


Abstract
If fear memory is expressed by a long-term potentiation (LTP) of synaptic transmission in the amygdala, then reversal of LTP (depotentiation) in this area of the brain may provide an important mechanism for amelioration of anxiety and post-traumatic stress disorder. Herein, we show that low-frequency stimulation (LFS) of the external capsule elicits a depotentiation in the lateral nucleus of the amygdala. The induction of depotentiation requires activation ofN-methyl-d-aspartate receptors and voltage-dependent calcium channels but is independent of adenosine A1 and metabotropic glutamate group II receptors. Extracellular perfusion or loading cells with protein phosphatase (PP) 2B (calcineurin) inhibitors prevents depotentiation. The same stimulating protocol applied to the amygdala in vivo attenuates the expression of fear memory measured with fear-potentiated startle and reduces conditioning-elicited phosphorylation of Akt and mitogen-activated protein kinase (MAPK). This is paralleled by an increase in the activity of calcineurin. In addition, application of calcineurin inhibitor blocks LFS-induced extinction of fear memory and MAPK dephosphorylation. Taken together, this study characterizes the properties of LFS-induced depotentiation in the amygdala and suggests an involvement of calcineurin cascade in synaptic plasticity and memory storage.
The prevention of fear development or, more importantly, the erasure of fear memory is a major challenge to neuroscientists and psychiatrists today. Fear conditioning is the process by which a cue comes to induce an elevated startle when it is consistently paired with an aversive stimulus, such as a foot shock (Pavlov, 1927; Davis, 2000; LeDoux, 2000). It represents not only an animal model of fear and anxiety but also a model of long-term neural plasticity and memory because, once conditioned, animals can be left untrained for at least 1 month without a loss of startle susceptibility (Campeau and Davis, 1995). Previous studies suggested that LTP of synapses from auditory thalamus and cortex to the LA underlay the encoding of fear memory (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997). If this is true, then depotentiation in these synapses may result in an attenuation of fear memory. In an analogous situation, it has been shown that administration of LFS to the amygdala in vivo blocked the development and expression of kindled seizures, a phenomenon termed quenching (Weiss et al., 1995). A recent report demonstrated that depotentiation could be induced at the EC-basolateral amygdala synapse (Aroniadou-Anderjaska et al., 2001); however, the underlying mechanism was not elucidated.
In the present study, we first show that depotentiation occurs in the amygdala that is capable of reversing tetanus-induced LTP. Depotentiation is dependent onN-methyl-d-aspartate (NMDA) receptor and VDCC activation and requires postsynaptic phosphatase activity. By behavioral assessment, we further demonstrate that administration of LFS to the EC or LA using depotentiation-like parameters attenuates the expression of fear memory. Quenching stimulation, which also increases the activity of calcineurin, reduces fear training-induced Akt and MAPK phosphorylation. Thus, quenching could be attributed to an increased calcineurin activity, which dephosphorylates key proteins in the amygdala and results in the extinction of conditioned fear.
Materials and Methods
Slice Preparation and Electrophysiological Recordings.
Male Sprague-Dawley rats, 4 to 5 weeks old, were decapitated and their brains rapidly removed and placed in cold oxygenated artificial cerebrospinal fluid (ACSF) solution. Subsequently, the brain was hemisected and cut transversely posterior to the first branch and anterior to the last branch of the superior cerebral vein. The resulting section was glued to the chuck of a Vibroslice tissue slicer (Campden Instruments, Silbey, UK). Transverse slices of 450 μm thickness were cut and the appropriate slices placed in a beaker of oxygenated ACSF at room temperature for at least 1 h before recording. ACSF solution had the following composition (in mM): 117 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 25 mM NaHCO3, 1.2 mM NaH2PO4, and 11 mM glucose. The ACSF was bubbled continuously with 95% O2/5% CO2 and had a pH of 7.4.
A single slice was transferred to the recording chamber, in which it was held submerged between two nylon nets and maintained at 32 ± 1°C. The chamber consisted of a circular well of a low volume (1–2 ml) and was perfused constantly at a rate of 2 to 3 ml/min. Extracellular field potentials were recorded by electrical stimulation of the external capsule, which contained fibers from the auditory cortex to the lateral amygdala, with a concentric bipolar stimulating electrode (SNE-100; Kopf Instruments, Bern, Germany). Electrical stimuli (150 μs in duration) were delivered at a frequency of 0.05 Hz. Intracellular recording microelectrodes were pulled from 1.0-mm microfiber capillary tubing on a Brown-Flaming electrode puller (Sutter Instruments, San Rafael, CA). The electrodes were filled with 4 M potassium acetate, with resistance ranging from 70 to 130 MΩ. Baseline field potentials were adjusted to ∼30 to 40% of the maximal responses. LTP was elicited by three trains of tetanus (100 Hz, 1 s, at 1.5-min intervals) at the same stimulation intensity used for baseline. In most experiments, bicuculline (1 μM) was present in the perfusion solution. Data were expressed as mean ± S.E. The data were analyzed by analysis of variance and Student's t test, and p < 0.05 was considered statistically significant.
Surgery and Quenching.
Rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and mounted on a stereotaxic apparatus. Two stimulating electrodes (MS303/2; Plastic Products, Roanoke, VA) were implanted bilaterally into the EC or LA. The coordinates were anteroposterior,−2.8 mm; mediolateral, ± 5.3 mm; dorsoventral, −7.8 mm, according to the method of Paxinos and Watson (1986). Three jewelry screws were implanted over the skull, serving as anchors, and the whole assembly was affixed on the skull with dental cement. The rats were monitored and handled daily and were given 5 to 7 days to recover. The animals were then subjected to training and behavioral tests performed the next day. Quenching stimulation (5 Hz for 3 min at the intensity of 0.1 mA) was given at 10 min after training (after training) or 1 h after testing (after testing).
Fear Conditioning.
Fear conditioning was measured using the potentiated startle paradigm (Cassella and Davis, 1986). Male Sprague-Dawley rats (100–150 g) were trained and tested in a startle chamber (San Diego Instruments) in which cage movement results in the displacement of an accelerometer. Startle amplitude was defined as peak accelerometer voltage within 200 ms after startle stimulus onset. The acoustic startle stimulus was a 50-ms burst of white noise at an intensity of 95 dB. The visual conditioned stimulus (CS) was a 3.7-s light flash produced by an 8-W fluorescent bulb attached to the back of a stabilimeter. The unconditioned stimulus (US), which serves as the aversive component necessary for fear conditioning, was a 0.6-mA foot shock with a duration of 0.5 s. On the training day, rats were placed in the startle chamber and received 10 CS-foot shock pairings. Unpaired control rats received the same number of CS and US presentation, but in an unpaired, pseudorandom fashion.
Western Blot Analysis.
Unless noted in the experiments, groups of sham control and LFS animals were sacrificed by decapitation at 10 min after training or 1 h after testing. The lateral and basolateral subregions of the amygdala were sonicated briefly in ice-cold buffer (50 mM Tris-HCl, pH 7.5, 0.3 M sucrose, 5 mM EDTA, 2 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml leupeptin, and 4 μg/ml aprotinin). After sonication, the samples were centrifuged at 7500 rpm for 15 min and the supernatant was obtained after pelleting the crude membrane fraction by centrifugation at 50,000 rpm for 1 h at 4°C. Protein concentration in the soluble fraction was then measured using a Bradford assay, with bovine serum albumin as the standard. Equivalent amounts of protein for each sample were resolved in 8.5% SDS-polyacrylamide gels, blotted electrophoretically to Immobilon-P transfer membrane (Millipore, Bedford, MA), and blocked overnight in Tris-buffered saline (50 mM Tris-HCl, pH 7.5, 150 mM NaCl) containing 3% bovine serum albumin. For detection of the phosphorylated forms of MAPK, blots were incubated with anti-phospho-ERK (New England Biolabs, Beverly, MA) antibodies. To control the content of the specific protein per lane, membranes were stripped with 100 mM β-mercaptoethanol and 2% SDS in 62.5 mM Tris-HCl, pH 6.8, for 30 min at 70°C and reprobed with a mouse monoclonal anti-pan-ERK (BD Transduction Laboratories, Lexington, KY) antibody. An enhanced chemiluminescence kit (Amersham Biosciences, Piscataway, NJ) was used for detection. Western blots were developed in the linear range used for densitometry. The density of the immunoblots was determined by an image analysis system installed with a software BIO-ID (Vilber Lourmat, Marne La Vallée, France). To assess for changes in the activation of MAPK, total kinase levels were first normalized to total protein levels. Activated kinase levels in trained animals were normalized to total kinase levels and then were expressed as a percentage of those in control animals.
Calcineurin Activity Assay.
Rats trained with light alone were sacrificed by decapitation immediately after trials. The LA and BLA were microdissected and frozen on dry ice. Phosphatase assay was performed according to the instructions of the calcineurin assay kit (Promega Corp., Madison, WI). Pooled LA and BLA areas were homogenized in ice-cold buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10 mM EGTA, 5 mM EDTA, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml leupeptin, and 4 μg/ml aprotinin) and centrifuged at 100,000 rpm for 1 h to remove particular matter. Supernatants were added to the reaction buffer from the kit and incubated the reaction at 30°C for 10 min. The reaction buffer contains 50 mM imidazole, pH 7.2, 0.2 mM EGTA, 10 mM MgCl2, 1 mM NiCl2, 50 μg/ml calmodulin, and 0.02% β-mercaptoethanol. FK-506 (1 μM) in the supernatant completely blocked Pi release, indicating that the measured phosphatase activity reflects calcineurin function. The enzyme activity was expressed in nanomoles of Pi released per minute per milligram of protein from the substrate. The calcineurin substrate sequence is RRA(pT)VA.
Results
Induction of LTP in the LA Neurons.
In amygdala slices of 4- to 6-week-old rats, delivery of three sets of tetanic stimulation (TS; 100 Hz for 1 s) to the EC at an interstimulus interval of 1.5 min produced a robust enhancement of synaptic responses in the LA neurons that persisted for more than 2 h. The slopes of fEPSP were 213.4 ± 18.2 and 200.1 ± 15.2% (n = 6) of pretetanus level at 1 and 2 h after the stimulation, respectively (Fig. 1). To examine whether LTP was NMDA-receptor dependent, 50 μMd-2-amino-5-phosphonovaleate (d-APV) was added to the perfusion medium 5 min before, during, and 10 min after TS. Consistent with previous reports, LTP was blocked byd-APV (Huang and Kandel, 1998). The slopes of fEPSP were 122.6 ± 4.9 and 91.6 ± 4.5% (p< 0.001, n = 6, unpaired t test) at 1 and 2 h after the stimulation.
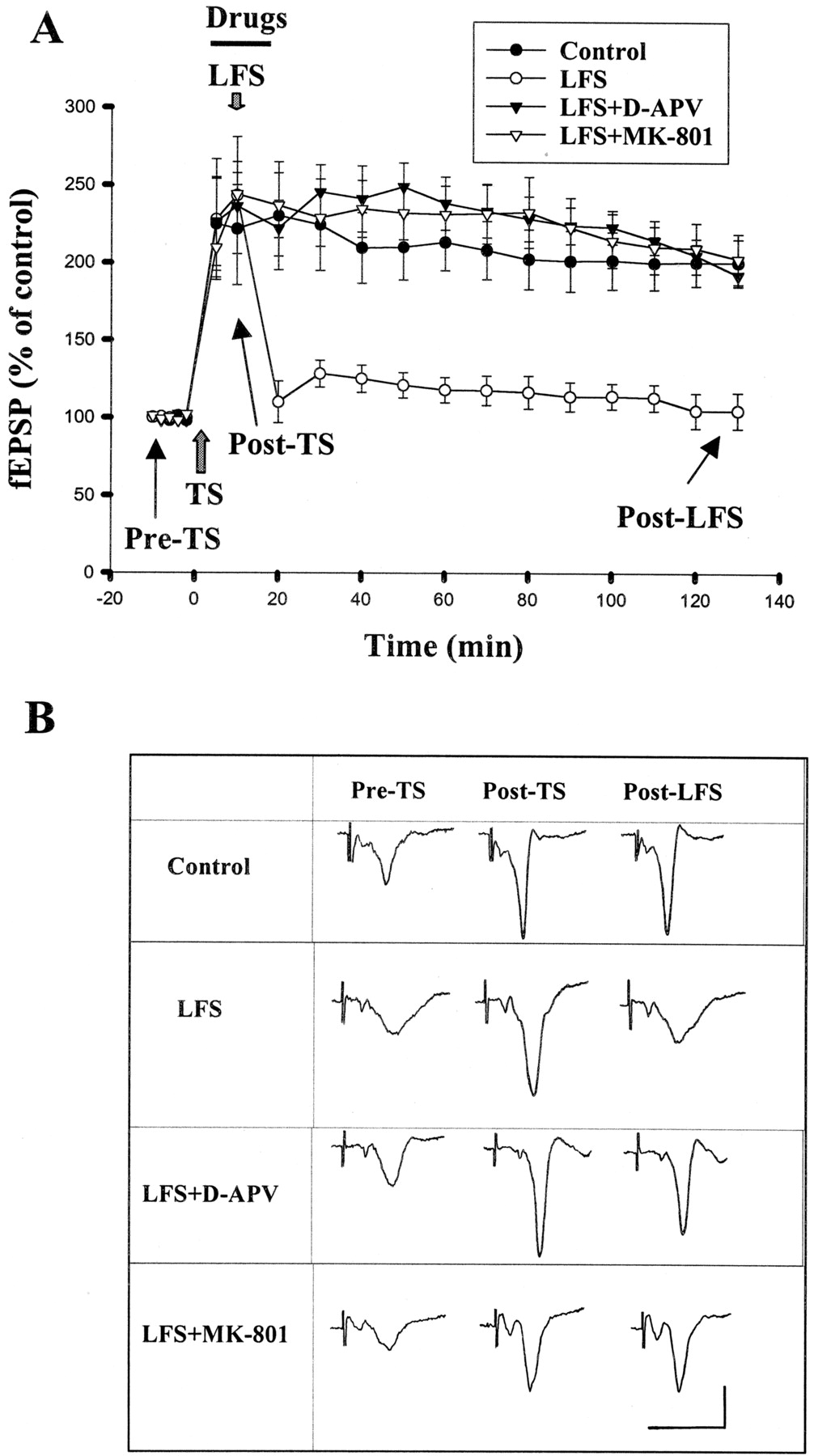
Depotentiation in the amygdala depends on the activation of NMDA receptors. A, the graph represents the mean ± S.E. slope of fEPSPs plotted against time. TS is marked by a gray up-arrow, and the horizontal bar represents the period of drugs application. LFS delivered in the presence of d-APV (50 μM) or MK-801 (10 μM) failed to induce depotentiation. B, sample traces taken at the times indicated in A. Calibration: 0.5 mV, 10 ms.
Depotentiation by LFS.
We attempted to establish a protocol for investigating depotentiation and found that, when delivered at 10 min after TS, reliable depotentiation could be induced by 15 min of stimulation at 1 Hz, 7.5 min at 2 Hz, or 3 min at 5 Hz. Because it is easier to handle and quench the animals (seeQuenching) with a shorter LFS protocol, 5 Hz stimulation for 3 min was used to elicit depotentiation. In six experiments, the slopes of fEPSP returned to 118.0 ± 8.1 and 104.9 ± 11.4% of pretetanus level at 1 and 2 h after TS, respectively (Fig. 1). In another set of experiments, in which LFS was applied at 1 h (instead of 10 min) after TS, this LFS protocol caused similar depotentiation. The fEPSP measured at 2 h after TS was 112.0 ± 10.1% (n = 6) of pretetanus value, which was not significantly different from that LFS applied at 10 min after TS (p > 0.1).
Several neurotransmitter systems have been implicated in the mediation of depotentiation. We first examined whether depotentiation is NMDA receptor-dependent by application of d-APV. Because NMDA receptor antagonists block the induction of LTP, d-APV was applied at 5 min after TS. LFS was then given at 5 min afterd-APV perfusion, and the drug was removed at the end of LFS. Figure 1 shows that d-APV (50 μM) blocked depotentiation. The fEPSPs measured at 1 and 2 h after TS were 231.7 ± 16.0 and 181.3 ± 7.1% (n = 6), respectively, of pretetanus value, which were significantly different from that without d-APV application (p < 0.01, unpaired t test). To further ensure NMDA dependence, LFS was delivered in the presence of the noncompetitive antagonist MK-801 (10 μM). Depotentiation was blocked in a similar way by MK-801. The fEPSPs measured at 1 and 2 h after TS were 234.1 ± 23.8 and 197.6 ± 16.1% (n= 6) of pretetanus value, respectively.
In hippocampal CA1 neurons, depotentiation has been shown to be blocked by adenosine A1 receptor antagonists, suggesting an involvement of this receptor (Larson et al., 1993; Fujii et al., 1997; but see De Mendonca et al., 1997). We tested whether this is the case in the amygdala by application of a selective adenosine receptor antagonist, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX). DPCPX (500 nM) was applied 5 min before LFS and removed from the perfusion medium immediately after LFS. Consistent with previous reports showing that there were significant amounts of endogenous adenosine present in the extracellular space (Dunwiddie et al., 1981), DPCPX increased the fEPSP by 28 ± 6%. The effect was reversible within 20 min of washing. As summarized in Table 1, DPCPX did not affect depotentiation. The fEPSPs measured at 1 and 2 h after TS were 123.1 ± 12.5 and 109.1 ± 12.5% (n = 6) of baseline, respectively, which was not significantly different from that without DPCPX treatment.
Effects of drugs treatments on LFS-induced depotentiation and synaptic transmission at EC-LA synapses
In basolateral amygdala neurons, primed facilitation of depotentiation was blocked by 2S-α-ethylglutamic acid (EGLU), suggesting the involvement of mGluR II (Li et al., 1998). To examine the involvement of mGluR II, EGLU (20 μM) was applied 5 min before and during LFS. EGLU did not affect depotentiation (Table 1). The fEPSPs measured at 1 and 2 h after TS were 131.7 ± 21.9 and 97.9 ± 17.2% (n = 6) of baseline, respectively.
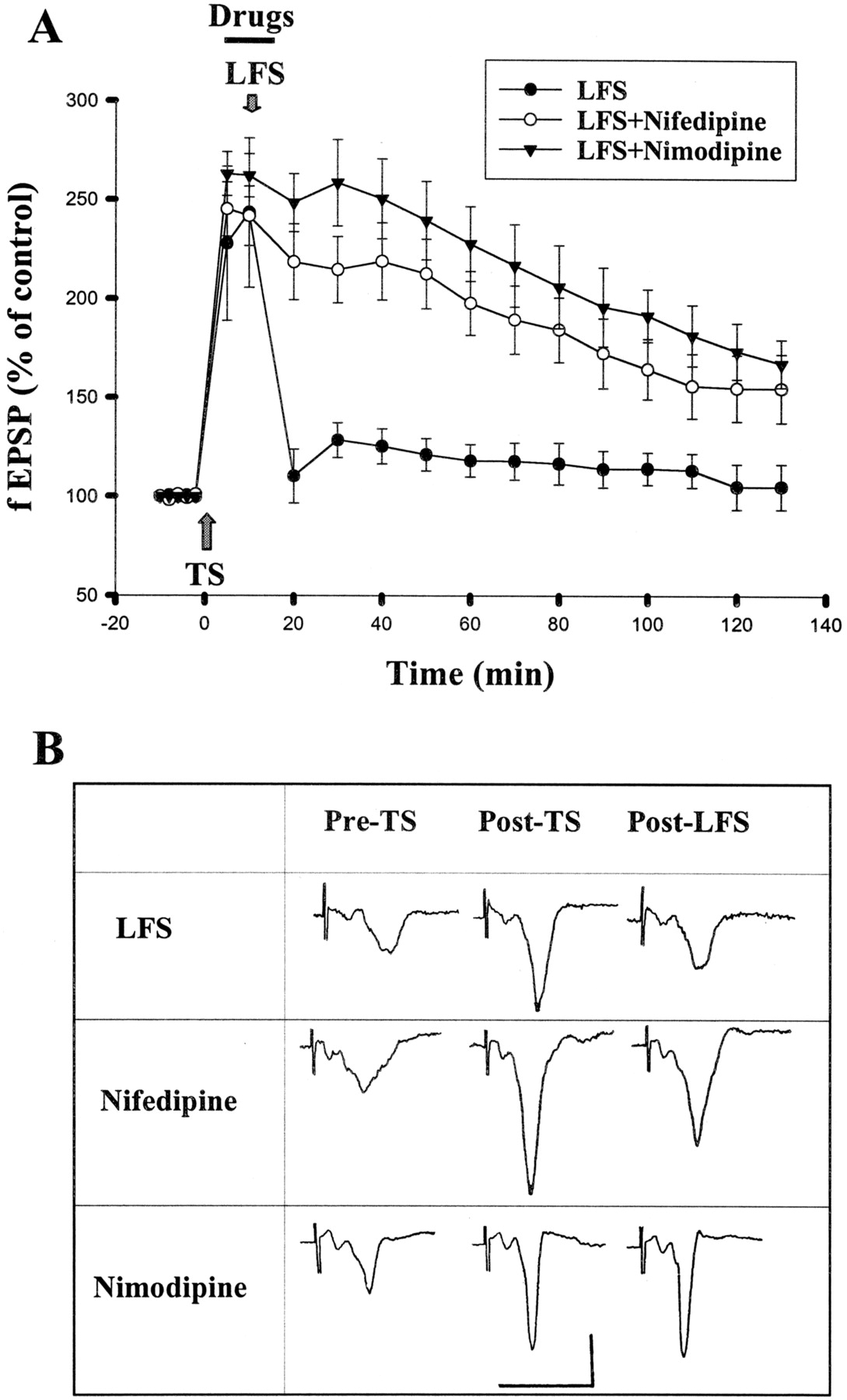
Aside from NMDA receptors, Ca2+ could enter the cell through VDCCs. Indeed, NMDA-independent forms of synaptic plasticity in several brain areas, including the amygdala (Weisskopf et al., 1999), depend on L-type VDCCs (Christie et al., 1997; Kapur et al., 1998; Morgan and Teyler, 1999). We therefore tested whether inhibition of L-type VDCCs affected amygdala depotentiation. Figure2 shows that blocking L-type VDCCs with nifedipine (20 μM) partially prevented LFS-induced depotentiation. The fEPSP slopes were 197.9 ± 16.1 and 154.9 ± 16.8% (n = 6) of baseline at 1 and 2 h, respectively, after the induction of LTP. We applied another selective L-type VDCC blocker, nimodipine. Nimodipine (2 μM) blocked depotentiation. The fEPSPs were 227.9 ± 18.9 and 173.7 ± 14.2% of baseline 1 and 2 h after TS, respectively, which was not statistically different from control without receiving LFS (p > 0.05, Fig. 2).
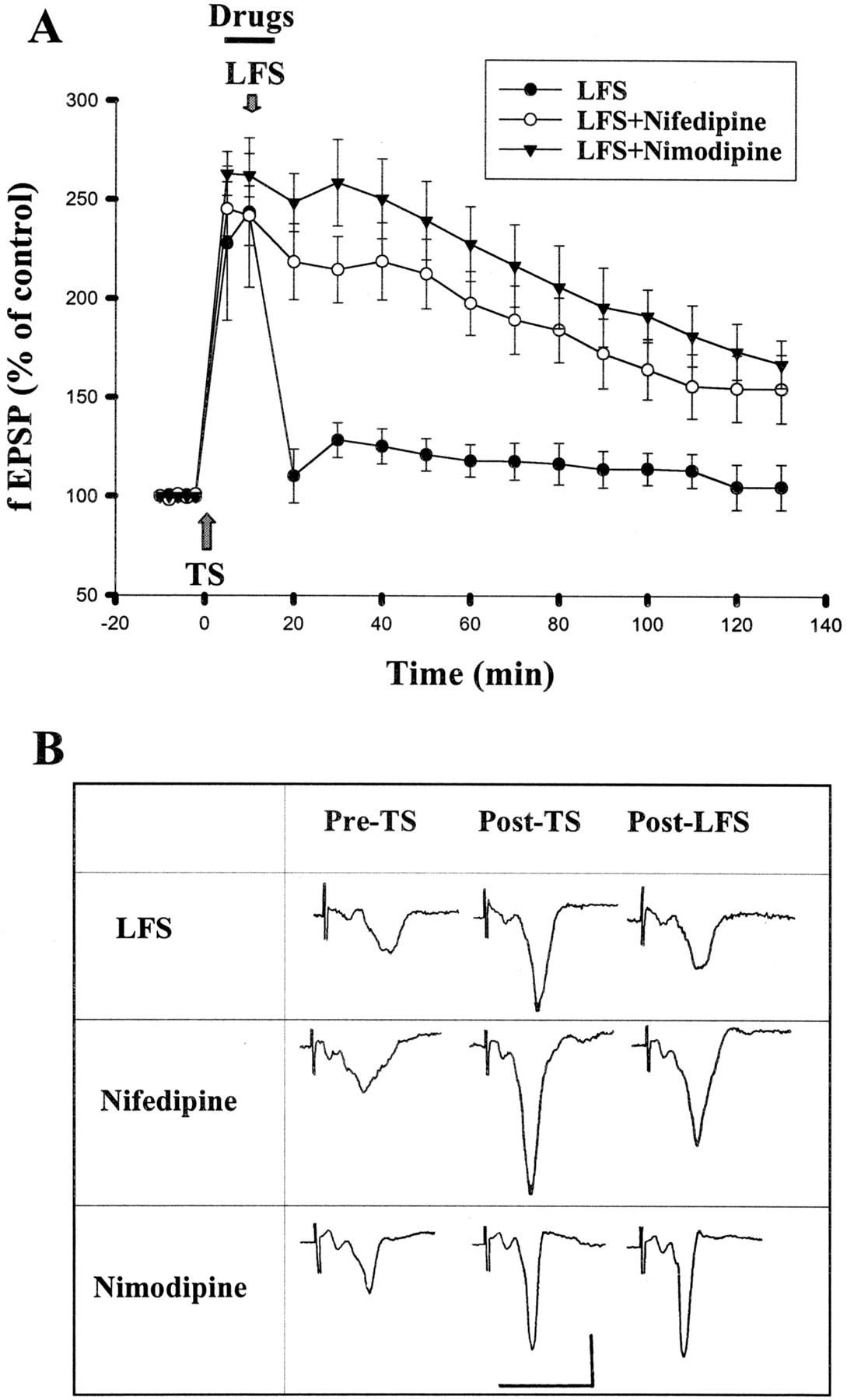
L-type calcium channel blockers inhibit depotentiation. A, application of nifedipine (20 μM) or nimodipine (2 μM) 5 min before and during LFS blocked depotentiation. B, sample traces taken at pre-TS, post-TS, and post-LFS. Calibration: 0.5 mV, 10 ms.
Involvement of Calcineurin in Synaptic Depotentiation in the Amygdala.
One likely signal pathway that is Ca2+-dependent and may be involved in the reversal of LTP is the serine/threonine protein phosphatase calcineurin (O'Dell and Kandel, 1994; Staubli and Chun, 1996; Zhuo et al., 1999). We examined whether amygdala depotentiation is associated with a change in phosphatase activity by application of specific calcineurin inhibitors. Figure 3 shows that cyclosporin A, an immunosuppressant that inhibits calcineurin in a complex with cyclophilin (Liu et al., 1991), blocked depotentiation in a concentration-dependent manner (F2,15 = 15.48,p < 0.001). In 10 μM cyclosporin A, the fEPSP slopes were 170.4 ± 16.1 and 146.3 ± 12.3% (n = 6) of baseline at 1 and 2 h, respectively, after the induction of LTP. In 100 μM, the fEPSP slopes were 207.8 ± 21.7 and 196.8 ± 12.4% (n = 6) of baseline at 1 and 2 h, respectively, after the induction of LTP, which was significantly different from LFS group (p < 0.01) but was not statistically different from the group not receiving LFS (p > 0.05). Similarly, another selective calcineurin inhibitor, FK-506 (100 μM), also blocked depotentiation (fEPSPs were 233.8 ± 10.9 and 190.4 ± 24.0% of baseline 1 and 2 h, respectively, after TS, n = 6) (Fig. 3). As a control, we examined whether basal synaptic strength is regulated by calcineurin. We found that both cyclosporin A and FK-506 had no significant effect on basal fEPSP (Table 1). The lack of effect of calcineurin inhibitors on basal synaptic strength may suggest that calcineurin either is not constitutively active at synapses during low levels of synaptic activity or is functionally expressed in adult but not young rats (Wang and Kelly, 1997).

Effect of calcineurin inhibitors on depotentiation. A, summary of experiments in which LFS was applied in the presence of cyclosporin A (100 μM) or FK-506 (100 μM). B, sample traces taken at pre-TS, post-TS, and post-LFS. Calibration: 0.5 mV, 10 ms.
To further define the locus of action of calcineurin inhibitors (pre- versus postsynaptic site), we used intracellular recording technique and loaded the LA neurons with FK-506 (200 μM). After impalement, the cells were allowed to stabilize for at least 30 min to allow FK-506 diffusion into the cells. Baseline responses were then obtained for a further 10 min before TS was delivered. In control experiments in which cells were recorded with normal recording solution with solvent added, delivery of TS to the EC produced an enhancement of synaptic responses in the LA neurons. The EPSP amplitude was 156.0 ± 17.8% (n = 6) at 1 h after the last TS. Figure4 shows that reliable depotentiation was induced by LFS given at 10 min after TS. The EPSP amplitude returned to 89.8 ± 5.9% (n = 6) of pretetanus level at 1 h after TS. By contrast, depotentiation was blocked in neurons loaded with FK-506. The EPSP amplitude was 156.9 ± 11.9% (n = 6) of baseline at 1 h after TS, which was not statistically different from control without receiving LFS (p > 0.05).

Effect of intracellular FK-506 on depotentiation. After impalement, the cells were allowed to stabilize for at least 30 min to allow FK-506 to diffuse into the cells. Baseline responses were then obtained for a further 10 min before TS was delivered. A, summary of six experiments in which the effect of intracellular FK-506 (200 μM) was compared with those of recorded with normal recording solution with solvent added. B, representative traces taken 10 min before and 60 min after TS. Calibration: 10 mV, 50 ms.
Quenching.
In the first set of experiments, rats were implanted bilaterally with stimulating electrodes into the EC or LA and were given 5 to 7 days to recover. The rats were then given 10 pairs of light (CS) and foot shock (US) and tested 24 h later (pre-LFS test). After initial training, animals exhibited fear of the light, which manifested as an increase in acoustic startle. They were subsequently divided into three groups: sham-operated control, 0.1-Hz LFS, and 5-Hz LFS. LFS groups were given low-frequency stimulation (0.1 or 5 Hz for 3 min) bilaterally 1 h after pre-LFS tests and were retested 23 h later, whereas the sham control group did not receive electrical stimulation. Figure 5A illustrated the mean startle potentiation in control and LFS groups. After LFS, the degree of potentiation was significantly reduced in rats of the 5-Hz post-LFS group compared with pre-LFS test (t 9 = 3.45, p < 0.01). In contrast, startle amplitudes in animals of the sham-operated and 0.1 Hz post-LFS groups were not significantly different from their pre-LFS tests (t 5 = 3.5,p = 0.34). The differential results obtained from 0.1- and 5-Hz stimulation could have be caused by the different number of stimulations. Therefore, additional control experiments were performed in which rats received three sessions of stimulation and each session consisted of three trains of 100-Hz stimulation at an intersession interval of 1 min. As shown in Fig. 5A, startle amplitude was not changed between pre- and poststimulation tests in these rats, indicating that 100-Hz stimulation was unable to reduce conditioned fear. Figure 5B illustrates the electrode tip locations in these experiments. Only rats with cannula tips at or within the boundaries of LA and BLA were included in the data analysis.
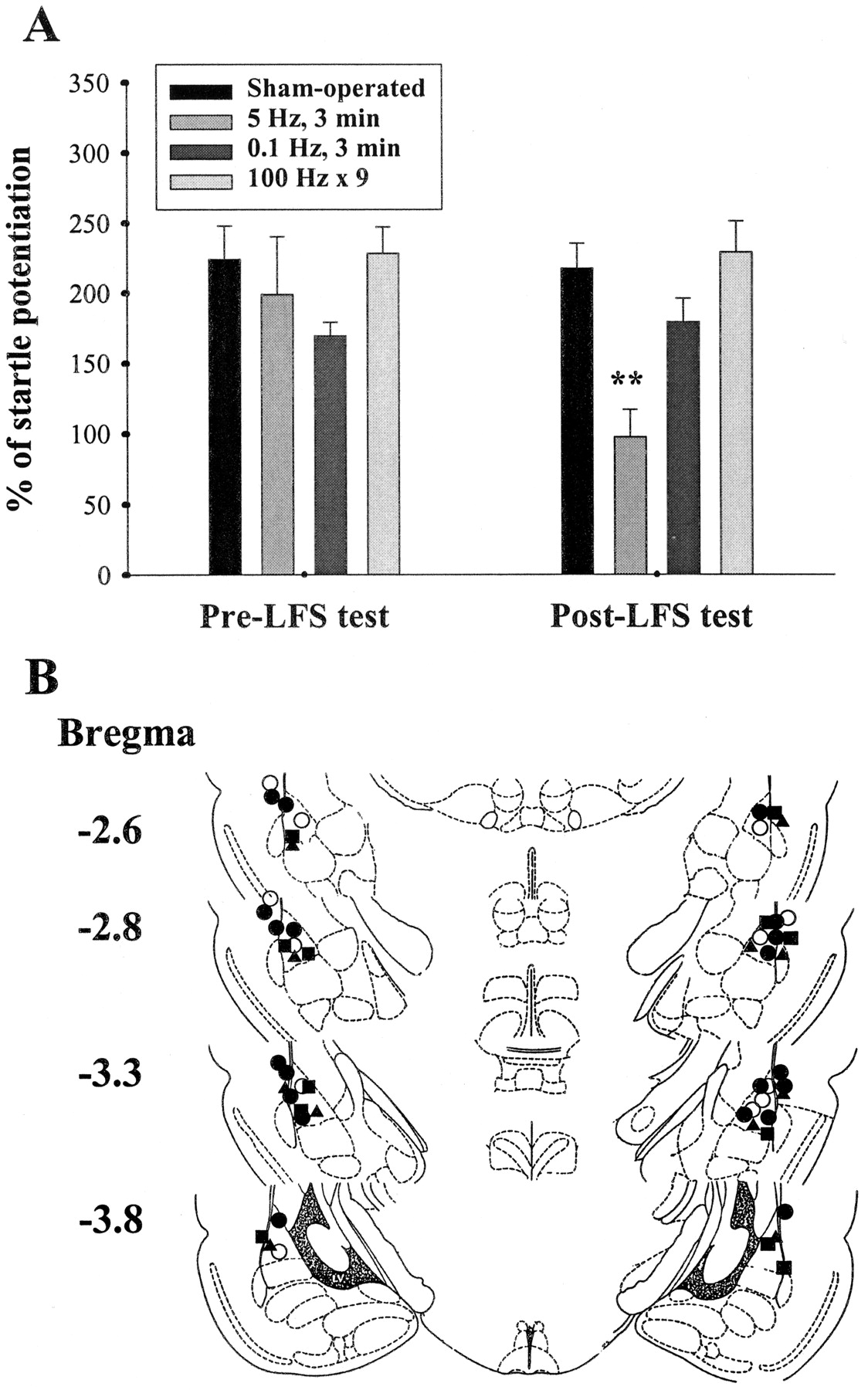
Quenching reduces fear-potentiated startle. A, comparison of startle potentiation between pre- and post-LFS tests. Startle potentiation was significantly reduced in the LFS group. **,p < 0.01 versus pre-LFS test. B, electrode tip placements in sham-operated control (○), 5-Hz LFS (●), 0.1-Hz LFS (▵), and 100-Hz (▪) rats in experiments shown in A.
Quenching Induces Dephosphorylation of Akt and MAPK.
We have recently shown that fear conditioning caused a selective activation of phosphoinositide-3 kinase in the amygdala (Lin et al., 2001). It is of interest to determine whether conditioning-elicited Akt phosphorylation is affected by quenching stimulation. Rats were implanted bilaterally with stimulating electrodes and trained with startle reflex paradigm. After training, the rats were divided into three groups; after-test, after-training, and sham-operated control groups. The group of after-training was given LFS 10 min after fear conditioning, whereas the group of after-test received LFS 1 h after pre-LFS test (Fig.6A). The sham-operated rats were similarly implanted with electrodes without receiving LFS. Figure 6B shows that LFS after training significantly reduced the degree of Akt phosphorylation (101.6 ± 4.3%, n = 6,p < 0.01 versus conditioned). Similarly, Akt phosphorylation was inhibited by LFS after pre-LFS test (100.9 ± 6.2%, n = 6; p < 0.01 versus conditioned). By comparison, Akt phosphorylation in sham-operated control rats was comparable with those of conditioned animals (p = 0.28, unpaired t test). No change in the immunoreactivity against Akt was detected, suggesting that the total amount of Akt was not altered (Fig. 6C).

Quenching stimulation reduces conditioning-induced Akt phosphorylation. A, behavioral procedure employed in the experiments B. B and C, representative blots and mean percentage ± S.E. of P-Akt (B) and Akt (C) immunoreactivity from unpaired control rats (lane 1), conditioned rats (lane 2), after test (lane 3), after training (lane 4), and sham-operated control rats (lane 5). **,p < 0.01 versus conditioned.
It has been shown that fear conditioning and LTP stimulation resulted in a selective activation of MAPK in the amygdala (Huang et al., 2000;Schafe et al., 2000; Lin et al., 2001). We next investigated the effect of quenching stimulation on conditioning-elicited phosphorylation of MAPK. As shown in Fig. 7, LFS after training significantly reduced the degree of MAPK phosphorylation (p42, 106.3 ± 12.4%; p44, 113.7 ± 23.2%; n = 6,p < 0.01 versus conditioned). Similarly, MAPK phosphorylation was inhibited by LFS after pre-LFS test (p42, 122.0 ± 13.3%; p44, 117.4 ± 21.3%; n = 6;p < 0.01 versus conditioned). In contrast, MAPK phosphorylation in sham-operated control rats (p = 0.85) or after 0.1-Hz stimulation (p = 0.71) was not significantly different from those of conditioned animals. These results suggest that fear training-induced MAPK phosphorylation was markedly attenuated after LFS. Figure 7 also shows that the immunoreactivity against MAPK was not changed, indicating that the total amounts of MAPKs were not altered.
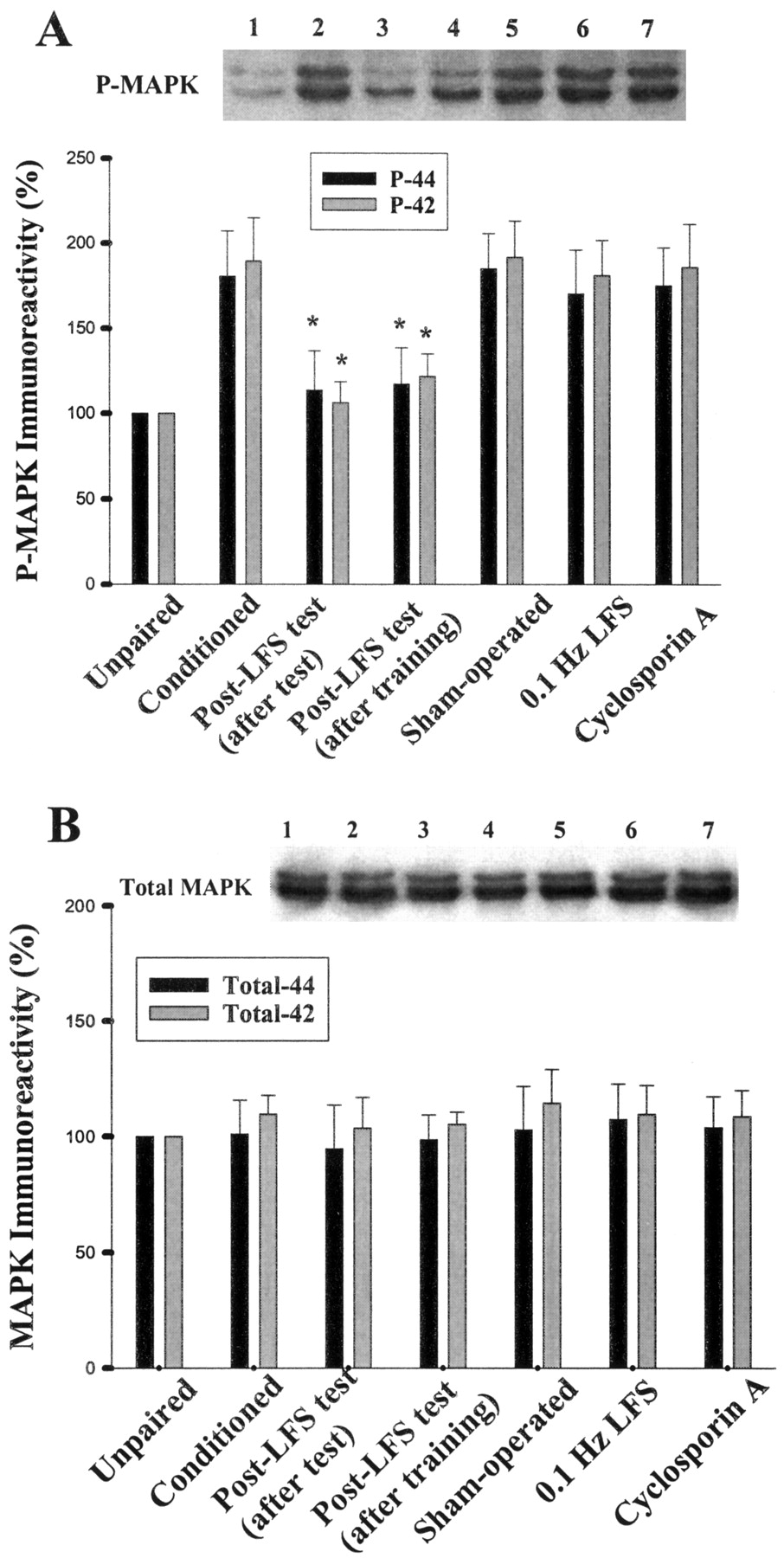
Effect of quenching on conditioning-induced MAPK activation. Representative blots and mean percentage ± S.E. of phosphorylated p42 and p44 (A), and total 42 and 44 (B) immunoreactivity from unpaired control (lane 1), conditioned (lane 2), after test (lane 3), after training (lane 4), sham-operated control (lane 5) and 0.1-Hz stimulation (lane 6) rats. Intravenous administration of cyclosporin A (20 mg/kg) before LFS blocked quenching-induced dephosphorylation of MAPK. *, p< 0.01 versus conditioned.
Calcineurin Inhibitor Blocks Both Extinction and Dephosphorylation.
It has been proposed that synaptic plasticity and memory storage is determined by the balance between protein phosphorylation and dephosphorylation mediated by cAMP-dependent protein kinase and phosphatases. Therefore, a decrease in the phosphorylated state of Akt and MAPK after quenching stimulation could result from a recruitment of phosphatase calcineurin. To examine this possibility, animals were intravenously administered cyclosporin A (1, 10, or 20 mg/kg) immediately after the pre-extinction tests. As shown in Fig. 8A, rats that received vehicle showed a significant decrease in the startle after quenching stimulation. In contrast, rats given intravenous infusion of cyclosporin A before stimulation exhibited less reduction. An analysis of variance revealed that cyclosporin A produced a dose-dependent inhibition of LFS-induced decrease in startle responses (F3,19 = 27.71, p < 0.001). In addition, a parallel experiment determined that cyclosporin A prevented quenching-induced dephosphorylation of MAPK (Fig. 7A). Thus, cyclosporin A blocked LFS-induced startle reduction at the same dose that inhibited MAPK dephosphorylation.

Quenching-induced reduction of fear memory is blocked by calcineurin inhibitor. A, intravenous administration of cyclosporin A (1, 10, or 20 mg/kg) dose-dependently antagonized LFS-induced reduction of fear memory. *, p < 0.01; **,p < 0.001 versus LFS. B, increase in the enzymatic activity of calcineurin after quenching. Calcineurin activity was assayed by measuring the released inorganic phosphate (Pi) from the phosphopeptide substrate in the LA and BLA area supernatants from unpaired control (lane 1, n = 8 rats), conditioned (lane 2, n = 8 rats), 5-Hz stimulation (lane 3, n = 8 rats), and 0.1-Hz stimulation (lane 4, n = 7 rats) rats. **, p < 0.001 versus conditioned.
Because depotentiation in this area of the brain is blocked by L-type Ca2+ channel inhibitors, we investigated whether L-type Ca2+ channel inhibitors also blocked quenching of conditioned fear. Rats were given verapamil (10 mg/kg, i.p.) at a dose sufficient to block NMDA receptor-independent LTP in the hippocampus (Morgan and Teyler, 1999) before LFS stimulation. Figure 8A showed that memory retention in verapamil-treated rats (205.2 ± 46.8%, n = 4) was significantly better than that of the LFS group.
Calcineurin Activity Is Increased after Quenching Stimulation.
We directly determined the involvement of calcineurin in quenching by measuring the released Pi from the phosphopeptide substrate that was insensitive to okadaic acid but could be blocked by FK-506. The rates of Pi released from LA and BLA in unpaired control and conditioned rats were 4.5 ± 0.3 (n = 8) and 4.8 ± 0.3 nmol/min/mg (n = 8), respectively. The value was increased to 6.9 ± 0.4 nmol/min/mg after quenching stimulation (n = 8, p < 0.001 versus unpaired or conditioned). By contrast, calcineurin activity was unaltered in rats given 0.1-Hz stimulation (5.0 ± 0.4, n = 7,p = 0.5 versus conditioned) (Fig. 8B).
Discussion
The major findings of this study are that 1) LFS is capable of eliciting depotentiation in the in vitro amygdala slices and, in parallel, reduces conditioned fear in whole animals; 2) inhibition of protein phosphatase 2B by calcineurin inhibitors blocks depotentiation in slices and prevents LFS-induced extinction of fear memory in animals; and 3) fear training-induced phosphorylation of Akt and MAPK in the rat amygdala is reduced after quenching stimulation, which is accompanied by an increase in the enzymatic activity of calcineurin. The paralleled effects of LFS (quenching) on depotentiation in the in vitro slices and on phosphorylation of protein kinases and retention of fear memory in vivo provide the direct evidence suggesting that synaptic plasticity and memory storage are regulated by protein phosphorylation and dephosphorylation in the amygdala.
Properties of Amygdala Depotentiation.
In hippocampal CA1 neurons, the reversal of LTP or depotentiation was thought to result from activation of NMDA or adenosine A1 receptors during LFS, which modulates Ca2+ entry or intracellular cAMP levels, leading to a decrease of previously enhanced responses (Fujii et al., 1991, 1997; Otmakhova and Lisman, 1998). We investigated the cellular mechanism of synaptic depotentiation at the EC-LA synapse, which carries axons from secondary auditory and perirhinal cortices to the amygdala and is important for mediation of fear conditioning (Davis et al., 1993; Campeau and Davis, 1995). We found that synaptic depotentiation at the EC-LA synapse was independent of adenosine A1 or mGluR II receptors but required activation of NMDA receptors, L-type Ca2+ channels, and calcineurin. In addition, loading of LA neurons with calcineurin inhibitor blocked depotentiation. Taken together, these results suggest that during LFS, Ca2+ enters through NMDA receptors and L-type Ca2+ channels, resulting in the activation of calcineurin (PP2B). Many protein kinases including Akt and MAPK are substrates of PP2A, and PP2A seems to be the major kinase phosphatase in eukaryotic cells that down-regulates activated protein kinases (Andjelkovic et al., 1996; Millward et al., 1999). Thus, calcineurin may dephosphorylate inhibitor-1, resulting in an activation of PP1 and PP2A. Active PP1 and 2A then inactivated protein kinases, which in their phosphorylated state were required for synaptic plasticity. This is consistent with genetic studies showing that depotentiation was abolished completely in mice lacking the predominant calcineurin isoform (Zhuo et al., 1999). Alternatively, calcineurin could directly act on ligand-gated ion channels independent of PP1 and PP2A (Yakel, 1997).
It is unclear why depotentiation in the amygdala neurons requires conjoint activation of NMDA receptors and VDCCs. However, calcium spikes in amygdala neurons were readily recruited by a single afferent stimulus that elicited NMDA EPSP (Calton et al., 2000). It is possible that by our stimulating protocol, when nifedipine or nimodipine blocked VDCCS, depotentiation was not induced because of insufficient calcium influx through NMDA receptors. Depotentiation occurs only when additional calcium entered through VDCCs that were evoked on top of NMDA depolarization.
In the lateral amygdala, LTP was dependent on NMDA receptors but not on VDCCs when it was induced by TS (Huang and Kandel, 1998; Lin et al., 2001). Conversely, LTP was dependent on VDCCs but not on NMDA receptors when induced by pairing presynaptic stimulation with postsynaptic depolarization (Weisskopf et al., 1999). Interestingly, in behavioral study, bilateral infusion of NMDA receptor antagonists into the LA impaired both short- and long-term memory of fear conditioning, whereas VDCC blockade selectively impaired long-term memory (Bauer et al., 2002). These results suggest the differential contribution of NMDA receptor and VDCC in different types of memory formation in the LA. It would be interesting for the future studies to determine the relative contribution of both types of Ca2+ influx in initiating second-messenger systems and/or gene expression that underlie memory extinction.
Quenching Effect.
Here we verify the hypothesis that depotentiation in the amygdala may weaken or extinguish memory performance by first showing that bilateral administration of LFS to the amygdala in vivo produced a profound effect on the expression of fear memory. Secondly, fear training-elicited Akt and MAPK phosphorylation was attenuated after quenching stimulation. This was paralleled by an increase in the enzymatic activity of calcineurin, suggesting that dephosphorylation of key proteins in the amygdala by serine/threonine phosphatase contributes to memory storage. Furthermore, converging evidence indicates that the AMPA receptor itself may be the critical substrate of calcineurin that mediates the expression of quenching extinction. Phosphorylation on Ser845 of the GluR1 subunit regulates the open-channel probability of AMPA receptors, whereas on Ser831, it increases the apparent single-channel conductance (Derkach et al., 1999; Banke et al., 2000). In this respect, it has been shown that LTD induction is associated with persistent dephosphorylation of GluR1 at Ser845 and dephosphorylation occurs at Ser831 when previously activated synapse is depotentiated (Lee et al., 2000). In addition, phosphorylation of GluR1 regulates synaptic trafficking of AMPA receptors and the enhancement of AMPA receptor endocytosis after LTD induction is blocked by calcineurin inhibitors (Beattie et al., 2000; Ehlers, 2000). Thus, LFS may initiate an influx of Ca2+ into cells through NMDA receptors or L-type Ca2+ channels (Huang and Kandel, 1998;Weisskopf et al., 1999), leading to the activation of calcineurin. Calcineurin induces dephosphorylation of GluR1, a process that decreases the synaptic function of AMPA receptors by altering channel properties and/or promoting the endocytosis. In conclusion, in a combination of behavioral, pharmacological, and biochemical experiments, we show for the first time that LFS elicits depotentiation in in vitro amygdala slices and reduces conditioned fear in whole animals; both effects seem to result from an increase in calcineurin activity.
Recent studies in human subjects demonstrated the relevance of fear conditioning to human fear and anxiety states. Gulf War veterans with post-traumatic stress disorder exhibit an exaggerated acoustic startle reflex (Morgan et al., 1996), suggesting that sensitized fear response is created by the stress of combat trauma. In the present study, we have characterized the molecular mechanisms underlying depotentiation in the amygdala and defined the parameters for quenching fear memory in an attempt to provide better pharmacological strategies for treating anxiety disorders.
Acknowledgments
We thank Drs. M. D. Lai and B. Mirasty for valuable discussion and comments on the manuscript.
Footnotes
- Received April 30, 2002.
- Accepted September 19, 2002.
-
This study was supported by National Science Council grant NSC89-2320-B006-011 and by Academic Excellence Program of the Ministry of Education grant 89-B-FA08-1-4, Taiwan, Republic of China.
Abbreviations
- LTP
- long-term potentiation
- LFS
- low-frequency stimulation
- PP
- protein phosphatase
- EC
- external capsule
- NMDA
- N-methyl-d-aspartate
- VDCC
- voltage-dependent calcium channel
- LA
- lateral nucleus of amygdala
- MAPK
- mitogen-activated protein kinase
- ACSF
- artificial cerebrospinal fluid
- CS
- conditioned stimulus
- US
- unconditioned stimulus
- ERK
- extracellular signal-regulated kinase
- BLA
- basolateral amygdala
- FK-506
- tacrolimus
- TS
- tetanic stimulation
- fEPSP
- field excitatory postsynaptic potential
- d-APV
- d-2-amino-5-phosphonovaleate
- MK-801
- dizocilpine maleate
- DPCPX
- 8-cyclopentyl-1,3-dipropylxanthine
- EGLU
- 2S-α-ethylglutamic acid
- mGluR II
- metabotropic glutamate group II receptor
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- GluR1
- glutamate receptor 1
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}