

Abstract
Blood–brain barrier (BBB) disruption occurs early enough to be within the thrombolytic time window, and this early ischemic BBB damage is closely associated with hemorrhagic transformation and thus emerging as a promising target for reducing the hemorrhagic complications of thrombolytic stroke therapy. However, the mechanisms underlying early ischemic BBB damage remain poorly understood. Here, we investigated the early molecular events of ischemic BBB damage using in vitro oxygen–glucose deprivation (OGD) and in vivo rat middle cerebral artery occlusion (MCAO) models. Exposure of bEND3 monolayer to OGD for 2 h significantly increased its permeability to FITC-labeled dextran and promoted the secretion of metalloproteinase-2 and -9 (MMP-2/9) and cytosolic translocation of caveolin-1 (Cav-1). This same OGD treatment also led to rapid degradation of tight junction protein occludin and dissociation of claudin-5 from the cytoskeleton, which contributed to OGD-induced endothelial barrier disruption. Using selective MMP-2/9 inhibitor SB-3CT (2-[[(4-phenoxyphenyl)sulfonyl]methyl]-thiirane) or their neutralizing antibodies or Cav-1 siRNA, we found that MMP-2 was the major enzyme mediating OGD-induced occludin degradation, while Cav-1 was responsible for claudin-5 redistribution. The interaction between Cav-1 and claudin-5 was further confirmed by coimmunoprecipitation. Consistent with these in vitro findings, we observed fluorescence tracer extravasation, increased gelatinolytic activity, and elevated interstitial MMP-2 levels in ischemic subcortical tissue after 2 h MCAO. Moreover, occludin protein loss and claudin-5 redistribution were detected in ischemic cerebromicrovessels. These data indicate that cerebral ischemia initiates two rapid parallel processes, MMP-2-mediated occludin degradation and Cav-1-mediated claudin-5 redistribution, to cause BBB disruption at early stroke stages relevant to acute thrombolysis.
Introduction
Hemorrhagic transformation (HT), particularly severe hemorrhage, is the most feared neurovascular complication of tissue plasminogen activator (tPA) thrombolysis, the only FDA-approved stroke treatment (Alberts, 1998; Derex and Nighoghossian, 2008). Since loss of blood–brain barrier (BBB) integrity is considered to be a precursor to HT (Latour et al., 2004), understanding the dynamics of BBB injury and the underlying molecular mechanisms are of particular importance for the prevention or treatment of HT. BBB damage is a progressive process, with an initial injury resulting from ischemia (del Zoppo et al., 1998; Simard et al., 2007), aggravated by reperfusion due to a mechanism of reperfusion injury (Hallenbeck and Dutka, 1990). In more than one decade, reperfusion-associated BBB injury has been a topic of intensive investigation, which leads to the identification of several mechanisms accounting for reperfusion BBB injury, such as oxidative damage, inflammatory injury, vascular activation, and dysregulated extracellular proteolysis (Rosenberg et al., 1998; Wang and Lo, 2003; Jung et al., 2010). In contrast, the initial BBB damage resulting from ischemia is a far less well studied topic.
Recent animal and human studies offer an intriguing observation that ischemic brain regions with compromised BBB at early ischemic stroke stages often undergo HT following thrombolytic reperfusion (Hjort et al., 2008; Kastrup et al., 2008; Kassner et al., 2009; Sun et al., 2010), which supports a close association between early ischemic BBB damage and posttreatment HT. Moreover, assessing early BBB damage with computed tomography appears promising for identifying stroke patients at high risk of postthrombolysis HT (Aviv et al., 2009). Thus, understanding early ischemic BBB is crucial for reducing thrombolysis-associated HT.
Tight junctions (TJs) are important BBB structural components that seal the gaps between adjacent endothelial cells and thus restrict paracellular permeability (Wolburg and Lippoldt, 2002). The transmembrane protein claudins and occludin are key molecules forming the seal (Förster, 2008). In animal stroke models with reperfusion, altered distribution or loss of TJ proteins is frequently seen in ischemic cerebral microvessels, resulting in compromised BBB integrity (Asahi et al., 2001; Y. Yang et al., 2007; Liu et al., 2009a; ElAli et al., 2011; Wang et al., 2011). And in these same studies, the gelatinases, matrix metalloproteinase-2 and 9 (MMP-2/9), have been shown to mediate the degradation of several TJ proteins including occludin, claudin-5, and zonula occludens-1. There are a few studies specifically designed to look at early ischemic BBB damage by using animal stroke models with limited reperfusion (10–30 min) (DiNapoli et al., 2008; Chen et al., 2009; Gerriets et al., 2009). However, the molecular events mediating this early ischemic BBB damage remain virtually unknown.
In the present study, we investigated the effect of early ischemia on MMP-2/9, TJ proteins, and BBB integrity using in vitro [oxygen–glucose deprivation (OGD)] and in vivo [middle cerebral artery occlusion (MCAO)] stroke models. Our results demonstrated that cerebral ischemia initiated two rapid parallel processes, MMP-2-mediated occludin degradation and caveolin-1-mediated redistribution of claudin-5, to cause BBB disruption in an early stroke stage relevant to acute stroke thrombolysis.
Materials and Methods
Cell culture.
Mouse brain microvascular endothelial cells bEND3 (American Type Culture Collection) were grown as a monolayer in DMEM with 15% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified incubators with 5% CO2 and 95% room air. The cells were subcultured into 60 mm dishes coated with type I collagen (Nakamuta et al., 2005) and allowed to grow to confluence before exposure to OGD for 2 h. After OGD treatment, cell toxicity was measured by lactate dehydrogenase (LDH) assay using a CytoTox 96 Nonradioactive Cytotoxicity Assay Kit (Promega). In addition to endothelial cells, we also tested the effect of OGD on MMP-2/9 secretion in two mouse neural cell lines, C8-D1A (astrocyte) and SH-SY5Y (neuron). Both cell lines were purchased from American Type Culture Collection. C8-D1A cells and SH-SY5Y cells were cultured in DMEM or DMEM/F12 (1:1) (Sigma-Aldrich) containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, respectively. These cells were allowed to grow to 80–90% confluence before exposure to 2 h OGD treatment.
OGD treatment.
To mimic acute ischemia-like conditions in vitro, bEND3 cells were exposed to OGD for 2 h as we described previously (Furuichi et al., 2005). In brief, confluent bEND3 cells were subjected to an ischemic injury by transferring cultures to glucose-free medium (DMEM without glucose) preequilibrated with 95% N2 and 5% CO2. Cells were then incubated in a humidified airtight chamber (Billups-Rothenberg) equipped with an air lock and flushed with 95% N2 and 5% CO2 for 15 min. The chamber was then sealed and kept at 37°C for another 105 min. The oxygen concentration was <0.2% as monitored by an oxygen analyzer (Sable Systems). Control cultures were incubated with normal DMEM medium without FBS for 2 h at 37°C in 95% air and 5% CO2. Immediately after OGD treatment, the conditioned media (CM) and cells were collected separately for further analyses.
Preparation of total cell lysate, subcellular fractions.
To reveal a redistribution of MMP-2/9, Caveolin-1 (Cav-1), and TJ proteins in OGD-treated bEND3 cells, three subcellular fractions including membranous, cytosolic, and cytoskeletal fractions were extracted using ProteoExtract Subcellular Proteome Extraction Kit (Calbiochem) according to manufacturer's instruction. The specificity of each fraction was confirmed using anti-cytochrome Cypor [membrane/organelle fraction (MF)], anti-calpain [cytosolic fraction (CF)], and anti-vimentin [actin cytoskeletal fraction (ACF)] antibodies. To extract total cell lysates, cells were lysed in RIPA buffer (Santa Cruz Biotechnology).
Endothelial cell monolayer permeability assay.
The effect of OGD on endothelial monolayer permeability to fluorescein isothiocyanate-conjugated 70 kDa dextran (FITC-dextran; Sigma-Aldrich) was assessed using Anopore membrane 24-well cell culture inserts with 0.2 μm pore size (Nunc). Cells were placed on the upper side of the insert and allowed to grow to confluence. FITC-dextran at a concentration of 3.5 μm (Eckle et al., 2008) was then added to the endothelial monolayer (luminal compartment) before exposing to 2 h OGD (see Fig. 1A, top panel). After OGD treatment, the contents of FITC-dextran in both luminal and abluminal compartments were determined using a fluorescence plate reader Spectramax M2e (Molecular Devices). Endothelial monolayer permeability was then assessed by calculating the apparent permeability coefficient (Papp) as previously described (Grabovac and Bernkop-Schnürch, 2006): Papp (in centimeters per second) = dQ/(dt*A*Co), where dQ was the amount of FITC-dextran getting into the abluminal compartment, dt was duration of OGD treatment, dQ/dt was the rate of transfer (in nanograms per second), A was surface area (in square centimeters), and Co was the initial concentration in the luminal chamber (in nanograms per cubic centimeter). To test whether MMP-2/9 and Cav-1 were implicated in OGD-induced endothelial barrier disruption, cells were treated with selective MMP-2/9 inhibitor 2-[[(4-phenoxyphenyl)sulfonyl]methyl]-thiirane (SB-3CT) (10 μm; Calbiochem) 2 h before and during OGD treatment or pretreated with Cav-1 siRNA for 48 h before OGD treatment.
siRNA transfection.
bEnd3 cells at 60–70% confluence were transfected with 80 pmol of Cav-1 siRNA (Santa Cruz Biotechnology; sc-29520) or scrambled control siRNA (Santa Cruz Biotechnology; sc-37007) using siRNA Transfection Reagent (Santa Cruz Biotechnology) according to the manufacturer's instruction. Forty-eight hours after transfection, cells were subjected to OGD treatment. Specific silencing was confirmed by Western blot.
Gel gelatin zymography.
After OGD treatment, MMP-2/9 in CM and cellular extracts (CEs) were analyzed by gelatin zymography as we described previously (Liu et al., 2007). In brief, equal amounts of CM or CEs (containing 400 μg of protein) were concentrated with gelatin-Sepharose 4B beads (GE Healthcare). The MMP-2/9 were then eluted from gelatin beads by incubating with elution buffer (10% DMSO in PBS) and electrophoretically separated on 10% SDS-polyacrylamide gels copolymerized with 1 mg/ml gelatin (Sigma-Aldrich) under non-reducing condition. Gels were washed in 2.5% Triton X-100 to remove SDS and then incubated for 48 h with a developing buffer containing 50 mm Tris, pH 7.6, 5 mm CaCl2, 0.2 mm NaCl, and 0.02% (w/v) Brij-35 at 37°C before staining with 0.125% Coomassie Blue R-250. Gels were destained to visualize gelatinolytic bands (MMP-2/9) on a dark blue background. The intensities of MMP-2/9 bands were analyzed using the Kodak 4000 image station (Carestream Molecular Imaging). A mixture of human MMP-2/9 (Millipore Bioscience Research Reagents) was used as gelatinase standards.
MMP-2 and -9 antibody neutralization.
To distinguish the role between MMP-2 and MMP-9 in mediating OGD-induced occludin degradation, we performed antibody neutralization experiments, as described previously (De Becker et al., 2007; Gong et al., 2008). In brief, 20 μg/ml control mouse IgG (Santa Cruz Biotechnology) or neutralizing antibodies (Millipore) against MMP-2, MMP-9, or both were added to confluent bEND3 cells right before 2 h OGD treatment. After OGD treatment, cells were lysed with RIPA buffer for assessing total occludin levels using Western blot.
Coimmunoprecipitation.
Coimmunoprecipitation was performed as described previously (Wen et al., 2010). Briefly, bEND3 cells were subjected to OGD treatment for 2 h and then lysed on ice in 1 ml of RIPA buffer. After preclearing with normal IgG, cell lysates (0.5 mg of protein) were incubated overnight at 4°C with 2 μg of anti-Cav-1 (Santa Cruz Biotechnology), followed by precipitation with 20 μl of Protein A/G Plus-Agarose (immunoprecipitation reagent) for 1 h at 4°C. The precipitated complexes were separated on SDS-polyacrylamide gels and immunoblotted with anti-claudin-5 to detect the presence of this protein in the complex, as described below.
Western blot.
Total cell lysates, subcellular fractions (CF, MF, and ACF) (20 μg of protein), and immunoprecipitates as prepared above were boiled and then electrophoresed in 12% SDS-PAGE acrylamide gels, transferred onto nitrocellulose membranes (Bio-Rad), and incubated for 1 h in TBS-T (Tris-buffered saline and 0.1% Tween 20) containing 5% nonfat milk. Membranes were then incubated overnight at 4°C with primary antibodies against Cav-1 (Santa Cruz Biotechnology; 1:500), claudin-5 (Invitrogen; 1:1000), or occludin (Invitrogen; 1:500), washed in TBS-T, and incubated for 1 h at room temperature with corresponding HRP-conjugated anti-rabbit or anti-mouse antibodies (Santa Cruz Biotechnology; 1:1000). The membranes were developed with the SuperSignal West Pico HRP substrate kit (Pierce) and photographed on a Kodak 4000 image station (Carestream Molecular Imaging). To control sample loading and protein transfer, the membranes were stripped and reprobed with β-actin antibody (Santa Cruz Biotechnology; 1:1000). For subcellular fraction samples, we noticed that the actin levels were comparable for each sample among different fractions, so we only used one normalizing loading control (CF actin) for all three subcellular fractions.
Real-time RT-PCR.
Total cellular RNA was isolated using Trizol reagents (Invitrogen) according to manufacturer's protocol. The 0.5 μg of RNA was reverse transcribed (RT) with random primers in a 20 μl final reaction volume using TaqMan Reverse Transcription Kits (Applied Biosystems). The 0.5 μl RT products were amplified with the 7900HT Fast Real-Time PCR System (Applied Biosystems) in a 10 μl final reaction volume using SYBR Green PCR Master Mix (Applied Biosystems) under the following conditions: 2 min at 50°C and 10 min at 95°C, followed by a total of 40 cycles of two temperature cycles (15 s at 95°C and 1 min at 60°C). Primers (Integrated DNA Technologies) for MMP-2, MMP-9, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were designed against known mouse sequences: MMP-2 (NM_008610), forward, 5′-TAACCTGGATGCCGTCGT-3′; reverse, 5′-TTCAGGTAATAAGCACCCTTG-3′; MMP-9 (NM_013599), forward, 5′-ACGACATAGACGGCATCCA-3′; reverse, 5′-GCTGTGGTTCAGTTGTGGTG-3′; GAPDH (NM_008084), forward, 5′-CAATGTGTCCGTCGTGGATCT-3′; reverse, 5′-GTCCTCAGTGTAGCCCAAGATG-3′. The fluorescence threshold value (Ct value) was calculated using the SDS Enterprise Database software (Applied Biosystems). The relative value of mRNA expression was calculated by the comparative ΔΔCt method described in our previous publication (Liu et al., 2007). In brief, mean Ct values were normalized to the internal control GAPDH and the difference was defined as ΔCt. The difference between the mean ΔCt values of treated and untreated cells was calculated and defined as ΔΔCt. The comparative mRNA expression level was expressed as 2−ΔΔCt.
Immunostaining of occludin and claudin-5 in OGD-treated bEND3 cells.
The bEND3 cells grown to confluence on collagen-coated coverslips were subjected to the indicated treatments. For immunostaining, cells were washed three times with PBS, fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100 for 5 min, and then blocked for 1 h at room temperature with 3% BSA plus 0.1% Tween 20 plus 5% goat serum. The cells were then incubated with anti-occludin or anti-claudin-5 primary antibodies (1:100 dilution; Invitrogen) overnight at 4°C, followed by incubation with FITC- or Cy3-conjugated anti-mouse or anti-rabbit second antibodies (1:200 dilution) for 60 min. After washing in PBS, the coverslips were mounted on glass slides with antifade solution Vectashield (Vector Laboratories). Images were acquired using an LSM 510 confocal laser-scanning microscope (Zeiss).
Rat model of middle cerebral artery occlusion.
The Laboratory Animal Care and Use Committee of the University of New Mexico approved all experimental protocols. Male Sprague Dawley rats (Charles River Laboratories) weighing 290–320 g were anesthetized with isoflurane (4% for induction, 1.75% for maintenance) in N2O/O2 (70:30%) during surgical procedures and the body temperature was maintained at 37.5 ± 0.5°C using a heating pad. The rats were subjected to 2 h of reversible MCAO using the intraluminal filament technique as previously described (Liu et al., 2009a). Briefly, a 4-0 silicone-coated monofilament nylon suture was inserted into the internal carotid artery and advanced along the internal carotid artery to ∼17–18 mm from the bifurcation, thereby blocking the ostium of the MCA. Because we aimed to investigate ischemia-induced early changes in MMP-2/9 and BBB permeability, rats were not reperfused when collecting microdialysis samples for MMP-2/9 measurement or reperfused for only 10 min when assessing BBB damage (as described below). For a total number of 38 rats included in this study, successful MCAO was confirmed by 2,3,5-triphenyltetrazolium chloride (TTC) staining of the 1-mm-thick brain coronal section 6 mm away from the tip of the front lobe as we described previously (Liu et al., 2008).
Evaluation of ischemia-induced BBB damage.
To visualize ischemia-induced BBB damage, all rats received femoral vein injections of 30 mg/kg body weight FITC-albumin (25 mg/ml in sterile PBS) at the end of 2 h MCAO. Then the rats were reperfused for 10 min to ensure sufficient circulation of fluorescent tracer to the ischemic brain and also minimize the effect of reperfusion on BBB permeability. At the end of reperfusion, rats were transcardially perfused with 250 ml of cold PBS to remove intravascular FITC-albumin. The brain was then removed and a 5-mm-thick brain region 7 mm away from the tip of the front lobe was rapidly frozen in methylbutanol prechilled in a −80°C freezer. Twenty-micrometer-thick cryosections were cut with a Leica cryostat (Leica Microsystems) and mounted for fluorescence microscopic observation at an excitation wavelength of 485 nm and a 528 nm filter for collecting fluorescence emission (Olympus IX-81; Olympus). The whole brain coronal section was automatically photographed (10× objective lens) using the Stereo Investigator software to visualize FITC-albumin leakage (BBB disruption), which appeared as green fluorescence on brain sections.
Evaluation of MMP-2/9, occludin, and claudin-5 changes in ischemic tissue with BBB damage.
In situ zymography and immunohistochemistry (IHC) were performed to analyze the gelatinolytic activities of MMP-2/9 and occludin/claudin-5 protein levels in ischemic tissue with compromised BBB, respectively. In these experiments, FITC-albumin was replaced with Texas Red-conjugated albumin (Texas Red-albumin; Sigma-Aldrich) to label ischemic tissue with BBB damage because FITC-albumin may interfere with in situ zymography analysis in which FITC-labeled DQ-gelatin is used as a substrate. At the end of 2 h MCAO and 10 min reperfusion, rats were transcardially perfused, and the same 5-mm-thick brain region was cut out and frozen in prechilled 2-methylbutane, as described above. Ten-micrometer-thick and 20-μm-thick cryosections were prepared for IHC or in situ zymography analysis, respectively, as described below.
The 10-μm-thick cryosections were fixed with 4% PFA for IHC analysis for occludin and claudin-5, as we described previously (Y. Yang et al., 2007; Liu et al., 2009a). In brief, nonspecific binding sites were blocked by preincubating tissue for 1 h at room temperature in PBS containing 0.1% Triton X-100, 1% BSA, and 5% goat serum. Sections were then incubated overnight with Alexa Fluor 488-conjugated claudin-5 antibody (1:100 dilution; Invitrogen) or anti-FITC-conjugated occludin antibody (1:100 dilution; Invitrogen) at 4°C. Immunostaining was visualized under LSM 510 confocal laser-scanning microscope (Zeiss), and images were taken from the ischemic region with Texas Red-albumin leakage and the mirrored regions on the nonischemic hemisphere.
The 20-μm-thick cryosections were used for analyzing gelatinolytic activities of MMP-2/9 by in situ zymography as we described previously (Liu et al., 2009a). In brief, the section was incubated for 2 h at 37°C in a reaction buffer containing 40 μg/ml FITC-labeled DQ-gelatin (Invitrogen). FITC-gelatin is cleaved by gelatinases, yielding peptides whose fluorescence is representative of the net gelatinolytic activity. At the end of incubation, sections were rinsed with PBS and mounted in Gel/Mount (Biomeda) for fluorescence microscopy (Olympus). Images were taken from the ischemic region with Texas Red-albumin extravasation and the mirrored region on the nonischemic hemisphere.
Isolation of cerebral microvessels.
Isolation of cerebral microvessels was performed as we described previously (Liu et al., 2009a). In brief, the hemispheric brain tissue was dissected and homogenized in ice-cold PBS. The homogenate was filtered through a 41 μm nylon mesh (Spectrum), and the nylon mesh was washed three times with PBS. Microvessels retained on the mesh were then washed off and pelleted by centrifugation at 4000 × g for 10 min at 4°C. The pellets were resuspended in 15% dextran T-500 and then added onto 20% dextran T-500, followed by centrifugation at 25,000 × g for 10 min at 4°C. The pellets were collected as the cerebral microvessels and stored at −80°C until further analysis. To determine the redistribution of claudin-5, we pooled the hemispheric microvessels isolated from three rats to get enough protein for preparing the subcellular fractions with ProteoExtract Subcellular Proteome Extraction Kit (Calbiochem).
In vivo microdialysis sampling.
Microdialysis sampling is a well established diffusion-based sample collection method for collecting molecules including MMPs from the interstitial space in a specific tissue or organ in situ (Liu et al., 2003; Nilsson and Dabrosin, 2006; Wang et al., 2009). The conventional MMP extracting method (tissue homogenization) is not able to separate intracellular MMPs from extracellular MMPs produced in the ischemic brain; therefore, we applied the in vivo microdialysis sampling to collect MMPs in the interstitial space of the ischemic brain tissue (see Fig. 7B). Before conducting MCAO surgery, two microdialysis guide cannulas were surgically placed separately into left and right striatum using the coordinates of 0.3 mm posterior to bregma, 4 mm lateral to the midline, and 7 mm below the surface of the brain according to a previous study in our laboratory (Liu et al., 2003). Then the guide cannulas were removed and rats were subjected to MACO surgery. Right after ischemia onset, rats were placed back to the stereotactic device for microdialysis probe implantation. Under isoflurane anesthesia, two identical PES probes (CMA 12; 4 mm; 100 kDa cutoff membrane) (CMA/Microdialysis), which were pre-perfused with 70% ethanol and then sterile water, were inserted into the left and right striatum along the guide cannulas. Then, the inlet and outlet tubing were connected to a CMA 102 microdialysis dual-channel pump (CMA/Microdialysis). Rats were under anesthesia during sample collection. Both probes were perfused with sterile artificial CSF with the following sequence: 5 μl/min for 10 min, 2 μl/min for 10 min (these perfusates were discarded), and then 2 μl/min for ∼90 min until the end of 2 h MCAO. The perfusate collected in the last 90 min of perfusion was used for measuring MMP-2/9 with zymography. The 5 μl/min flush for the first 10 min was performed ensure that fluid lines were open after the implantation of the probe. Additionally, as microdialysis sampling is an invasive procedure, any solutes released due to the surgery or probe insertion into the tissue will be flushed out with a higher flux as mass removal during microdialysis sampling is directly correlated with perfusion flow rate. After completion of the dialysate collection, rats were killed and the brains were removed and sectioned at 1 mm intervals. The probe placement was confirmed by visual observation.
In addition, to determine whether MMP-2/9 induction contributed to MMP-2/9 changes in the interstitial space of the ischemic striatum, we performed another set of experiments to assess MMP-2/9 mRNA expression in the striatal tissue after 2 h MCAO. Striatal tissue was dissected, and total RNA was isolated using Trizol reagents and subjected to real-time RT-PCR analysis as described above. Primers for rat MMP-2, MMP-9, and housekeeping gene rpl 32 were the same as described in our previous study (Liu et al., 2007): MMP-2 (U65656), forward, 5′-GATCTGCAAGCAAGACATTGTCTT-3′; reverse, 5′-GCCAAATAAACCGATCCTTGAA-3′; MMP-9 (U24441), forward, 5′-GTAACCCTGGTCACCGGACTT-3′; reverse, 5′-ATACGTTCCCGGCTGATCAG-3′; rpl 32 (NM_013226), forward, 5′-AGACCTGAATGTGAAGGAAG-3′; reverse, 5′-CCTTGGGATTGGTGACTCTGA-3′.
Statistical analysis.
All data were presented as means ± SE. Differences between groups were assessed by Student's t test or ANOVA followed by Tukey's post hoc test as indicated in the figure legends. A value of p ≤ 0.05 was considered statistically significant.
Results
Ischemia rapidly disrupts the endothelial barrier in vitro and in vivo
In view of the close association between ischemia-induced initial BBB damage and the hemorrhagic complications of acute stroke thrombolysis (Hjort et al., 2008; Kastrup et al., 2008; Kassner et al., 2009; Sun et al., 2010), we sought to understand how cerebral ischemia affects BBB integrity within the established 3 h thrombolytic time window in vitro and in vivo. Brain endothelial monolayers challenged with OGD are frequently used as an in vitro model of ischemic BBB damage (Benchenane et al., 2005; Zhu et al., 2010). We exposed confluent brain microvascular endothelial cells (bEND3) grown on 24-well cell culture inserts to 2 h OGD without reoxygenation. The barrier integrity of the endothelial monolayer in control condition or after 2 h OGD treatment was assessed by measuring the transfer rate (Papp) of FITC-dextran from the luminal compartment to the abluminal compartment (Fig. 1A, top panel). OGD treatment significantly sped up the passage of FITC-dextran across the endothelial monolayer, indicating disruption of its barrier function (Fig. 1A, bottom panel). To determine whether cytotoxicity contributed to OGD-induced permeability increase of the endothelial monolayer, we also evaluated cytotoxicity with a LDH assay kit and found that OGD under our experimental conditions did not increase LDH release into the cell culture supernatant (data not shown), indicating that OGD-induced endothelial monolayer barrier dysfunction was not due to a direct toxic effect on endothelial cells.

Ischemia causes rapid disruption of the BBB in vitro and in vivo. A, Top panel, Schematic representation of the in vitro BBB model (bEND3 monolayer grown on an insert) with FITC-dextran loaded in the luminal compartment. Bottom panel, The endothelial monolayer permeability was assessed by calculating the transfer rate of FITC-dextran from luminal compartment to abluminal compartment and was expressed as apparent permeability coefficient (Papp) (in centimeters per second). Exposure of bEND3 monolayer to OGD for 2 h significantly increased its permeability to FITC-dextran. *p < 0.05 versus control cultures, Student's t test; n = 6. B, Cerebral ischemia rapidly induced BBB disruption in the ischemic brain of all tested rats (n = 6). Representative fluorescence micrographs of brain cryosections from four different rats revealed that 2 h MCAO induced FITC-albumin extravasation (bright green fluorescence) in the subcortical regions in the ischemic hemisphere. No FITC-albumin leakage was observed in other brain regions. Error bars indicate SEM.
We also examined the effect of cerebral ischemia on BBB integrity on an in vivo rat model of 2 h MCAO. Since postischemic reperfusion can exacerbate BBB damage (Simard et al., 2007), we reperfused the MCAO rats for a brief 10 min duration to allow sufficient circulation of FITC-albumin to the ischemic brain region, while minimizing the impact of reperfusion on the BBB. As seen on the fluorescent micrographs of representative brain cryosections (Fig. 1B), 2 h MCAO consistently induced FITC-albumin extravasation in the ischemic hemisphere, and the leakages were invariably seen in the subcortical regions. These in vitro and in vivo data clearly indicate that ischemia causes BBB disruption at an early stroke stage (within 2 h after ischemia onset) relevant to acute stroke thrombolysis.
OGD triggers occludin loss and claudin-5 redistribution in endothelial cells
Altered distribution of TJ proteins, particularly claudins and occludin, or their loss are frequently seen in the compromised BBB following cerebral ischemia and reperfusion (Y. Yang et al., 2007; McColl et al., 2008; Liu et al., 2009a). To determine whether the rapid disruption of endothelial barrier function observed above was due to a quick disturbance on TJ proteins by ischemia, we investigated the effect of 2 h OGD on two important transmembrane TJ proteins, claudin-5 and occludin (Fig. 2A–D). Total cellular extracts of control bEND3 cells exhibited readily detectable protein bands of occludin and claudin-5 on the immunoblots. Occludin exhibited as a doublet of 60 and 65 kDa on Western blot, which represents two different isoforms of occludin monomers (McCaffrey et al., 2007). Following 2 h OGD insult, bEND3 cells manifested a dramatic reduction in the total protein level of occludin (Fig. 2A). Surprisingly, OGD treatment did not change the total protein levels of claudin-5 (Fig. 2B). In addition to total protein levels, redistribution of TJ proteins, particularly their dissociation from the cytoskeletal framework, also contributes to BBB compromise (Song et al., 2007). To monitor the redistribution of occludin and claudin-5 in OGD-treated cells, we prepared subcellular fractions to detect the translocation of occludin and claudin-5 between subcellular compartments. As shown in Figure 2C, OGD treatment led to a significant increase in claudin-5 levels in the detergent soluble fractions including the CF and MF and a concurrent decrease in its level in the detergent-resistant ACF, indicating a dissociation of claudin-5 from the cytoskeletal framework under OGD condition. Unlike claudin-5, occludin was uniformly decreased in all tested subcellular fractions but did not exhibit any redistribution changes after OGD insult (data not shown). The characterization of the subcellular fractions was verified by the presence of specific marker protein in each fraction (Fig. 2D). These data suggest that OGD triggers rapid loss of occludin protein and rapid dissociation of claudin-5 from the cytoskeleton framework.

OGD induces rapid degradation of occludin and redistribution of claudin-5 in bEND3 cells. After exposing bEND3 cells to OGD for 2 h, total cell lysates and extracts of subcellular fractions were prepared and subjected to Western blot analysis for tight junction proteins occludin and claudin-5. As a loading control, the blots were stripped and reprobed with β-actin antibody. A, B, Two hour OGD induced a significant reduction in total occludin protein level but did not change the total protein level of claudin-5. C, OGD induced redistribution of claudin-5 between subcellular compartments, as reflected by a remarkable reduction in claudin-5 levels in the ACF and a significant increase of its levels in the CF and MF. The blots for CF were stripped and reprobed with β-actin antibody. D, The specificity of each fraction was confirmed with anti-calpain (CF), anti-Cypor (MF), and anti-vimentin (ACF) antibodies. *p < 0.05 versus control, Student's t test; n = 4. Error bars indicate SEM.
MMP-2/9 contribute to occludin loss, but not claudin-5 redistribution in OGD-treated endothelial cells
We next examined the mechanisms underlying the rapid changes of TJ proteins under OGD condition. MMP-2/9 have been shown to proteolytically disrupt TJ proteins under various pathological conditions including ischemic stroke (Y. Yang et al., 2007; McColl et al., 2008; Liu et al., 2009a; Lischper et al., 2010). Therefore, we asked whether MMP-2/9 were involved in OGD-induced disturbance in TJ proteins. To test this possibility, we first examined whether OGD could affect MMP-2/9 levels in bEND3 cells in an ischemic duration as short as 2 h. Since MMP-2/9 are synthesized intracellularly and secreted into the extracellular space to digest matrix components (Taraboletti et al., 2006), we assessed MMP-2/9 levels in the CM using gel gelatin zymography, the most widely used method for detecting MMP-2/9, which reveals proteolytic bands on zymogram gels derived from both pro- and active forms of MMP-2/9 (Min et al., 2006). Under control conditions, latent MMP-9 and both latent (top band) and active (bottom band) forms of MMP-2 were detected in the CM of bEND3 cells, according to standard MMP-2/9 bands (Fig. 3A). Following 2 h OGD treatment, MMP-2/9 levels were significantly increased in the CM, while their activation patterns did not change (Fig. 3A). For this reason, we did not separately label the latent and active MMPs on the zymogram gels, but rather called them together as MMP-2 or MMP-9.

OGD-induced occludin degradation is MMP-2/9 dependent. A, OGD rapidly elevated MMP-2/9 levels in conditioned media. After exposure of bEND3 cells to OGD for 2 h, a significant increase in MMP-2/9 levels was detected in the conditioned medium on gelatin zymograms when compared with the control cultures (Ctrl). Active MMP-2 (the bottom band), but no active MMP-9, was seen on zymogram gels. MMP-2 or -9 levels were quantified by measuring the sum intensity of their latent and active bands. *p < 0.05 versus Ctrl, Student's t test; n = 5. Std, Standard human MMP-2/9. B, Selective MMP-2/9 inhibitor SB-3CT completely inhibited OGD-induced occludin degradation. bEND3 cells were treated with SB-3CT (10 μm) 2 h before and during 2 h OGD. Occludin protein in total cellular extracts was detected with Western blot. β-Actin served as a loading control. *p < 0.05 versus vehicle (DMSO) plus Ctrl, #p < 0.05 versus vehicle plus OGD, ANOVA; n = 5. C, MMP-2 neutralizing antibody (M2) completely inhibited OGD-induced occludin degradation, while no significant effects were observed for IgG or MMP-9 neutralizing antibody (M9). bEND3 cells were treated with 20 μg/ml control mouse IgG, MMP-2 or MMP-9 neutralizing antibodies, or both (M2,9) during 2 h OGD. Occludin protein in total cellular extracts was detected with Western blot. β-Actin served as a loading control. *p < 0.05 versus control (Ctrl), #p < 0.05 versus OGD alone (−) or OGD plus IgG, ANOVA; n = 4. D, Representative confocal micrographs showed MMP-dependent degradation of occludin. Control bEND3 cells revealed a circumcellular immunostaining of occludin, which was significantly reduced after exposing cells to OGD for 2 h. SB-3CT treatment completely inhibited occludin reduction in OGD-treated cells. Experiments were repeated three times with similar results. Scale bar, 20 μm. E, Inhibition of MMP-2/9 with SB-3CT had no effect on OGD-induced claudin-5 redistribution. Claudin-5 proteins in subcellular fractions were detected with Western blot. *p < 0.05 versus vehicle plus Ctrl, ANOVA; n = 5. Error bars indicate SEM.
Next, we investigated whether OGD-induced alteration in occludin and claudin-5 was dependent on the proteolytic activity of MMP-2/9. Using the highly selective, mechanism-based potent MMP-2/9 inhibitor SB-3CT (Brown et al., 2000), we found that inhibition of MMP-2/9 completely abolished OGD-induced occludin reduction in bEND3 cells (Fig. 3B). Worthy of note, SB-3CT-treated control bEND3 cells also exhibited a slight increase (not statistically significant) in occludin level compared with vehicle-treated cells (DMSO; final concentration of 1/1000 in volume ratio), suggesting that MMP-2/9 may play a role in maintaining the normal turnover of occludin under control conditions. To further distinguish between the roles of MMP-2 and MMP-9 in mediating OGD-induced occludin degradation, we selectively immunodepleted MMP-2, MMP-9, or both using neutralizing antibodies. The blocking of MMP-2, but not MMP-9, by neutralizing antibody significantly inhibited OGD-induced occludin degradation compared with control IgG (Fig. 3C). To test effectiveness of MMP-9 neutralizing antibody, we incubated bEND3 extracts with active MMP-9 (5 μg/ml; R&D Systems) in the presence or absence of 20 μg/ml MMP-9 neutralizing antibody and found that the presence of MMP-9 antibody significantly inhibited active MMP-9-induced occludin degradation (data not shown). These results indicate that MMP-2 is the major gelatinase contributing to occludin degradation after 2 h OGD, which is in agreement with our observation that the active form of MMP-2, but not active MMP-9, was detected in OGD-treated bEND3 cells (Fig. 3A).
To further confirm a gelatinase-dependent degradation of occludin under OGD conditions, we performed immunofluorescence staining to visualize the changes of occludin in OGD-treated cells in the presence of SB-3CT. Confocal microscopic images revealed a circumcellular staining for occludin protein in control bEND3 cells, regardless of whether they were treated with vehicle or SB-3CT (Fig. 3D, left panels). Consistent with the Western blot results, exposure of cells to OGD for 2 h significantly reduced the immunostaining of occludin, and this reduction was completely inhibited when cells were pretreated with SB-3CT (Fig. 3D, right panels). It is worthy of note that SB-3CT-treated control cells appeared to exhibit a slight stronger immunostaining (brighter fluorescence) for occludin than vehicle-treated control cells (Fig. 3D, left panels). In contrast to occludin, OGD-induced changes in claudin-5 were not dependent on MMP-2/9 because SB-3CT had no effect on its redistribution between subcellular fractions (Fig. 3E). In summary, these data indicate that OGD rapidly elevates extracellular MMP-2/9 levels and that the active MMP-2 is the key effector molecule responsible for the rapid loss of occludin protein, but not for claudin-5 protein redistribution, in endothelial cells under ischemic condition.
OGD stimulates MMP-2/9 secretion, but not their expression, in endothelial cells
We next asked how OGD led to a rapid increase (within 2 h) in extracellular MMP-2/9 on bEND3 cultures. The extracellular MMP-2/9 levels are largely dependent on their synthesis and secretion. Therefore, we assessed the effect of OGD on intracellular MMP-2/9 levels and their mRNA expression under same experimental condition in an attempt to dissect these two pathways (promoting synthesis or secretion) that could potentially contribute to the observed rapid increase in secreted MMP-2/9 in OGD-treated bEND3 cells. After 2 h OGD treatment, the MMP-2/9 levels in the CM and the whole CEs were analyzed with gel gelatin zymography. As shown in Figure 4A, OGD-induced MMP-2/9 increase in the CM was accompanied by a remarkable decrease in their levels in the CEs, indicating increased MMP-2/9 secretion under OGD condition. To rule out the possibility that OGD could rapidly stimulate MMP-2/9 synthesis, which in turn contributed to their increase in the CM, we measured MMP-2/9 mRNA expression using real-time RT-PCR. Indeed, 2 h OGD did not affect MMP-2/9 mRNA expression (Fig. 4B), further supporting a secreting mechanism. Further supporting a secreting mechanism was our findings that incubation of bEND3 cells with mRNA synthesis inhibitor actinomycin D (Act D) at 2 μg/ml or protein synthesis inhibitor cycloheximide (CHX) at 100 μg/ml 1 h before and during 2 h OGD treatment did not significantly affect MMP-2/9 increase in the CM in OGD-treated cells (Fig. 4C,D). These results indicate that OGD rapidly elevates extracellular MMP-2/9 levels through promoting their secretion from the preexisting intracellular MMP-2/9 pool, rather than stimulating their de novo synthesis. They also indicate that new protein synthesis is not required to mediate OGD-induced MMP-2/9 secretion.

OGD elevates extracellular MMP-2/9 levels through promoting their secretion from the preexisting intracellular pool in bEND3 cells. A, Gelatin zymography analysis showed that 2 h OGD markedly increased MMP-2/9 levels in the CM, which was accompanied by a significant reduction in their levels in whole CEs. *p < 0.05 versus control, Student's t test; n = 5. Std, Human MMP-2/9 standards. B, Real-time RT-PCR analysis showed that 2 h OGD did not change MMP-2/9 mRNA expression in bEND3 cells. n = 5. C, D, Inhibition of mRNA synthesis with Act D or inhibition of protein synthesis with CHX did not significantly affect OGD-induced MMP-2/9 secretion in bEND3 cells. Cells were treated with Act D (2 μg/ml) or CHX (100 μg/ml) or vehicle (DMSO) 1 h before and during 2 h OGD treatment. *p < 0.05 versus control (Ctrl), ANOVA; n = 5. E, Two hour OGD also stimulates MMP-2/9 secretion from astrocytic cell line C8-D1A (left panel) and neuronal cell line SH-SY5Y (right panel), reflected by increased MMP-2/9 in CM and concurrent reduction of their levels in CEs. Experiments are repeated four times with similar results. Error bars indicate SEM.
In addition to brain microvascular endothelial cells, astrocytes and neurons, the two most numerous neural cell types, are also important cellular sources of MMP-2/9 in the brain (Yong et al., 1998; Liu et al., 2007). Therefore, it would be interesting to know whether OGD treatment could trigger rapid MMP-2/9 secretion in these two cell types. To test this, astrocytic cell line C8-D1A and neuronal cell line SH-SY5Y were exposed to 2 h OGD, and MMP-2/9 levels in the CM and CE were analyzed with gel gelatin zymography. Similar to what we observed for bEND3 cells, OGD induced rapid MMP-2/9 secretion in both cell types, reflected by increased levels of MMP-2/9 in the CM and a concomitant reduction of their levels in the CE (Fig. 4E).
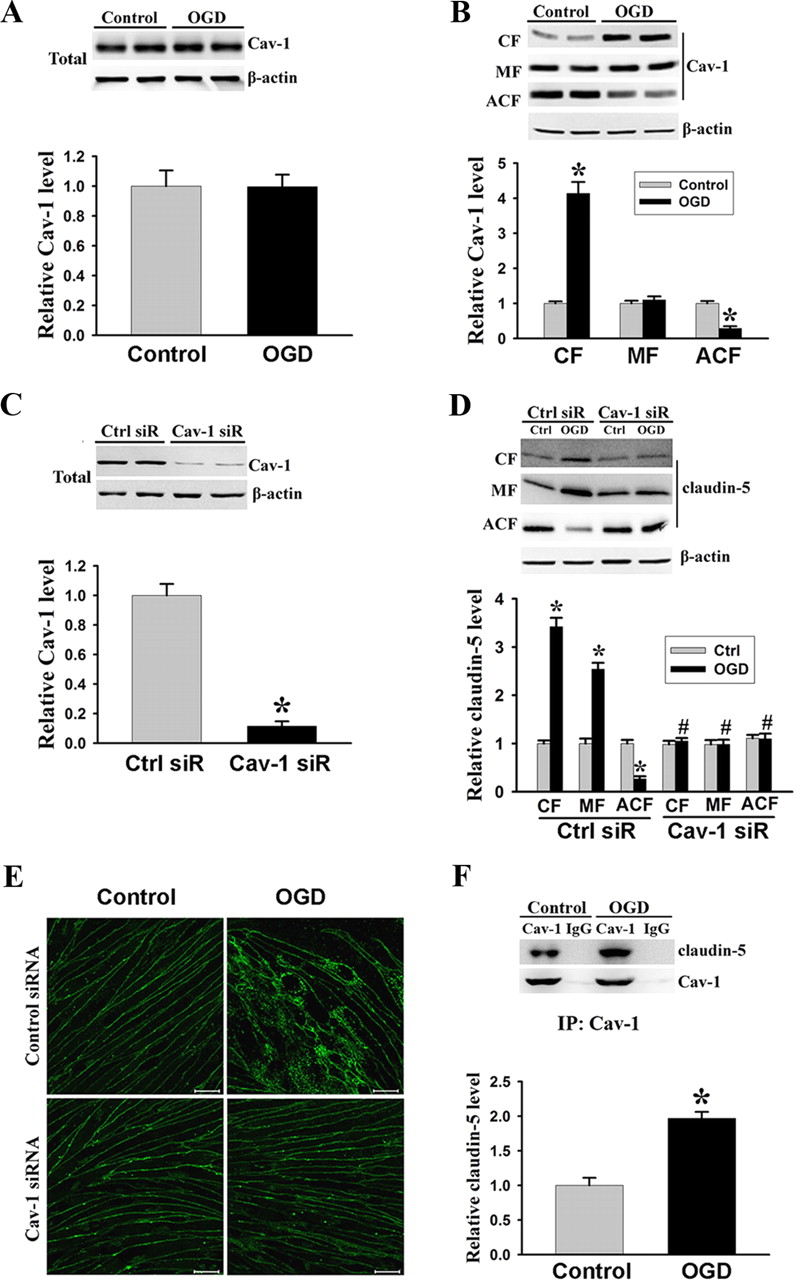
Cav-1 mediates OGD-induced redistribution of claudin-5 in endothelial cells
Cav-1 has been shown to play an important role in endocytosis, vesicular trafficking, and signal transduction (Lisanti et al., 1994; Okamoto et al., 1998; Smart et al., 1999), and more recently, it was found to be implicated in inflammatory BBB injury via mediating the internalization of TJ proteins (Nag et al., 2007; Stamatovic et al., 2009). Therefore, we speculated that Cav-1 might mediate OGD-induced dissociation of claudin-5 from the cytoskeletal framework. To investigate this possibility, we studied the interaction between these two proteins. First, we examined the changes of Cav-1 protein in the total cellular extracts as well as in subcellular fractions following 2 h OGD treatment. Western blot analysis showed that OGD did not affect Cav-1 protein levels in total cellular extracts (Fig. 5A) as well as in the MF (Fig. 5B), but significantly stimulated its translocation from the cytoskeleton to the cytosol, as reflected by increased Cav-1 protein level in the CF and its decrease in the ACF (Fig. 5B). Interestingly, these changes of Cav-1 were similar to what we saw above for claudin-5 under the same OGD condition (Fig. 2B,C). To further determine whether there was an association between these two events triggered by OGD, we next applied siRNA approach to knock down Cav-1 protein expression before exposing bEND3 cells to OGD treatment. The ability of Cav-1 siRNA to specifically knock down Cav-1 protein was verified in Figure 5C, in which a ∼90% reduction in Cav-1 protein level was seen on immunoblots at 48 h after Cav-1 siRNA transfection. We then examined the effects of Cav-1 siRNA on OGD-induced claudin-5 redistribution using Western blot and immunofluorescence staining. Notably, in bEND3 cells with Cav-1 knocked down by siRNA, OGD could no longer trigger the redistribution of claudin-5 among subcellular fractions (i.e., claudin-5 increase in CF/MF and its decrease in ACF) (Fig. 5D). Additionally, immunocytochemistry analysis further confirmed these immunoblotting results (Fig. 5E). Confocal microscopic images revealed a circumcellular claudin-5 staining in control bEND3 cells, regardless of whether they were treated with control siRNA or Cav-1 siRNA (Fig. 5E, left panels), which was similar to the staining pattern for occludin observed above. OGD treatment interrupted the circumcellular staining pattern for claudin-5, with increased claudin-5 staining in the cytosol (Fig. 5E, right top panel). Knockdown of Cav-1 completely prevented the disturbance on claudin-5 distribution induced by OGD (Fig. 5E, right bottom panel).

Cav-1 mediates OGD-induced claudin-5 dissociation from the cytoskeleton in endothelial cells. A, B, Western blot analysis showed that exposing bEND3 cells to OGD for 2 h had no effects on the total protein levels of Cav-1, but triggered its redistribution among subcellular fractions, as reflected by increased Cav-1 level in the CF and decreased level in the ACF. *p < 0.05 versus control, Student's t test; n = 5. C, Cav-1 siRNA effectively knocked down Cav-1 protein expression in bEND3 cells. Western blot analysis showed that incubation cells with Cav-1 siRNA (Cav-1 siR) for 48 h significantly (∼90%) reduced Cav-1 protein levels. *p < 0.05 versus control siRNA (Ctrl siR), Student's t test; n = 5. D, Knockdown of Cav-1 with siRNA prevented OGD-induced claudin-5 redistribution in bEND3 cells. Following OGD treatment, claudin-5 levels in CF, MF were increased, while its level in ACF was markedly reduced, which was inhibited by Cav-1 siRNA. *p < 0.05 versus control (Ctrl); #p < 0.05 versus Ctrl siR plus OGD, ANOVA; n = 5. E, Representative confocal microscope images revealed a circumcellular immunostaining of claudin-5 in control bEND3 cells. Two hour OGD treatment did not change the immunostaining intensity of claudin-5, but significantly disturbed its normal distribution pattern. Knockdown of Cav-1 with siRNA inhibited claudin-5 redistribution induced by OGD. Experiments were repeated three times with similar results. Scale bar, 20 μm. F, Representative immunoblot of coimmunoprecipitation of Cav-1 and claudin-5 from whole-cell lysates of control cultures or OGD-treated cells with anti-Cav-1 antibody or normal IgG (top panel). Bottom panel, OGD enhanced interaction of Cav-1 with claudin-5. *p < 0.05 versus control, Student's t test; n = 4. Error bars indicate SEM.
To investigate more directly the interaction of Cav-1 with claudin-5, we performed coimmunoprecipitation experiments. We found that claudin-5 coimmunoprecipitated with Cav-1 and that OGD enhanced the interaction of Cav-1 with claudin-5 by approximately onefold above the control (Fig. 5F). Collectively, these data demonstrate that Cav-1 directly interacts with claudin-5, and this interaction is crucial for mediating claudin-5 dissociation from the cytoskeleton framework in OGD-treated endothelial cells.
Knockdown of Cav-1 or inhibiting MMP-2/9 reduces OGD-induced disruption of the endothelial barrier
In view of the critical role of claudin-5 and occludin in maintaining normal endothelial barrier function, we next examined whether MMP-mediated occludin degradation and Cav-1-dependent redistribution of claudin-5 were responsible for OGD-induced endothelial barrier disruption (Fig. 1A). We pretreated bEND3 cells with specific MMP-2/9 inhibitor SB-3CT or Cav-1 siRNA before exposing the cells to 2 h OGD. As expected, scrambled control siRNA or vehicle (DMSO; final concentration of 1/1000 in volume ratio) did not affect endothelial monolayer permeability to FITC-dextran for both control cultures and OGD-treated cells when compared with the results in Figure 1A. Notably, SB-3CT or Cav-1 siRNA alone significantly reduced the permeability of OGD-treated endothelial monolayer to FITC-dextran, and their combination treatment completely prevented endothelial barrier compromise (Fig. 6). These results indicate that OGD-induced early changes in tight junction proteins occludin and claudin-5 are responsible for rapid loss of endothelial barrier function under ischemic condition.

Inhibition of MMP-2/9 with SB-3CT or knockdown of Cav-1 with siRNA reduces OGD-induced BBB disruption in vitro. The permeability of FITC-dextran across bEND3 monolayers was significantly increased after 2 h exposure to OGD, which was partially inhibited by pretreating cells with MMP-2/9 inhibitor SB3-CT or Cav-1 siRNA. Combination of SB-3CT and Cav-1 siRNA completely preserved the endothelial barrier integrity of OGD-treated endothelial monolayer. The endothelial monolayer permeability was assessed by calculating the transfer rate of FITC-dextran from luminal compartment to abluminal compartment, and was expressed as apparent permeability coefficient (Papp) (in centimeters per second). *p < 0.05 versus control; #p < 0.05 versus vehicle-OGD cultures; φp < 0.05 versus control siRNA (Ctrl siR) plus OGD; &p < 0.05 versus Cav-1 siRNA (Cav-1 siR) plus OGD or SB-3CT plus OGD, ANOVA; n = 6. Error bars indicate SEM.
Two hour MCAO induces MMP-2/9 increase in tissue interstitial space, occludin loss, and claudin-5 redistribution in ischemic cerebromicrovessels
To validate our findings in vitro, we next examined the effects of 2 h MCAO on MMP-2/9, TJ protein occludin, and claudin-5 in ischemic brain tissue. First, we performed in situ zymography on cryosections obtained from brain tissue injected with Texas Red-albumin to examine the gelatinolytic activity of MMP-2/9. As shown in Figure 7A, increased gelatinolytic activity (bright green fluorescence) was only observed in ischemic striatal tissue that showed concurrent extravasation of Texas Red-albumin. No tracer leakage and weak gelatinolytic activity were observed in nonischemic striatal tissue. To demonstrate a MMP-secreting mechanism in vivo, we assessed extracellular MMP-2/9 levels in the ischemic brain tissue using in vivo microdialysis sampling. We inserted the microdialysis probes into the striatal tissue because the striatum was referred to as an “ischemic core” region that exhibited very low residual cerebral blood flow after MCAO (Butcher et al., 1990) and higher susceptibility of its microvasculature to ischemic injury (Nagaraja et al., 2011), and our results showed BBB disruption in this region after 2 h MCAO (Fig. 1B). Figure 7B showed the schematic diagram for microdialysis sampling. Gel gelatin zymography analysis showed that 2 h MCAO induced a significant increase in MMP-2 in the interstitial space of the ischemic striatal tissue compared with its nonischemic counterpart. MMP-2 seemed to be the major form of the secreted gelatinases in the ischemic striatum because MMP-9 was barely seen on the zymogram gels (Fig. 7C). To determine whether 2 h MCAO upregulated MMP-2/9 expression, we extracted total RNA from the striatal tissue and analyzed MMP-2/9 mRNA expression. As shown in Figure 7D, 2 h MCAO did not change MMP-2/9 mRNA expression in the ischemic striatum. These in vivo data suggest that cerebral ischemia leads to a rapid accumulation of extracellular MMP-2 in the ischemic brain through promoting their secretion, rather than stimulating their synthesis in an early stroke stage relevant to acute stroke thrombolysis.

Cerebral ischemia rapidly increases extracellular MMP-2/9 levels and their activities in ischemic striatum. After 2 h MCAO, the gelatinolytic activity of MMP-2/9 and their extracellular levels were analyzed by in situ zymography and microdialysis sampling/gel zymography, respectively. A, In situ zymography was performed on cryosections obtained from brain tissue injected with Texas Red-albumin. Increased gelatinolytic activity of MMP-2/9 was found in the ischemic striatal tissue (bright green fluorescence), where Texas Red-albumin leakage concurrently occurred. No tracer leakage and weak gelatinolytic activity were seen in the corresponding nonischemic striatal tissue. Scale bar, 50 μm. Experiments were repeated four times with similar results. B, Schematic diagram of in vivo microdialysis sampling, by which MMP-2/9 in the interstitial space of the nonischemic (Non-I) and ischemic (I) striatum were collected during 2 h MCAO. C, Gel gelatin zymography analysis of collected dialysates showed that MMP-2/9, particularly MMP-2, were significantly increased in the interstitial space of the ischemic striatum. *p < 0.05 versus Non-I, Student's t test; n = 4. D, MMP-2/9 mRNA expression was not changed in the ischemic striatal tissue after 2 h MCAO (n = 6). Total RNA was extracted from nonischemic and ischemic striatal tissues and mRNA expression was analyzed by real-time RT-PCR. Error bars indicate SEM.
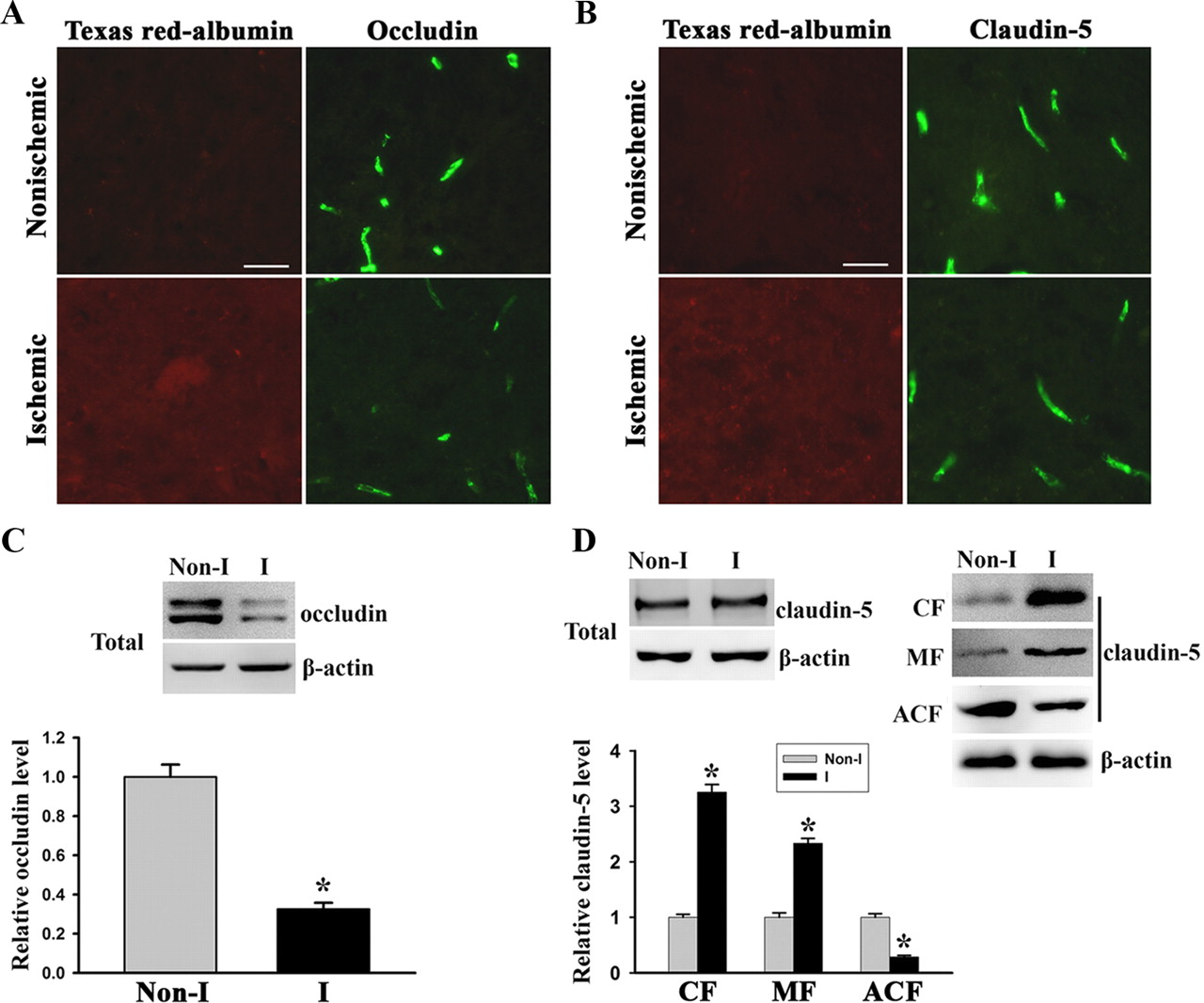
Next, IHC and immunoblotting were conducted to detect the changes of TJ protein claudin-5 and occludin in the ischemic brain after 2 h MCAO. We performed IHC on cryosections obtained from brain tissue injected with Texas Red-albumin and found that the immunostaining of occludin (Fig. 8A), but not claudin-5 (Fig. 8B), was significantly reduced in ischemic striatal tissue that showed Texas Red-albumin extravasation, when compared with nonischemic striatal tissue. However, we did not see subcellular redistribution of claudin-5 even under higher magnification (63× objective) (data not shown), which was observed in OGD-treated bEND3 cells (Fig. 5E). To confirm these IHC results, we performed Western blot to detect the changes of occludin and claudin-5 in cerebral microvessels isolated from ischemic brain tissue. Consistent with the IHC results, 2 h MCAO induced a significant reduction in occludin protein levels (Fig. 8C), while it did not change the total levels of claudin-5 protein (Fig. 8D, left top panel). Notably, through measuring claudin-5 contents in subcellular fractions of microvascular extracts, we found that 2 h MCAO induced increases in claudin-5 in the CF and MF, while it decreased its contents in the ACF (Fig. 8D, right top and bottom panels). These results indicate that cerebral ischemia leads to rapid occludin degradation and claudin-5 redistribution in vivo.

Cerebral ischemia induces rapid occludin degradation and claudin-5 redistribution in vivo. After 2 h MCAO, the protein levels of occludin and claudin-5 in cerebral microvessels were analyzed by IHC or Western blot. A, B, Immunostaining for occludin and claudin-5 was performed on cryosections obtained from brain tissue injected with Texas Red-albumin. Immunostaining (green) for occludin and claudin-5 were clearly seen on the microvessels of the nonischemic tissue, where no Texas Red-albumin leakage was observed. In the ischemic hemisphere, tracer leakage was accompanied by reduced occludin staining on the microvessels, while no appreciable changes were observed for claudin-5 staining. Scale bar, 25 μm. Experiments were repeated four times with similar results. C, D, Cerebral microvessels were isolated from nonischemic (Non-I) and ischemic (I) hemispheric tissue after 2 h MCAO. Total microvascular extracts and subcellular fractions were prepared for analyzing occludin and claudin-5 protein levels with Western blot. As a loading control, the blots were stripped and reprobed with β-actin antibody. MCAO induced a significant reduction in occludin levels in total microvascular extracts (C). *p < 0.05 versus Non-I, Student's t test; n = 6. MCAO did not change total claudin-5 levels in cerebral microvascular extracts (top left panel) but led to a remarkable reduction in claudin-5 level in the ACF and its significant increases in the CF and MF (top right and bottom panels). *p < 0.05 versus Non-I, Student's t test; n = 4. Error bars indicate SEM.
Discussion
Early ischemic BBB damage that occurs within the time window of thrombolysis for acute ischemic stroke is being increasingly appreciated because it carries significant prognostic information and may help determine treatment options (thrombolytic therapy or not) (Bang et al., 2007; Hjort et al., 2008; Kastrup et al., 2008; Tanne, 2008; Kassner et al., 2009; Sun et al., 2010). In the present study, we used in vitro OGD and in vivo MCAO models to elucidate how the BBB is damaged at such an early stroke stage. Our data demonstrate that cerebral ischemia initiates two rapid parallel processes, MMP-2-mediated occludin degradation and Cav-1-mediated redistribution of claudin-5, to cause BBB disruption in early ischemic stage. Moreover, our data demonstrate that ischemia rapidly elevates extracellular MMP-2/9 levels through promoting secretion, while not their production.
BBB damage is a common event in ischemic stroke, presenting as a progressive process, with an initial damage resulting from cerebral ischemia (del Zoppo et al., 1998; Simard et al., 2007), aggravated by reperfusion due to a mechanism of reperfusion injury (Hallenbeck and Dutka, 1990; Warach and Latour, 2004). Accumulating evidence suggests that the timing of BBB damage can be early enough to be within the 3–4.5 h thrombolytic time window (Hacke et al., 2008; Del Zoppo et al., 2009). In animal stroke models, BBB damage or even severe microvascular disruption was observed in the subcortical region of the ischemic hemisphere 2.5 h after ischemia onset (Chen et al., 2009; Sun et al., 2010). In stroke patients, BBB disruption was found to be evident at a median estimated time of 3.8 h from stroke onset (Latour et al., 2004; Warach and Latour, 2004). The importance of this early ischemic BBB damage is increasingly appreciated because animal and human stroke studies offer an intriguing observation that ischemic brain tissue with compromised BBB in early ischemic stroke stages often develop ICH during thrombolytic reperfusion (Bang et al., 2007; Hjort et al., 2008; Kastrup et al., 2008; Kassner et al., 2009; Sun et al., 2010). These results reveal a close association between early ischemic BBB damage and postthrombolysis ICH. Surprisingly, despite its close link to postthrombolysis ICH, how the BBB is damaged at such an early stroke remains virtually unknown. Data presented in this study provide a novel mechanistic understanding of early ischemic BBB damage.
We show here that 2 h of ischemia (OGD or MCAO) without reoxygenation or a brief 10 min reperfusion induced BBB disruption both in vitro and in vivo, as reflected by increased permeability to fluorescence-labeled tracers. In the MCAO rats, FITC-albumin leakage invariably occurred in the ischemic subcortical regions (frequently seen in the ventromedial striatum and the preoptic area), indicating that the BBB in these regions suffers the earliest and the most, probably due to very low residual cerebral blood flow to these regions when the MCA is occluded (Butcher et al., 1990; Nagaraja et al., 2011). The mechanism(s) accounting for this early ischemic BBB damage is unclear. Animal stroke studies have demonstrated that the gelatinases MMP-2/9 are induced in the ischemic brain and critically contribute to BBB disruption at relative late stroke stages during postischemic reperfusion (Romanic et al., 1998; Heo et al., 1999; Wang et al., 2003; Gasche et al., 2006; Kamada et al., 2007; Y. Yang et al., 2007; Liu et al., 2009b; Jin et al., 2010). Increased MMP-2 and/or MMP-9 expression has also been observed in vitro after exposing endothelial cells to OGD followed by reoxygenation or to prolonged OGD treatment (24 h) (M. Z. Yang et al., 2007; Slevin et al., 2009). In the present study, we investigated how ischemia affected MMP-2/9 in the early ischemic stage. Our data that exposure of endothelial cells to OGD for 2 h led to MMP-2/9 increase in the CM and a remarkable reduction in their intracellular contents suggest that OGD may elevate extracellular MMP-2/9 levels through promoting secretion. Further support for this secreting mechanism is provided by the observation that OGD did not upregulate MMP-2/9 mRNA expression and that inhibiting de novo mRNA or protein synthesis had no effect on OGD-induced MMP-2/9 increase in the CM. These cellular findings were confirmed in vivo because 2 h MCAO increased gelatinolytic activities and MMP-2/9 levels, particularly MMP-2 levels, in the interstitial spaces of the ischemic striatum, while it did not upregulate their mRNA expression. In the setting of ischemic stroke, the stimulators for MMP-2/9 production are mainly produced during the phase of reperfusion, which include inflammatory cytokines (Ellison et al., 1999; Rosenberg and Mun-Bryce, 2004), serine proteases (tPA and thrombin) (Kolev et al., 2003; Wang et al., 2003; Yepes et al., 2003), and free radicals (Floyd, 1999; Jian Liu and Rosenberg, 2005). Therefore, in the early ischemic stage, promoting MMP-2/9 secretion may represent the only and rapid way to elevate extracellular MMP-2/9 levels, which is strongly supported by our data. Under our experimental conditions, 2 h OGD did not induce detectable cytotoxicity (data not shown), so it is not likely that the increased MMP-2/9 levels in the conditioned media are due to a passive release from the injured endothelial cells. However, this possibility may exist in the setting of 2 h MCAO because tissue infarction is evident on TTC-stained brain slices (data not shown). Worthy of note, our in vivo observation that MMP-2 is the major gelatinase secreted by ischemic brain tissue after 2 h MCAO is consistent with the emerging notion that MMP-2 is an early mediator in ischemic brain injury, while MMP-9, being an inflammatory molecule (normally existing at a low basal level), is induced and involved in brain injury at relatively late stroke stages (Y. Yang et al., 2007). The knowledge about MMP-2/9 intracellular trafficking and mode of their secretion remains quite limited, although there is evidence supporting a vesicular secretion mechanism in both cancer cells (Schnaeker et al., 2004; Taraboletti et al., 2006) and neural cells (Sbai et al., 2008, 2010). Future studies are warranted to further explore the mechanisms underlying enhanced MMP-2/9 secretion under ischemic condition.
We next investigated whether and how ischemia-induced MMP-2/9 secretion contributed to early ischemic BBB damage. MMP-2/9 have been shown to mediate BBB disruption through degrading TJ proteins in animal stroke models with reperfusion (Asahi et al., 2001; Date et al., 2006; Y. Yang et al., 2007; Liu et al., 2009a; Zhang et al., 2009). Similarly, here we found that 2 h OGD induced a significant reduction in TJ protein occludin, and this change was completely blocked by MMP-2/9 inhibitor SB-3CT and by MMP-2 neutralizing antibody; however, blocking of MMP-9 with its neutralizing antibody only showed marginal effects. These results indicate that MMP-2 is the enzyme responsible for occludin degradation under our experimental conditions, and this may be explained by our observation that active MMP-2, but not active MMP-9, was detected in the CM. Surprisingly, under the same experimental condition, another transmembrane TJ protein claudin-5 responded to OGD very differently, in which OGD did not cause claudin-5 degradation but stimulated its dissociation from the cytoskeletal framework in a MMP-2/9-independent manner. On the in vivo MCAO model, we also found that cerebral ischemia rapidly induced occludin degradation and claudin-5 redistribution in the ischemic cerebral microvessels. It is worth pointing out that, due to technical difficulty, we were unable to detect claudin-5 redistribution by ICH on cryosections, but we clearly showed its translocation from the cytoskeletal subcellular fraction to cytosolic and membranous fractions in isolated ischemic microvessels. At the TJs, although occludin and claudin-5 closely interact with each other to seal the gaps between adjacent endothelial cells, their fates seem to be very different when exposed to MMP-2/9 as incubation of microvascular or endothelial extracts with purified MMP-2 or -9 led to proteolytic degradation for occludin, but not claudin-5 (Giebel et al., 2005; Liu et al., 2009a). However, under more complicated pathological conditions, such as ischemic stroke, MMP-2 and/or -9 have been reported to mediate claudin-5 disruption in the ischemic brain at early and late reperfusion phases following cerebral ischemia (Y. Yang et al., 2007; McColl et al., 2008). In addition to MMP-2/9, caspase-3-mediated rapid disruption of zonula occludens-1 and claudin-5 has recently reported in a coculture neurovascular unit model after 30 min of OGD treatment, and this disruption is not accompanied by apoptotic cell death (Zehendner et al., 2011).
Redistribution of claudin-5 occurs in endothelial cells in response to various stimuli (Andras et al., 2003; Hawkins and Davis, 2005; Song et al., 2007; Larson et al., 2008; Kondo et al., 2009). What mediates claudin-5 redistribution is not clear. Cav-1 has been shown to be an important mediator involving in endocytosis, vesicular trafficking, and signal transduction (Lisanti et al., 1994; Okamoto et al., 1998; Smart et al., 1999), and Stamatovic et al. (2009) recently reported caveolae-associated internalization of claudin-5 in endothelial cells challenged by chemokine CCL2. In this study, we found that OGD stimulated translocation of Cav-1 from the cytoskeleton to the cytosol, which paralleled to the changes observed for claudin-5. Notably, knockdown of Cav-1 with siRNA completely abolished OGD-induced claudin-5 redistribution, implicating a causal link between these two events. Taking this a step further, we performed coimmunoprecipitation experiments and found that claudin-5 coimmunoprecipitated with Cav-1 and their interaction appeared to be enhanced under ischemic condition. Currently, we do not know the mechanisms by which ischemia triggered Cav-1 translocation and what the fates are for the redistributed Cav-1 and claudin-5 (recycling or degradation?). Clearly, more work is required to understand the trafficking of these proteins under ischemic condition.
Last, our results that inhibiting MMP-2/9 with SB-3CT or knockdown of Cav-1 with siRNA alone partially preserved endothelial barrier function and their combination led to a full preservation in OGD-treated endothelial monolayer support the notion that MMP-2-dependent occludin degradation and Cav-1-mediated redistribution of claudin-5 are two parallel molecular events, which can be quickly initiated by ischemia to mediate early ischemic BBB damage. It is worth pointing out that our data do not exclude that cerebral ischemia may also trigger rapid changes in other tight junction proteins such as ZO-1, JAM-1 (junctional adhesion molecule) (Wolburg and Lippoldt, 2002), adherens junctions (Vorbrodt and Dobrogowska, 2004), and other BBB structural components including astrocytic endfeet (Lo et al., 2005), pericytes (Zozulya et al., 2008), and cerebral mast cells (Mattila et al., 2011).
In summary, our results define a novel mechanism underlying early BBB damage induced by ischemia. We propose that ischemia triggers two rapid and parallel processes, MMP-2-mediated occludin degradation and Cav-1-mediated dissociation of claudin-5 from the cytoskeleton, to cause early ischemic BBB disruption. Findings in the present study may provide clues for developing new treatment strategies to protect the BBB against ischemic damage and thus safely expand the time window for thrombolytic reperfusion.
Footnotes
This work was supported by National Institutes of Health Grants P20 RR15636 and R01 AG031725, and American Heart Association Grant 10BGIA3190010. The images in this paper were generated in the University of New Mexico and Cancer Center Fluorescence Microscopy Shared Resource, funded as detailed on http://hsc.unm.edu/crtc/microscopy/Facility.html.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Wenlan Liu, College of Pharmacy, University of New Mexico Health Sciences Center, Albuquerque, NM 87131. weliu{at}salud.unm.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}