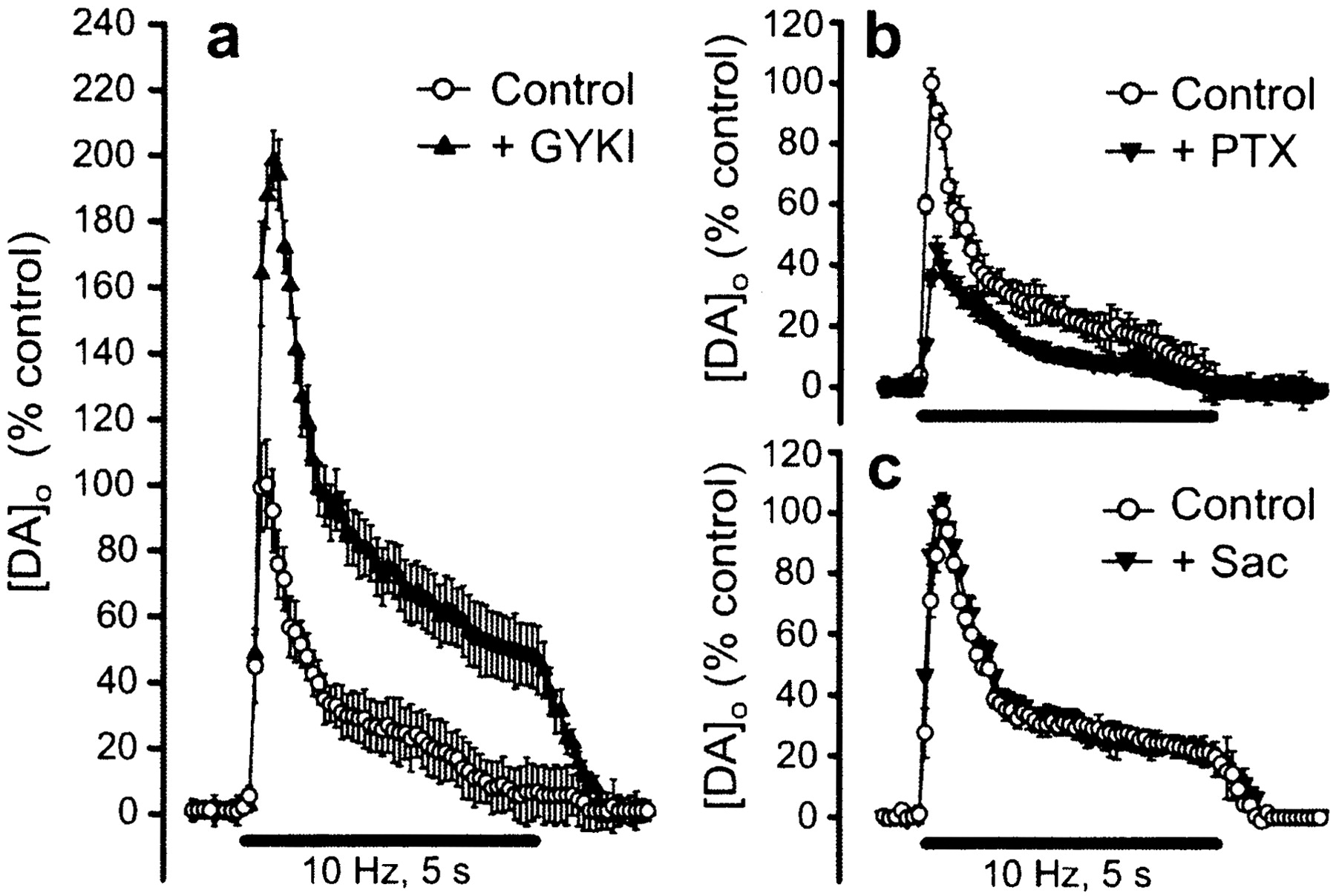
In initial studies of possible modulation of DA release by glutamate, we examined the effect of selective receptor antagonists on evoked [DA]o in striatal slices. Blockade of AMPA-type glutamate receptors with the selective antagonist GYKI-52466 caused a profound increase in evoked [DA]o(198% of control; p < 0.001; n = 6) (Fig. 1a). In contrast, the NMDA-receptor antagonist AP-5 had no effect on evoked [DA]o (p > 0.05;n = 11). This presumably reflected the mild stimulation used and the presence of physiological levels of Mg2+, both of which have been shown to prevent NMDA receptor-mediated excitation in the striatum in vitro (Jiang and North, 1991; Kita, 1996).

Modulation of evoked DA release by glutamate and GABA receptor activation. a, Application of the AMPA-receptor antagonist GYKI-52466 (GYKI; 50 μm) significantly increased evoked [DA]o in the striatum (p < 0.001;n = 6); mean peak [DA]o in control records was 1.57 ± 0.12 μm (n = 6). b, The GABAA antagonist picrotoxin (PTX; 100 μm) caused a significant decrease in evoked [DA]o (p < 0.001; n = 6), whereas the GABABantagonist saclofen (c, Sac; 50 μm) had no effect (p > 0.05;n = 8). a–c, Data are mean ± SEM, illustrated as percentage of same-site control. DA release was elicited by pulse-train stimulation (10 Hz). Solid barsindicate stimulation period.
The absence of AMPA receptors on DA terminals (Bernard and Bolam, 1998;Chen et al., 1998) indicates that AMPA receptor-dependent inhibition of DA release must be indirect, via activation of a secondary inhibitory pathway. We therefore tested the possible involvement of a GABAergic circuit using GABA-receptor antagonists. A role for GABA in this process presupposes that GABA normally inhibits DA release; GABA receptor blockade would be expected to increase evoked [DA]o in striatum, as we found previously for somatodendritic DA release in the substantia nigra pars compacta (Chen and Rice, 2002). Unexpectedly, however, blockade of striatal GABAA receptors with picrotoxin caused a decrease in evoked [DA]o (by 46%;p < 0.001 vs control; n = 6) (Fig.1b), whereas the GABAB antagonist saclofen had no effect (p > 0.05;n = 8) (Fig. 1c). This net excitatory influence of GABA, therefore, could not mediate glutamatergic inhibition of DA release. Furthermore, the paucity of GABA receptors on DA terminals (Fujiyama et al., 2000) suggests that the influence of GABA is also indirect.
Glutamate-dependent inhibition of DA release is mediated by H2O2
The lack of involvement of conventional GABAergic circuitry in AMPA receptor-dependent inhibition of DA release suggested that another, perhaps unconventional, pathway was involved. We reported previously that endogenously generated H2O2 can reversibly inhibit DA release in striatum (Chen et al., 2001, 2002). Because studies in cultured cells show that ionotropic glutamate-receptor activation can increase production of H2O2and other reactive oxygen species (ROS) (Bondy and Lee, 1993;Lafon-Cazal et al., 1993; Dugan et al., 1995; Reynolds and Hastings, 1995; Bindokas et al., 1996; Carriedo et al., 2000), we tested whether glutamate-dependent H2O2modulated DA release in striatal slices. In these experiments, DA release was evoked in the presence of an H2O2-metabolizing enzyme, either catalase or GSHPx, and then AMPA receptors were blocked with GYKI-52466.
In the presence of active catalase, the GYKI-52466-dependent increase in evoked [DA]o was prevented completely (p > 0.05; GYKI-52466 vs control;n = 6) (Fig.2a). When catalase was heat inactivated, however, GYKI-52466 caused the usual ∼200% increase in evoked [DA]o in paired slices (p < 0.001; n = 6) (Fig.2b). Application of exogenous GSHPx also prevented the effect of GYKI-52466 (Fig. 2c), with the expected GYKI-52466-induced increase in evoked [DA]o in heat-inactivated GSHPx (p < 0.001;n = 8) (Fig. 2d). Because catalase and GSHPx are high-molecular-weight proteins (catalase, 240 kDa; GSHPx, 125 kDa), they are unlikely to enter cells and therefore must remain in the extracellular space, although their action might not be limited to that compartment.

Modulation of striatal DA release by glutamate depends on the availability of H2O2.a, Active catalase (Cat; 500 IU/ml) abolished the effect of GYKI-52466 (50 μm) on evoked [DA]o (p > 0.05;n = 6); b, in the presence of heat-inactivated catalase (I-Cat), application of GYKI-52466 caused the usual increase of evoked [DA]o(p < 0.001; n = 6).c, Active GSHPx (3 IU/ml) also prevented the effect of GYKI-52466 on evoked [DA]o (p> 0.05; n = 8); d, in the presence of heat-inactivated GSHPx (I-GSHPx), GYKI-52466 again caused a significant increase in evoked [DA]o(p < 0.001; n = 8).a–d, Solid bars indicate stimulation period.
To confirm the involvement of H2O2 in DA modulation, we examined the effect of manipulating H2O2 availability by using MCS to inhibit endogenous GSHPx activity (Ying et al., 1999; Sokolova et al., 2001; Chen et al., 2002) and then adding exogenous catalase to oppose this. Consistent with the expected effect of elevated H2O2 levels, MCS caused an ∼40% decrease in evoked [DA]o (Fig.3a). This suppression was reversed when catalase was added in the continued presence of MCS (Fig.3a). These data demonstrate the H2O2 specificity of both manipulations and hence confirm a modulatory role for H2O2. The data also indicate that changes in local H2O2 availability can increase or decrease DA release.

Modulation of striatal DA release by endogenous H2O2 is AMPA-receptor dependent.a, Average evoked [DA]o under control conditions and in the presence of the GSHPx inhibitor MCS (1 mm). Application of MCS caused a 40% decrease in evoked [DA]o from 1.52 ± 0.07 to 0.89 ± 0.04 μm (p < 0.001;n = 7); inset shows identifying DA voltammograms recorded at the peak of the response under control conditions (solid line) and after MCS (dashed line). In the continued presence of MCS, added catalase (Cat; 500 IU/ml) reversed the MCS-induced suppression of evoked [DA]o (to 1.62 ± 0.12 μm;p > 0.05; MCS plus catalase vs control;n = 7). b, The AMPA-receptor antagonist GYKI-52466 (GYKI; 50 μm) caused a significant increase in evoked [DA]o(p < 0.001; n = 5). In the continued presence of GYKI-52466, the effect of MCS in evoked [DA]o was prevented (p > 0.05 vs GYKI-52466 alone; n = 5). c, DA release elicited by single-pulse stimulation (1 p,arrow) was unaffected by GYKI-52466 (50 μm), indicating a lack of glutamate-dependent regulation (p > 0.05 vs control;n = 4). d, Effect of GSHPx inhibition by MCS on DA release evoked by single-pulse and 30-pulse train stimulation (10 Hz). Average maximum [DA]o evoked by single-pulse stimulation was 1.43 ± 0.08 μm(n = 5) and was not affected by MCS (p > 0.05; n = 5). During pulse-train stimulation, however, MCS caused a decrease in evoked [DA]o (p < 0.001;n = 5) (indicated by dashed line), which reversed during MCS washout (p > 0.05 wash vs control; n = 5). a,b, d, Solid bar indicates pulse-train stimulation period.
We next tested whether the primary source of H2O2 generation was downstream from AMPA-receptor activation. If this were true, the effect of MCS should be lessened in the presence of GYKI-52466. Indeed, when AMPA receptors were blocked by GYKI-52466, the MCS-induced decrease in evoked [DA]o was lost (p> 0.05; n = 5; GYKI-52466 plus MCS vs GYKI-52466 alone) (Fig. 3b).
To examine whether repetitive stimulation is required for H2O2 modulation of DA release, we compared the influence of MCS on [DA]o elicited by single-pulse and 30-pulse stimulations. An essential first step in this study was to determine whether [DA]o evoked by a single pulse was affected by AMPA-receptor activation. In contrast to the marked effect of GYKI-52466 on [DA]o evoked by pulse-train stimulation (Fig. 1a), single-pulse evoked release was not altered by AMPA-receptor blockade (p > 0.05;n = 4) (Fig. 3c). When single-pulse and pulse-train stimulations were alternated in a given slice, MCS caused the usual reversible decrease in evoked [DA]oduring pulse-train stimulation (p < 0.001; MCS vs control or wash; n = 5). In contrast, maximum [DA]o evoked by a single pulse was unaltered in the presence of MCS in the same slice (p > 0.05; n = 5) (Fig. 3d). The lack of effect of MCS on single-pulse-evoked release shows that regulation of DA release by H2O2 is dynamic rather than tonic. Together with the data in Figure 3b, these findings also confirm a requirement for AMPA-receptor activation in generating modulatory H2O2.
H2O2 acts directly to modulate DA release, not DA uptake
Absolute [DA]o reflects the net effect of both release and uptake. We determined whether H2O2 acts by inhibiting DA release or by increasing DA uptake via the DA transporter (DAT) by examining the effect of MCS on evoked [DA]o in the presence of the DAT inhibitor GBR-12909. Inhibition of the DAT caused a significant increase in maximum evoked [DA]o and prolonged the [DA]o time course, as expected (Chen and Rice, 2001); however, uptake inhibition had no effect on the usual ∼40% decrease in evoked [DA]o induced by MCS (Fig.4a).

The inhibitory effect of H2O2 on DA release is direct and species independent. a, H2O2 modulates DA release but not uptake. In the presence of the DA transport inhibitor GBR-12909 (GBR; 2 μm), inhibition of GSHPx by MSC (1 mm) decreased maximum evoked [DA]o from 4.40 ± 0.17 to 2.75 ± 0.08 μm (p < 0.001;n = 4). b, Effect of ascorbate on evoked [DA]o under control conditions and during GSHPx inhibition. Stable evoked DA release was elicited under control conditions, and then ascorbate (Asc; 400 μm) was applied. The presence of ascorbate did not alter evoked [DA]o (p > 0.05;n = 5), nor did it interfere with the usual depression of evoked [DA]o by MCS (1 mm;p < 0.001; n = 5).c, Average evoked [DA]o in rat striatum in control and in the presence of MCS. Application of MCS caused a reversible 35% decrease in evoked [DA]o in rat dorsal striatum, from a maximum of 1.80 ± 0.15 to 1.17 ± 0.07 μm (p < 0.001;n = 7). a–c, Solid bars indicate stimulation period.
To assess whether inhibition of DA release is a direct effect of H2O2 or is mediated by the hydroxyl radical (·OH), which can be formed from the interaction of H2O2 and trace metal ions (Cohen, 1994; Avshalumov et al., 2000), we examined the effect of physiological levels (400 μm) of the primary extracellular antioxidant ascorbate (Rice, 2000). Ascorbate had no effect on control evoked [DA]o(p > 0.05; n = 5). Moreover, the inhibitory effect of endogenous H2O2 was unaffected by ascorbate (p < 0.001; MCS plus ascorbate vs ascorbate alone; n = 5) (Fig. 4b). This indicates that the action of H2O2 is not mediated by ·OH, which is readily scavenged by ascorbate (Cohen, 1994; Avshalumov et al., 2000; Rice, 2000). In addition, because of known species differences of antioxidant regulation (Rice, 2000), we investigated the effect of MCS on DA release in slices of rat striatum. The species independence of H2O2-mediated inhibition of synaptic DA release was shown by a reversible decrease in evoked [DA]o when GSHPx was inhibited by MCS in rat striatum (p < 0.001; MCS vs control or wash;n = 7) (Fig. 4c), which was comparable with that seen in guinea pig (Fig. 3a).
GABA-dependent modulation of DA release is also mediated by H2O2
Having established that the inhibitory effect of AMPA-receptor activation on DA release was mediated by H2O2, we tested whether the apparently excitatory role of GABA acting at GABAA receptors was also H2O2 dependent. Strikingly, active catalase completely prevented the effect of picrotoxin on DA release (p > 0.05; n = 6) (Fig.5a); in heat-inactivated catalase, however, picrotoxin caused the usual decrease in evoked [DA]o (p < 0.001;n = 6) (Fig. 5b).

GABA opposes the inhibitory effect of H2O2 on synaptic DA release in striatum.a, The decrease in evoked [DA]o by the GABAA-receptor antagonist picrotoxin (PTX; 100 μm) was prevented by catalase (Cat) (p > 0.05; n = 6);b, in heat-inactivated catalase (I-Cat), picrotoxin induced a decrease in evoked [DA]o(p < 0.001; n = 6).c, Application of GYKI-52466 (GYKI; 50 μm) significantly increased evoked [DA]o(p < 0.001; n = 7).d, In the continued presence of GYKI-52466, the effect of picrotoxin was completely prevented (p> 0.05; n = 7), showing that the influence of GABA on DA release requires AMPA-receptor activation. a–d,Solid bars indicate stimulation period.
We also hypothesized that glutamate and GABA acted on the same source of H2O2, with GABA opposing glutamate-dependent H2O2generation. If this were true, GABAA-receptor activation should have no effect when glutamate receptors were blocked. In experiments to test this, GYKI-52499 caused the expected increase in evoked [DA]o (p < 0.001; n = 7) (Fig. 5c); the continued presence of GYKI-52466 also completely prevented the effect of picrotoxin on evoked [DA]o(p > 0.05; n = 7) (Fig.5d).
H2O2 inhibits DA release by activating K+ channels
In many cell types, including neurons (Seutin et al., 1995), exogenous H2O2 activates a K+ conductance that causes reversible membrane hyperpolarization. In pancreatic β-cells, the source has been identified as ATP-sensitive potassium (KATP) channels, which can be blocked by the sulfonylurea agent tolbutamide (Krippeit-Drews et al., 1999). We therefore tested whether endogenous H2O2 inhibited DA release by the same mechanism. Consistent with KATP-channel involvement, tolbutamide (200 μm) caused a significant increase in evoked [DA]o (p < 0.01;n = 8) (Fig.6a). Moreover, tolbutamide completely prevented the inhibitory effect of MCS on DA release (p > 0.05; n = 8) (Fig.6a). Modulation of DA release by AMPA and GABAA receptors also required K+-channel activation; neither GYKI-52466 (Fig. 6b) nor picrotoxin (Fig. 6c) altered evoked [DA]o in the presence of tolbutamide (p > 0.05; n = 6 for GYKI-52466; n = 8 for picrotoxin).

H2O2-dependent modulation of DA release is mediated by sulfonylurea-sensitive K+ channels. a, Tolbutamide (Tolb; 200 μm) caused a significant increase in evoked [DA]o (p< 0.01; n = 8); in the continued presence of tolbutamide, the usual suppression of DA release by MCS (1 mm) was prevented (p > 0.05;n = 8). b, Tolbutamide prevented the usual increase in DA release with GYKI-52466 (GYKI; 50 μm) (p > 0.05;n = 6). c, Tolbutamide prevented the usual decrease in DA release by picrotoxin (PTX; 100 μm) (p > 0.05;n = 8). a–c, Solid bars indicate stimulation period.
Discussion
This report introduces H2O2 as a diffusible messenger that mediates glutamatergic inhibition. Only one previous report described inhibition after direct glutamate-receptor activation, with a hyperpolarization of midbrain DA neurons induced by metabotropic-receptor activation at glutamate synapses on these cells (Fiorillo and Williams, 1998). The present results are more surprising, with glutamate-dependent inhibition of DA release mediated by ionotropic receptors, which are not expressed on DA terminals (Bernard and Bolam, 1998; Chen et al., 1998). Several previous studies suggested a net inhibitory effect of glutamate on DA release in striatum; however, the mechanism was unresolved (Moghaddam and Gruen, 1991; Keefe et al., 1993; Wu et al., 2000). Here, we show that AMPA-receptor activation causes H2O2generation and subsequent opening of sulfonylurea-sensitive K+ channels. Furthermore, GABA, acting at GABAA receptors, enhances DA release by opposing AMPA receptor-dependent H2O2 generation. Thus, H2O2 effectively reverses the expected consequences of glutamatergic excitation and GABAergic inhibition on DA release.
As a neutral, membrane-permeable molecule (Ramasarma, 1983), H2O2 shares characteristics with the known diffusible messengers nitric oxide (NO) and carbon monoxide (CO) (Dawson and Snyder, 1994; Kiss and Vizi, 2001). We show that H2O2, like NO (Kiss and Vizi, 2001), mediates a nonclassic form of neuronal communication by which glutamate and GABA affect release sites that do not receive direct synaptic input. Significantly, the present in vitrodata are consistent with previous in vivo studies showing an increase in striatal [DA]o with local glutamate-antagonist application (Moghaddam and Gruen, 1991; Keefe et al., 1993). The similarity of these results suggests that the increase in [DA]o seen in vivo reflects local glutamate–DA interactions rather than requiring extrastriatal circuitry and that H2O2generation is the underlying mechanism both in vivo andin vitro.
AMPA receptor-dependent H2O2 generation
Historically, H2O2 and other ROS were viewed as toxic metabolic “byproducts” that must be eliminated to prevent oxidative stress. This has been challenged by recent studies showing that ROS can regulate signaling pathways (Klann and Thiels, 1999) and modulate synaptic transmission (Pellmar 1987;Chen et al., 2001) and plasticity (Auerbach and Segal, 1997; Klann and Thiels, 1999).
In most tissues, the primary source of ROS is mitochondrial respiration. Incomplete reduction of O2 produces the superoxide radical (·O2−), which is converted to H2O2 by superoxide dismutase (Boveris and Chance, 1973; Cohen, 1994). Production of H2O2 by mitochondria can exceed 2% of oxygen consumed (Boveris and Chance, 1973). Thus, any activity-dependent increase in oxygen consumption, including striatal stimulation (Kennedy et al., 1992), would enhance H2O2 production (Boveris and Chance, 1973). Moreover, generation of ROS, including H2O2, can be induced by ionotropic glutamate-receptor activation (Bondy and Lee, 1993;Lafon-Cazal et al., 1993; Dugan et al., 1995; Reynolds and Hastings, 1995; Bindokas et al., 1996; Carriedo et al., 2000) but not metabotropic-receptor activation (Bondy and Lee, 1993).
The present studies provide the first evidence that endogenous glutamate, acting at AMPA receptors, generates H2O2: not only was AMPA receptor-dependent modulation of DA prevented by catalase or GSHPx (Fig. 2), but the suppression of DA release that accompanies GSHPx inhibition by MCS was also prevented by AMPA-receptor blockade (Fig.3b). These data imply that AMPA-receptor activation is necessary to generate modulatory H2O2, although confirmation awaits direct detection, e.g., by ROS imaging (Sah and Schwartz-Bloom, 1999).
H2O2-dependent inhibition of DA release
The first step in understanding the mechanism by which H2O2 depresses evoked [DA]o was the demonstration that H2O2 modulates release rather than uptake: MCS-induced suppression of evoked [DA]o was unaltered when the DAT was inhibited by GBR-12909 (Fig. 4a). In contrast, modulation of [DA]o by NO is DAT mediated, such that the enhancing effect of NO is prevented when the DAT is inhibited pharmacologically (Kiss and Vizi, 2001). The present findings also indicate that H2O2 acts directly and not via secondary ·OH formation, because physiological levels of the ·OH scavenger ascorbate (Cohen, 1994; Avshalumov et al., 2000; Rice, 2000) neither altered evoked [DA]o nor prevented the inhibitory effect of MCS. These data, combined with our previous findings that tissue DA content and [DA]o in striatum are stable during brief exposure to H2O2(Chen et al., 2001), also eliminate an alternative explanation that decreased [DA]o might reflect H2O2-dependent DA oxidation.
The lack of effect of MCS on [DA]o evoked by single-pulse stimulation shows that changing H2O2 availability by MCS has no effect on tonic regulation of DA release. Rather, H2O2 formed during the first pulse of a train inhibits DA release elicited by subsequent pulses. This is also supported by the lack of effect of AMPA-receptor blockade on single-pulse-evoked [DA]o. The efficacy of MCS on DA release during pulse-train stimulation also indicates that H2O2-mediated inhibition is fast: a decrease in evoked [DA]o in MCS versus control can be seen within the first three to five pulses (300–500 msec) of a 10 Hz train (Fig. 3). Thus, dynamic changes in H2O2 generated during neuronal activation can depress DA release on a physiological time scale.
Previous studies showed that exogenous H2O2 can cause membrane hyperpolarization by activating K+channels (Seutin et al., 1995; Krippeit-Drews et al., 1999). This suggested a mechanism by which endogenous H2O2 might inhibit DA release, which we tested using the sulfonylurea-receptor antagonist tolbutamide. Significantly, tolbutamide completely prevented DA release modulation by AMPA- and GABAA-receptor blockade and by MCS (Fig. 6). Thus, glutamate-generated H2O2 inhibits DA release by activating sulfonylurea-sensitive K+channels; sensitivity to blockade by tolbutamide implicates KATP channels (Stanford and Lacey, 1995;Krippeit-Drews et al., 1999; Liss et al., 1999). The present data are the first to demonstrate K+-channel activation by endogenous H2O2.
Where is modulatory H2O2 generated?
The absence of ionotropic glutamate receptors on DA terminals (Bernard and Bolam, 1998; Chen et al., 1998), together with the prevention of glutamate-dependent modulation of DA by extracellular peroxidase enzymes, indicates that glutamate-dependent H2O2 must be generated in non-DA cells. Statistically, the most likely cells are medium spiny neurons, which constitute 90–95% of striatal neurons (Kemp and Powell, 1971). This is supported by the pattern of sensitivity of DA release to glutamate antagonists, which mirrors the electrophysiological responsiveness of spiny neurons (Jiang and North, 1991; Kita, 1996). Moreover, glutamate synapses can be closely apposed to DA synapses on the dendrites of the these cells (Smith and Bolam, 1990; Bernard and Bolam, 1998; Chen et al., 1998), placing them in an ideal position to modulate DA release via diffusible H2O2. Other striatal cells that express AMPA receptors, e.g., cholinergic interneurons (Bernard and Bolam, 1998; Chen et al., 1998), could contribute. However, these are sparse and lack DA synapses, making it less likely that the level and lifetime of H2O2generated would be sufficient to affect DA release at relatively distant synapses.
Significantly, GABAA receptors are also expressed on the dendrites of medium spiny neurons (Fujiyama et al., 2000). Thus, GABAergic input is well positioned to oppose AMPA receptor-mediated excitation and consequent H2O2 generation. Indeed, the decrease in DA release usually seen with picrotoxin was prevented by AMPA-receptor blockade and by catalase (Fig. 5). The involvement of GABAA but not GABABreceptors (Fig. 1) also implicates medium spiny neurons in DA modulation, because GABA input to these cells is mediated predominantly by GABAA receptors (Jiang and North, 1991; Kita, 1996). A role for GABAA receptors in DA modulation contradicts a previous study (Wu et al., 2000); the high-frequency stimulus used previously, however, may have masked the GABA-dependent modulation of DA release revealed here. Together, the present findings indicate that glutamate and GABA act on the same pool of H2O2. Like a brake when there is no motion, however, GABA has no direct influence on DA release but rather counters the extent to which glutamate-receptor activation generates H2O2, thereby “fine-tuning” this process.
Our working hypothesis, therefore, is that regulation of striatal DA release by glutamate and GABA involves a triad of DA, glutamate, and GABA synapses, separated by a few micrometers on the dendrites of medium spiny neurons (Smith and Bolam, 1990; Bernard and Bolam, 1998; Chen et al., 1998; Fujiyama et al., 2000) but bound together functionally by diffusible H2O2. Mitochondria are a likely source of glutamate-dependent H2O2 generation (Dugan et al., 1995; Reynolds and Hastings, 1995; Bindokas et al., 1996; Carriedo et al., 2000), although other metabolic pathways could contribute (Lafon-Cazal et al., 1993).
Implications
We show that glutamate-dependent H2O2 is a diffusible messenger that can modulate DA release on a physiologically relevant time scale. This previously unknown inhibitory intermediate provided the key to understanding regulation of striatal DA release by glutamate and GABA. Given recent studies implicating DA–glutamate interactions in striatal plasticity underlying addictive behaviors (Everitt and Wolf, 2002), the findings have implications for reward and motor pathways. We anticipate that analogous patterns of modulation occur throughout the CNS; indeed, H2O2 and other ROS contribute to neuron–glial signaling in hippocampus (Atkins and Sweatt, 1999).
Finally, it should be noted that regulation of neurotransmission by H2O2 is a double-edged sword: if H2O2 generation and regulation became imbalanced, whether acutely or chronically, the consequence would be oxidative stress. Significantly, oxidative stress has been implicated in nigrostriatal degeneration in Parkinson's disease (Cohen, 1994; Sonsalla et al., 1997; Olanow and Tatton, 1999;Xu et al., 2002) and more recently as a causal factor in schizophrenia (Do et al., 2000; Yao et al., 2001). Thus, the present findings reveal a normal regulatory pathway that, if disrupted, could contribute to DA-system pathology.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant NS-36362. We appreciate helpful discussions about sulfonylurea-sensitive K+ channels with Jochen Roeper and William A. Coetzee and about mitochondrial localization in striatal cells with J. Paul Bolam.
Correspondence should be addressed to Dr. M. E. Rice, Department of Physiology and Neuroscience, New York University School of Medicine, 550 First Avenue, New York, NY 10016. E-mail: margaret.rice{at}nyu.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}