


Abstract
TAK-242 (resatorvid), a small-molecule–specific inhibitor of Toll-like receptor (TLR) 4 signaling, inhibits the production of lipopolysaccharide-induced inflammatory mediators by binding to the intracellular domain of TLR4. Cys747 in TLR4 has been identified previously as the binding site of TAK-242. However, the mechanism by which TAK-242 inhibits TLR4 signaling after binding to TLR4 remains unknown. The present study demonstrated, using coimmunoprecipitation, that TAK-242 interferes with protein-protein interactions between TLR4 and its adaptor molecules. Among 10 different human TLRs, TAK-242 selectively bound to TLR4. The time course of the inhibitory effect of TAK-242 on inflammatory mediator production corresponded to that of the binding of TAK-242 to TLR4. TAK-242 inhibited the association of TLR4 with Toll/interleukin-1 receptor domain-containing adaptor protein (TIRAP) or Toll/interleukin-1 receptor domain-containing adaptor protein inducing interferon-β-related adaptor molecule (TRAM) in human embryonic kidney (HEK) 293 cells overexpressing TLR4, MD-2, and TIRAP or TRAM, respectively. TAK-242 inhibited the TIRAP-mediated activation of nuclear factor κB (NF-κB) and the TRAM-mediated activation of NF-κB and interferon-sensitive response element in HEK293 cells stably expressing TLR4, MD-2, and CD14. The activation of endogenous interleukin-1 receptor-associated kinase in RAW264.7 cells was also inhibited by TAK-242 treatment. These findings suggest that TAK-242 binds selectively to TLR4 and subsequently disrupts the interaction of TLR4 with adaptor molecules, thereby inhibiting TLR4 signal transduction and its downstream signaling events. This work proposes a novel paradigm of a small molecule capable of disrupting protein-protein interactions.
Introduction
Toll, the initial member of the Toll-like receptor (TLR) family, was identified as a gene product essential for the development of embryonic dorsoventral polarity in Drosophila melanogaster. Later, it was also shown to play a critical role in the antifungal response of flies (Lemaitre et al., 1996). The immunological defense role of TLRs has been expanded in mammals to enable the broad recognition of microbial pathogens. In 1998, TLR4 was identified as the signaling receptor for lipopolysaccharide (LPS) or endotoxin from the outer membrane of Gram-negative bacteria (Poltorak et al., 1998). TLR4 is also required for the activation of proinflammatory cellular signaling pathways in response to endogenous molecules (Mollen et al., 2006). Accumulating evidence has implicated the activation or suppression of TLR4 in the development and progression of various inflammatory diseases (Schröder and Schumann, 2005; Gribar et al., 2008). Thus, TLR4 is an excellent therapeutic target for the treatment of inflammatory diseases. In fact, several small-molecule compounds that regulate TLR signaling have already been investigated (Bartfai et al., 2003; Lee et al., 2007).
To date, 11 TLRs have been identified in the human genome, and 13 TLRs have been identified in mammals. TLRs are type I integral membrane glycoproteins characterized by extracellular domains containing leucine-rich repeat motifs and a cytoplasmic domain homologous to that of the interleukin 1 (IL-1) receptor, termed the Toll/IL-1 receptor (TIR) domain (O'Neill and Dinarello, 2000). The TIR domain is also shared by the D. melanogaster Toll, IL-1 receptor accessory protein and cytoplasmic adaptors such as myeloid differentiation primary response gene 88 (MyD88), TIR domain-containing adaptor protein (TIRAP), TIR domain-containing adaptor protein inducing interferon-β (TRIF; also known as Toll/interleukin-1 receptor-containing adaptor molecule-1), TRIF-related adaptor molecule (TRAM; also known as Toll/interleukin-1 receptor-containing adaptor molecule-2), and sterile α- and armadillo-motif-containing protein (O'Neill and Bowie, 2007). TLR signaling is mediated through the TIR domain, the oligomerization of which initiates the recruitment of TIR domain-containing adaptor proteins. Homotypic interactions between receptor and the adaptor TIR domains are believed to mediate signal transduction. The subsequent kinase activation results in the induction of transcription factors such as nuclear factor-κB (NF-κB) and interferon regulatory factor followed by the production of cytokines and interferon for the immune response. Thus, the TIR domain plays a central role in TLR signaling.
TAK-242, a cyclohexene derivative, is a novel small-molecule compound that selectively inhibits TLR4 signaling (Ii et al., 2006; Kawamoto et al., 2008). We reported previously that TLR4 is indeed the target protein of TAK-242 and that TAK-242 binds to the TIR domain of TLR4 via Cys747 (Takashima et al., 2009). The TIR domain contains a central fully parallel five-strand β-sheet (βA through βE), five helices (αA through αE), and connecting loops (Xu et al., 2000; Khan et al., 2004). Sequence alignment predicts that in TLR4, Cys747 corresponds to a location in helix αC′ in the TIR domain (Xu et al., 2000; Fitzgerald et al., 2001). In the TIR domain of IL-1 receptor accessory protein-like, helix αC′ forms a part of the dimer interface (Khan et al., 2004). Likewise, Cys707 in helix αC′ in the TIR domain of TLR1 plays a critical role in TLR1/TLR1 homodimer formation (Khan et al., 2004). Furthermore, residues from αC-helix are contained in the dimer interaction surface in the TIR domain of TLR10 (Nyman et al., 2008). Therefore, the cysteine residue in helix αC or αC′ of the TIR domain is predicated to be involved in the dimer interface and is functionally relevant. Although the site to which TAK-242 binds is located in the dimer interface of the TIR domain as described above, TAK-242 did not affect the dimerization of TLR4 in a protein fragment complementation assay (Takashima et al., 2009). Therefore, the precise mode of action by which TAK-242 inhibits TLR4 signaling after binding to the receptor has remained uncertain. The present study attempted to clarify the molecular mechanism of TAK-242 for inhibiting TLR4 signal transduction. Our results described in this article suggest that TAK-242 inhibits TLR4 signaling by disrupting the interactions of TLR4 with its adaptor molecules.
Materials and Methods
Cells and Reagents.
Human embryonic kidney (HEK) 293 cells were purchased from Dainippon Sumitomo Pharma Co Ltd. (Osaka, Japan). HEK293 cells were cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum (FCS; TRACE Scientific, Melbourne, Australia), and 50 μg/ml gentamicin. HEK293 cells stably expressing TLR4, MD-2, and CD14 (HEK293-hTLR4/MD2-CD14) were purchased from InvivoGen (San Diego, CA). HEK293-hTLR4/MD2-CD14 cells were cultured in DMEM supplemented with 10% FCS, 10 μg/ml blasticidin, 50 μg/ml HygroGold, and 100 μg/ml Normocin (all from InvivoGen). Murine macrophage RAW264.7 cells were purchased from American Type Culture Collection (Manassas, VA). RAW264.7 cells were cultured in RPMI 1640 medium (Nikken Bio Medical Laboratories, Kyoto, Japan) containing 10% FCS and 10 μg/ml kanamycin at 37°C under a humidified atmosphere with 5% CO2.
Murine resident peritoneal macrophages were obtained by peritoneal lavage with ice-cold RPMI 1640 medium from 10-week-old pathogen-free female BALB/c mice (Charles River Japan Inc., Kanagawa, Japan). Cells were plated in a 96-well plate at a density of ∼5 × 105 cells/well in RPMI 1640 supplemented with 10% FCS and incubated for 2 h at 37°C in a humidified incubator containing 5% CO2. Then the cells were washed with RPMI 1640 containing 10% FCS to eliminate nonadherent cells. The animal experiments conducted in this study were approved by animal experiment ethics committee of Takeda Pharmaceutical Company Limited.
TAK-242 [resatorvid, ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate] (Fig. 1), was synthesized at Takeda Pharmaceutical Company Limited (Osaka, Japan). TAK-242 was dissolved in N,N-dimethylformamide. LPS (from Escherichia coli serotype O111:B4) was purchased from Sigma-Aldrich (St. Louis, MO). Recombinant mouse IFN-γ was purchased from Genzyme (Cambridge, MA). 3-[(4-Methylphenyl)sulfonyl]-(2E)-propenenitrile (BAY 11-7082) was purchased from Calbiochem (La Jolla, CA). BAY 11-7082 was dissolved in dimethyl sulfoxide.

Structure of TAK-242 (resatorvid).
Expression Vectors.
Expression vectors for FLAG-TLR4 and FLAG-TLR2 were cloned into pcDNA3.1Zeo (Invitrogen, Carlsbad, CA) and pFLAG-CMV-1 (Sigma-Aldrich), respectively. pUNO-hTLR1,3,5-10-hemagglutinin (HA) and pUNO-hMD2 were purchased from InvivoGen. pNifty-luc encoding a firefly luciferase-linked NF-κB reporter gene and phRL-TK encoding Renilla reniformis luciferase as an internal control were purchased from InvivoGen and Promega (Madison, WI), respectively. pISRE-TA-Luc encoding a firefly luciferase-linked interferon-sensitive response element (ISRE) reporter gene was purchased from Clontech (Mountain View, CA). Expression vectors for FLAG-TIRAP, FLAG-TRAM, and FLAG-TRIF were cloned into pFLAG-CMV-1. All the plasmids were prepared using the EndoFree Plasmid Maxi Kit (QIAGEN, Valencia, CA).
Measurement of Nitrite and Cytokine Concentrations in Culture Supernatants.
Resident mouse peritoneal macrophages were treated with TAK-242 and washed and then stimulated with 1 ng/ml LPS and 1 U/ml IFN-γ in RPMI 1640 medium containing 10% heat-inactivated FCS, 100 U/ml penicillin G, and 100 μg/ml streptomycin for 4 h (for TNF-α and IL-6 assay) or 20 h (for NO assay). Using 2,3-diaminonaphthalene (Dojindo Laboratories, Kumamoto, Japan), the production of NO was estimated by measuring the amount of nitrite, a stable metabolite of NO, using a fluorometric method (Misko et al., 1993). The concentrations of IL-6 and TNF-α in the culture supernatants were determined using the specific enzyme-linked immunosorbent assay kits (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK).
Radiolabeling of the Cells.
HEK293 cells were transiently transfected with expression vectors in a 10-cm dish (BD Biosciences Discovery Labware, Bedford, MA) using Lipofectamine or Lipofectamine LTX with Plus Reagents (Invitrogen).
Two days after transfection, the cells were incubated with [3H]TAK-242 (100 nM) at 37°C in DMEM supplemented with 1% FCS. Then, the cells were washed with ice-cold Dulbecco's phosphate-buffered saline (Invitrogen) and lysed with lysis buffer containing 10 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Nonidet P40, 0.05% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (Dojindo, Kumamoto, Japan), 30 mM sodium fluoride (NaF), 1 mM sodium orthovanadate (Na3VO4), and Protease inhibitor cocktail (Sigma-Aldrich) for 15 min on ice. The cell extracts were centrifuged at 12,000g for 10 min at 4°C, and the supernatant was collected as the total cell lysate.
Immunoprecipitation.
Immunoprecipitation was performed as described elsewhere (da Silva Correia and Ulevitch, 2002). In brief, total cell lysates were precleared three times for 20 min at 4°C with 60 μl of protein A Sepharose 6MB beads (GE Healthcare) and mixed with anti-FLAG M2 monoclonal antibody (mAb) (Sigma-Aldrich), anti-HA-tag mAb (InvivoGen), or anti-hTLR4 HTA125 mAb (IMGENEX, San Diego, CA) for 3 h at 4°C under constant agitation. The immune complexes were allowed to bind to 60 μl of protein A Sepharose 6MB beads at 4°C overnight, and the beads were washed three times with lysis buffer. The washed beads were resuspended in Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA) containing 5% 2-mercaptoethanol (Wako, Osaka, Japan) and boiled for 10 min. Samples were centrifuged, and the supernatants were obtained as immunoprecipitates.
Western Blot Analysis and Autoradiography.
The immunoprecipitates were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidene difluoride membranes (ATTO, Tokyo, Japan). After the membranes were blocked with Tris-buffered saline (50 mM Tris, 138 mM NaCl, and 2.7 mM KCl, pH 8.0) with 3% (w/v) nonfat milk (Sigma-Aldrich) at 4°C, the membranes were incubated with anti-FLAG M2 mAb, anti-HA-tag mAb, or anti-TIRAP FL-235 antibody (Ab) (Santa Cruz Biotechnology, Santa Cruz, CA) for 2 h and with horseradish peroxidase-conjugated anti-mouse IgG (GE Healthcare) or horseradish peroxidase-conjugated anti-rabbit IgG (GE Healthcare) for 1 h at room temperature. Specific bands were detected using enhanced chemiluminescence or ECL Advance Western blotting detection reagents (GE Healthcare). Chemiluminescent images were captured and digitized using LAS-3000 (Fujifilm, Tokyo, Japan). For autoradiography, the membranes were dried and the amount of bound 3H was revealed using BAS-2500 (Fujifilm).
Reporter Gene Assay.
HEK293 cells and HEK293-hTLR4/MD2-CD14 cells were seeded at a density of 2 to 3 × 104 cells/well in 96-well plates and incubated overnight. The cells were transfected with plasmids using Lipofectamine LTX with Plus Reagents (Invitrogen) according to manufacturer's instructions. For the NF-κB assays, each transfection contained 15 ng of pNiFty, 15 ng of phRL-TK as an internal control, the plasmid DNA of interest, and empty vector as filler DNA. For the ISRE assays, each transfection contained 30 ng of pISRE-TA-Luc, 1 ng of TRIF expression vector, 15 ng of phRL-TK as an internal control, the plasmid DNA of interest, and empty vector as filler DNA. To assess the constitutive activation of the reporter genes, the cells were treated with vehicle or test compounds 6 h after transfection and incubated overnight. The cells were lysed, and the luciferase activities were determined using Dual-Glo Luciferase Assay System (Promega). Data are presented as the mean ± S.E. of triplicate well readings, normalized to the activity of R. reniformis luciferase.
In Vitro Interleukin-1 Receptor-Associated Kinase-1 Kinase Assay.
Interleukin-1 receptor-associated kinase (IRAK)-1 kinase assay was essentially conducted as described previously (Nakayama et al., 2004) with a slight modification. The total cell lysates of RAW264.7 cells from a 10-cm dish (3–4 mg of total protein) were incubated with anti-IRAK-1 H-273 Ab (Santa Cruz Biotechnology) for 3 h at 4°C with rotation. Then, protein A Sepharose 6MB beads were added, and the samples were further incubated overnight at 4°C with rotation. The immunoprecipitated IRAK-1 complexes were washed four times with lysis buffer containing 50 mM HEPES, pH 7.5 (AppliChem, Darmstadt, Germany), 150 mM NaCl, 1 mM EDTA, 1% Nonidet P40, 20 mM β-glycerophosphate (Alfa Aesar, Ward Hill, MA), 1 mM Na3VO4, 1 mM NaF, 1 mM benzamidin (AppliChem), 5 mM para-nitrophenyl phosphate (Sigma-Aldrich), 1 mM dithiothreitol (Wako), 1 mM phenylmethylsulfonyl fluoride (MP Biomedicals, Solon, OH), and protease inhibitor cocktail (Sigma-Aldrich) and twice with kinase buffer (20 mM HEPES, pH 7.5, 20 mM MgCl2, 20 mM β-glycerophosphate, 20 mM para-nitro phenylphosphate, 1 mM EDTA, 1 mM Na3VO4, and 1 mM benzamidin). Twenty-five microliters of kinase buffer was then added to each tube; supplemented with 5 μM ATP, 1 μg of myelin basic protein (Wako), and 1 μl of [32P]ATP (6000 Ci/mmol; PerkinElmer Life and Analytical Sciences, Waltham, MA); and incubated at 37°C for 30 min. Ten microliters of Laemmli sample buffer was added, and the samples were subjected to SDS-PAGE analysis. The gel was dried, and the intensity of 32P was revealed using BAS-2500 (Fujifilm).
Results
TAK-242 Selectively Binds to TLR4.
TAK-242 has been shown previously to inhibit TLR4 signaling selectively in vivo and in vitro (Ii et al., 2006; Sha et al., 2007; Kawamoto et al., 2008). Although TAK-242 binds to TLR4 among TLRs 2, 3, 4, 5, and 9 (Takashima et al., 2009), binding activity of TAK-242 to TLRs 1, 6, 7, 8, and 10 has not been confirmed. To give further insights into the mechanism of action of TAK-242, we analyzed the binding of [3H]TAK-242 to human TLRs expressed in HEK293 cells. Among 10 human TLRs, TAK-242 selectively bound to TLR4 (Fig. 2).

Selective binding of [3H]TAK-242 to TLR4 among 10 human TLRs. HEK293 cells were transfected with expression vectors encoding FLAG-TLR2, FLAG-TLR4, TLR1-HA, TLR3-HA, and TLR5-10-HA. After 2 days of transfection, the cells were incubated with [3H]TAK-242 (100 nM) at 37°C for 6 h. After washing, the cells were lysed, and the cell lysates were immunoprecipitated with anti-FLAG M2 mAb or anti-HA-tag mAb. The immunoprecipitates were separated using SDS-PAGE and analyzed using immunoblotting with anti-FLAG M2 mAb or anti-HA-tag mAb. Radioactive images of the immunoprecipitates were analyzed using autoradiography.
Inhibitory Effect on Production of Inflammatory Mediators Corresponds to Binding of TAK-242 to TLR4.
TAK-242 has a quick onset of action and provides significant benefits in a mouse model of endotoxin shock (Sha et al., 2007). To clarify the characteristics of this quick action, we examined the time course of the inhibitory effect of TAK-242 on the production of inflammatory mediators in vitro. Peritoneal macrophages were pretreated with various concentrations of TAK-242, washed, and then stimulated with LPS and IFN-γ. TAK-242 inhibited the productions of NO and cytokines from the cells in a concentration-dependent manner. Pretreatment with TAK-242 (1 μM) for only 5 min showed a sufficient inhibitory effect on the production of TNF-α, IL-6, and NO (Fig. 3A). At lower concentrations, longer pretreatment increased the efficacy.

Comparison of the time courses of inhibitory effect on inflammatory mediator productions with that of binding to TLR4. A, time- and concentration-dependence of the inhibitory effect of TAK-242 on the productions of TNF-α, IL-6, and NO by mouse peritoneal macrophages stimulated with LPS and IFN-γ. Cells were incubated with TAK-242 at the indicated times, washed, and stimulated with LPS and IFN-γ. The concentrations of TNF-α and IL-6 in the culture supernatants were determined using specific enzyme-linked immunosorbent assay. The concentrations of nitrite in the culture supernatants were determined using a fluorometric method. B, time course of [3H]TAK-242 binding to TLR4. HEK293 cells were transfected with plasmids encoding FLAG-TLR4 and MD-2. Two days after the transfection, the cells were incubated with [3H]TAK-242 (100 nM) at 37°C. Total cell lysates were immunoprecipitated with anti-hTLR4 HTA125 mAb and subjected to immunoblotting using anti-FLAG M2 mAb. Radioactive images were revealed using autoradiography.
To determine whether the onset of the inhibitory effect of TAK-242 is correlated with its binding to TLR4, we investigated the time course of [3H]TAK-242 (100 nM) binding to TLR4. The binding experiment in HEK293 cells overexpressing TLR4/MD-2 demonstrated rapid [3H]TAK-242-TLR4 complex formation at as early as 1 min, followed by a subsequent increase thereafter (Fig. 3B).
TAK-242 Affects Associations of TIRAP and TRAM with TLR4.
We hypothesized that TAK-242 might interfere with protein-protein interactions between TLR4 and its adaptor molecules. Because TIRAP and TRAM directly interact with TLR4 and serve as a bridge for the recruitment of MyD88 and TRIF, respectively (O'Neill and Bowie, 2007), we tested the interactions between TLR4 and TIRAP/TRAM in cells using coimmunoprecipitation studies. HEK293 cells were transfected with plasmids encoding FLAG-TLR4, MD-2, and FLAG-TIRAP/ FLAG-TRAM and immunoprecipitated with anti-hTLR4 HTA125 mAb. As shown in Fig. 4A, FLAG-TIRAP was detected in the anti-hTLR4 HTA125 immunoprecipitated complex, in agreement with the results of previous studies (Fitzgerald et al., 2001). Treatment with TAK-242 inhibited the coprecipitation of TIRAP with TLR4 in a concentration-dependent manner (Fig. 4A). Likewise, cotransfected TRAM was detected in the immunoprecipitates, consistent with the results of previous reports (Fitzgerald et al., 2003; Oshiumi et al., 2003). TAK-242 inhibited the association of TRAM with TLR4 at concentrations similar to those at which it inhibited the association of TIRAP with TLR4 (Fig. 4B).

Inhibitory effect of TAK-242 on coimmunoprecipitation of adaptor molecules with TLR4. HEK293 cells were transfected with expression vectors encoding FLAG-TLR4, MD-2, and FLAG-TIRAP (A) or expression vectors encoding FLAG-TLR4, MD-2, and FLAG-TRAM (B). Six hours after transfection, the cells were incubated with various concentrations of TAK-242 at 37°C. The following day, the cells were washed and lysed. The cell lysates were immunoprecipitated with anti-hTLR4 HTA125 mAb. Total cell lysates (TCL) and immunoprecipitates (IP) were separated using SDS-PAGE and analyzed using immunoblotting (IB).
TAK-242 Inhibits NF-κB and ISRE Activation Mediated by Adaptor Molecules.
To test the functional consequences of the impaired interaction of the adaptor molecules TIRAP and TRAM with TLR4, the effects of TAK-242 on TIRAP- or TRAM-mediated NF-κB and ISRE reporter gene activation were investigated. First, we examined the influence of TIRAP/TRAM overexpression on NF-κB/ISRE activation. TIRAP overexpression in HEK293 cells constitutively activated NF-κB but not the ISRE reporter gene. TRAM overexpression constitutively activated both NF-κB and ISRE (Supplemental Fig. S1, top). Likewise, TIRAP overexpression in HEK293-hTLR4/MD2-CD14 cells constitutively activated NF-κB but not the ISRE reporter gene, whereas TRAM overexpression constitutively activated both NF-κB and ISRE, although the NF-κB activation was less marked than that mediated by TIRAP overexpression (Supplemental Fig. S1, bottom). Each effect was dependent on the concentration of the plasmid that was used. The activations of NF-κB and ISRE in HEK293 cells that are deficient in TLR4 expression (Latz et al., 2002) were approximately 10- and 5-fold less potent than those in HEK293-hTLR4/MD2-CD14 cells, respectively (Supplemental Fig. S1). In consideration of these results, we tested the effect of TAK-242 on TIRAP-mediated NF-κB activation and TRAM-mediated NF-κB and ISRE activation. TAK-242 did not inhibit TIRAP-mediated NF-κB activation or TRAM-mediated NF-κB and ISRE activation in intact HEK293 cells (Fig. 5, top). In HEK293-hTLR4/MD2-CD14 cells, in contrast, TAK-242 inhibited TIRAP-mediated NF-κB activation and TRAM-mediated NF-κB and ISRE activation in a concentration-dependent manner (Fig. 5, bottom).

Inhibitory effect of TAK-242 on NF-κB and ISRE activation mediated TIRAP/TRAM overexpression. For the NF-κB assay, the cells were transfected with 10 ng of TIRAP or TRAM expression vector, 15 ng of NF-κB-Luc plasmid pNiFty, and 15 ng of plasmid phRL-TK. For the ISRE assay, the cells were transfected with 10 ng of TRAM expression vector, 30 ng of ISRE-Luc plasmid, 1 ng of TRIF expression vector, and 15 ng of plasmid phRL-TK. Six hours after transfection, the cells were treated with TAK-242 and incubated overnight. Then, the cells were lysed, and the luciferase activities were measured. The results are shown as the mean ± S.E. Determinations were made in triplicate.
TAK-242 Has a Unique Molecular Mechanism of Action.
To demonstrate its unique mode of action, we compared the inhibitory activity of TAK-242 with other small-molecule inhibitors that would modulate TLR4 signaling pathway. Despite our efforts to find a small molecule of inhibiting NF-κB and ISRE activations, few compounds showed inhibitory activity without affecting cell viability in the examination using HEK293-hTLR4/MD2-CD14 cells with overexpression of TIRAP/TRAM. Under this condition, a pharmacological NF-κB inhibitor BAY 11-7082 inhibited TIRAP- and TRAM-mediated NF-κB activation but did not inhibit TRAM-mediated ISRE activation (Fig. 6A). Unlike TAK-242, BAY11-7082 did not affect the association of TIRAP with TLR4 (Fig. 6B) or the association of TRAM with TLR4 (Fig. 6C).

Comparison of TAK-242 with BAY 11-7082. A, inhibitory effect of BAY 11-7082 on NF-κB activation mediated TIRAP/TRAM overexpression. The reporter assay protocol was as described in Fig. 5. B and C, influence of BAY 11-7082 on coimmunoprecipitation of adaptor molecules with TLR4. HEK293 cells were transfected with expression vectors encoding FLAG-TLR4, MD-2, and FLAG-TIRAP (B), or expression vectors encoding FLAG-TLR4, MD-2, and FLAG-TRAM (C). Six hours after the transfection, the cells were incubated with TAK-242 (1 μM) or BAY 11-7082 (3 μM) at 37°C. The following day, the cells were washed and processed as described in the legend to Fig. 4.
TAK-242 Inhibits LPS-Induced Activation of Endogenous IRAK-1.
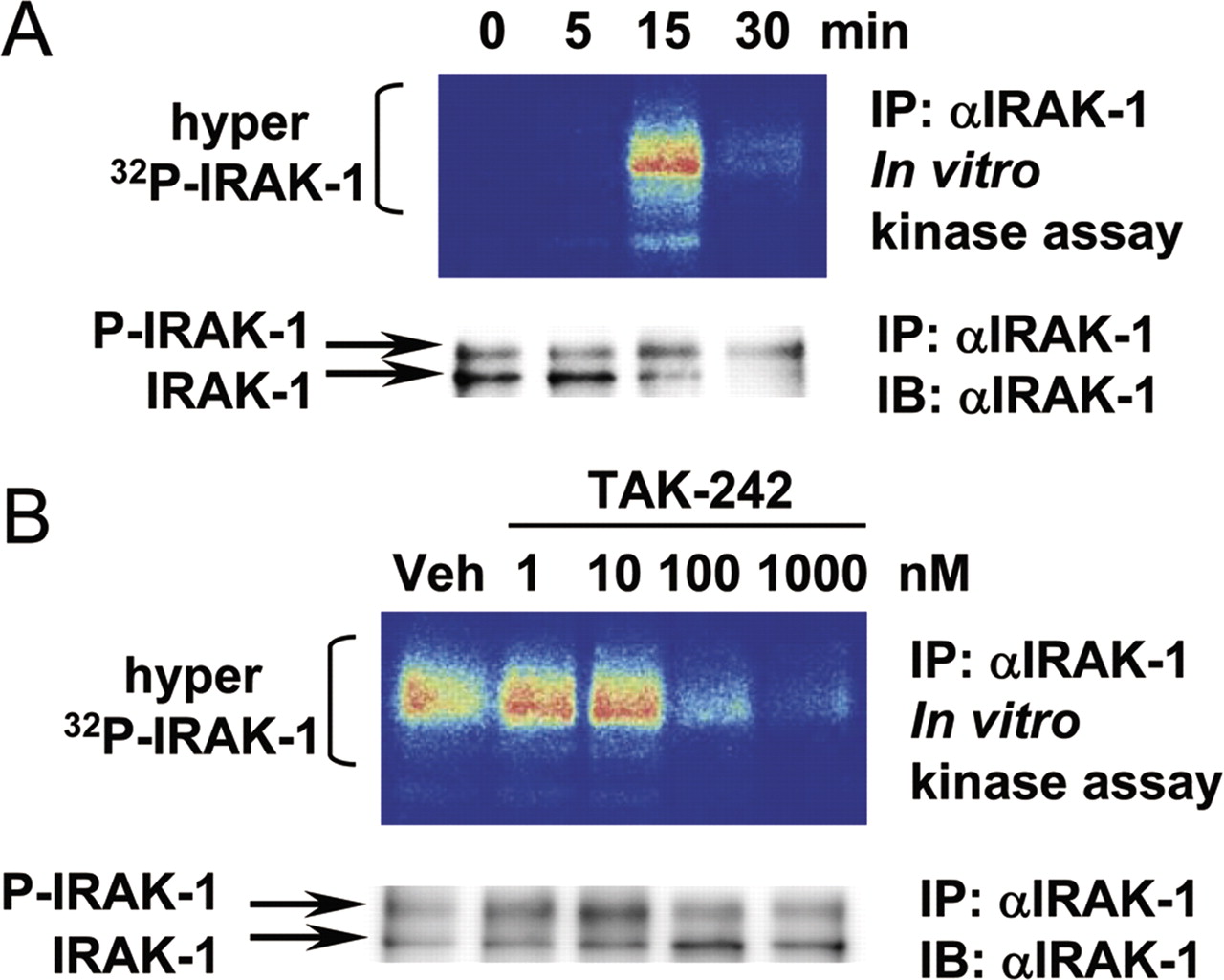
As further proof of the effect of TAK-242 on early events in TLR4 signaling, we examined the endogenous IRAK-1 response in RAW264.7 cells after LPS stimulation using in vitro kinase assay. IRAK-1 acts downstream of TIRAP/MyD88 in the MyD88-dependent pathway (Medvedev et al., 2002). LPS increased IRAK-1 activation and resulted in the hyperphosphorylation of IRAK-1, reflected by a shift in the apparent molecular mass from approximately 80 kDa to more than 100 kDa. IRAK-1 phosphorylation was markedly up-regulated at 15 min and decreased at 30 min after stimulation (Fig. 7A, top). The degradation of IRAK-1 was confirmed using immunoblotting at 30 min after LPS stimulation (Fig. 7A, bottom). We observed a concentration-dependent suppression of IRAK-1 phosphorylation by TAK-242 at 15 min after LPS stimulation in RAW264.7 cells (Fig. 7B).

Inhibitory effect of TAK-242 on LPS-induced activation of endogenous IRAK-1 in RAW264.7 cells. A, kinetic analysis of the phosphorylation of IRAK-1 upon LPS stimulation. The cells were stimulated with LPS (100 ng/ml) for various amounts of time. B, concentration-dependence of the inhibitory effect of TAK-242 on the activation of IRAK-1. The cells were pretreated with various concentrations of TAK-242 for 15 min. Then, the cells were stimulated with LPS (100 ng/ml) for 15 min. IRAK-1 proteins were immunoprecipitated (IP) using anti-IRAK-1 H-273 Ab. IRAK-1 kinase activities were determined using an in vitro kinase assay as described under Materials and Methods. The immunoprecipitated IRAK-1 protein levels were analyzed using immunoblotting (IB).
Discussion
We have demonstrated for the first time that a small-molecule compound TAK-242 disrupts the interactions of TLR4 with its adaptor molecules, TIRAP and TRAM. These findings helped to clarify the mechanism underlying TLR4 inhibition by TAK-242 after binding to TLR4.
Because TAK-242 exhibited a specific binding to TLR4 among the 10 TLRs (Fig. 2), the binding region of TAK-242 and its vicinity in the TIR domain of TLR4 might have a distinctive shape among other TLRs. This idea is supported by observations that TLR4 uses bridging adaptors, TIRAP and TRAM, unlike other TLRs. The binding selectivity of TAK-242 to TLR4 shown in Fig. 2 is consistent with the results reported in previous functional assays (Ii et al., 2006; Sha et al., 2007; Kawamoto et al., 2008).
To gain additional information about the mode of action of TAK-242, kinetic analysis of the production of inflammatory mediators and [3H]TAK-242 binding was performed. In the pretreatment experiments, TAK-242 exhibited a time- and concentration-dependent inhibition of the LPS-induced production of inflammatory mediators. Pretreatment for only 5 min at a concentration of 1 μM significantly inhibited the production of inflammatory mediators (Fig. 3A), and the binding of [3H]TAK-242 to TLR4 was observed at as early as 1 min (Fig. 3B). These results suggest that the inhibitory effect of TAK-242 may result from the direct action of TAK-242 on TLR4 without requiring the induction of intermediate gene expression or de novo protein synthesis.
TAK-242 binds directly to TLR4 via Cys747 in the TIR domain of TLR4 (Takashima et al., 2009). TAK-242 contains an α, β-unsaturated carbonyl group that functions as a Michael acceptor and reacts with biological nucleophiles, such as the sulfhydryl group on cysteine residues. With this chemical feature in mind, we presumed that TAK-242 reacts with TLR4 to form a complex containing a covalently attached cyclohexene ring at position 747. In the case of human TLR2, conformational differences exist between wild type and a C713S mutant, in which the mutation exists within the dimer interface (Tao et al., 2002). Because TAK-242 binds to the equivalent cysteine residue in TLR4, the binding of TAK-242 may cause a conformational change in the TIR domain of TLR4, subsequently affecting the functional recruitments of the adaptor molecules and kinases to the signaling complex.
TLR4 signals via two distinct sets of adaptor molecules, TIRAP and MyD88, and TRAM and TRIF. These adaptor molecules are allowed to recruit and form a postreceptor signaling complex after the dimerization of TLR4. Moreover, TIRAP and TRAM are predicted to bind at the same site of the TLR4 dimer interface (Núñez Miguel et al., 2007). Because Cys747, the binding site of TAK-242, is predicted to be located at the dimer interface, we hypothesized that TAK-242 may affect the associations of TIRAP/TRAM with TLR4. Coimmunoprecipitation studies revealed that TAK-242 inhibited the association of TIRAP with TLR4 and the association of TRAM with TLR4 (Fig. 4). Although it is not clear whether the binding of TIRAP and TRAM is mutually exclusive or whether a single activated TLR4 dimer can recruit both adaptor proteins, the disruption of any one of these interactions may affect the other protein-protein interactions that are involved. These observations provide strong evidence that TAK-242 modulates the formation of the signaling complex containing the proximal elements in TLR4 signaling.
TIRAP is required for the rapid activation of NF-κB in a MyD88-dependent signaling pathway (Fitzgerald et al., 2001; Horng et al., 2001). On the other hand, TRAM stimulates a TRIF-dependent pathway, leading to the activation of interferon response factor-3 and the late activation of NF-κB (Fitzgerald et al., 2003; Oshiumi et al., 2003). Because interferon response factor-3 binds to ISRE, we used an ISRE reporter system (Wietek et al., 2003) to examine the effect of TAK-242 on the TRIF-dependent pathway. NF-κB and ISRE were markedly activated upon TIRAP and TRAM overexpressions in HEK293-hTLR4/MD2-CD14 cells compared with HEK293 cells (Supplemental Fig. S1), suggesting that the interaction of these adaptors with TLR4/MD-2 is critical for TLR4 signaling and the activation of downstream genes. TLR4 along with two other proteins, MD-2 and CD14, are widely known to be capable of detecting the presence of LPS and activating the downstream signaling pathway (Triantafilou and Triantafilou, 2002). TAK-242 inhibited TIRAP-mediated NF-κB activation and TRAM-mediated NF-κB and ISRE activation in the presence of TLR4/MD-2 (Fig. 5, bottom). In addition, TAK-242 also inhibited LPS-induced NF-κB and ISRE activations in HEK293-hTLR4/MD2-CD14 cells (Supplemental Fig. S2). These results support our observation that TAK-242 impairs the ability of TLR4 to associate with adaptor molecules and blocks subsequent signal transduction.
To our knowledge, no small-molecule inhibitors of protein-protein interactions of TIR domain of TLR4 have yet been identified except TAK-242. To confirm this, we sought to find other small-molecule inhibitors that block TIRAP/TRAM-induced NF-κB/ISRE activation in HEK293-hTLR4/MD2-CD14 cells. An NF-κB inhibitor, BAY 11-7082, which inhibits IκB kinase activity to prevent the phosphorylation and subsequent degradation of IκB (Pierce et al., 1997) was found to inhibit TIRAP/TRAM-induced NF-κB activation (Fig. 6A). Luteolin (Lee et al., 2009) and isoliquiritigenin (Park and Youn, 2010) have been reported to inhibit TRIF-dependent signaling pathway; however, these flavonoids did not inhibit TRAM-induced ISRE activation in HEK293-hTLR4/MD2-CD14 cells without affecting cell viability under overnight culture condition (data not shown). We could hardly find other inhibitors that affect TIRAP/TRAM-induced NF-κB/ISRE activation in HEK293-hTLR4/MD2-CD14 cells despite our evaluations of inhibitors including IRAK-1/4 inhibitor 1-(2-(4-morpholinyl)ethyl)-2-(3-nitrobenzoylamino)benzimidazole, p38 MAPK inhibitor 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole (SB203580), MEK1/2 inhibitor 1,4-diamino-2,3-dicyano-1,4-bis(methylthio)butadiene (U0126), and JNK inhibitor 1,9-pyrazoloanthrone (SP600125). Differently from TAK-242, BAY 11-7082 did not appreciably affect the association of TIRAP with TLR4 and the association of TRAM with TLR4, although it inhibited both TIRAP- and TRAM-mediated NF-κB activations (Fig. 6). These results suggest that inhibition of IκB phosphorylation does not result in inhibition of TLR4-adaptor associations and that TAK-242 directly interferes with the associations of adaptor molecules.
The TIR domain of activated TLR4 forms a scaffold for the recruitment of downstream signaling components (Laird et al., 2009; Motshwene et al., 2009). LPS stimulation triggers the recruitment of TIRAP/MyD88 to activated TLR4. This action is then followed by the recruitment of the serine/threonine kinases IRAK-1 and IRAK-4, resulting in the phosphorylation of IRAK-1 both by autophosphorylation and by IRAK-4 (Burns et al., 2003; Kollewe et al., 2004). Using an in vitro kinase assay, endogenous IRAK-1 was found to be activated in response to LPS in RAW264.7 cells (Fig. 7A). TAK-242 inhibited the phosphorylation of IRAK-1 (Fig. 7B), suggesting that the scaffold upon which IRAK-1 functions to initiate the LPS responses cannot be correctly formed.
A schematic illustrations of the TLR4 signaling is shown in Fig. 8A, and the mode of action of TAK-242 is shown in Fig. 8B. TAK-242 interferes with the interaction of TIRAP and TLR4, impairing IRAK-1 activation and in turn inhibiting NF-κB activation and cytokine gene expression in response to LPS. TAK-242 also interferes with the interaction of TRAM and TLR4, inhibiting both NF-κB and ISRE activation and cytokine/interferon gene expression. The observation that a small-molecule compound is capable of regulating protein-protein interactions is noteworthy. Our findings may open up new possibilities of small molecules in developing pharmacological strategies for disrupting protein-protein interactions. However, to fully understand the physical basis whereby TAK-242 disturbs signaling complex formation and intracellular signal transduction, a crystal structure analysis of the TLR4-TAK-242 complex is needed.

Schematic illustration of the TLR4 signaling pathway (A) and mechanism of action of TAK-242 (B). TLR4 signaling can be divided into two distinct signaling pathways, namely MyD88- and TRIF-dependent pathway. TAK-242 interferes with interactions between TLR4 and its adaptor molecules, TIRAP and TRAM.
Authorship Contributions
Participated in research design: Matsunaga, Tsuchimori, Matsumoto, and Ii.
Conducted experiments: Matsunaga and Ii.
Contributed new reagents or analytic tools: Matsunaga.
Performed data analysis: Matsunaga and Ii.
Wrote or contributed to the writing of the manuscript: Matsunaga, Tsuchimori, Matsumoto, and Ii.
Acknowledgments
We thank Dr. K. Takashima and Dr. T. Kawamoto for providing expression vectors. Valuable discussions with Dr. K. Okonogi, Dr. Y. Iizawa, Dr. T. Kitazaki, and Dr. H. Odaka are greatly appreciated. We are grateful to Dr. H. Nagaya for helpful discussions during the preparation of the manuscript.
Footnotes
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.068064.
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- TLR
- Toll-like receptor
- LPS
- lipopolysaccharide
- IL
- interleukin
- IFN
- interferon
- TNF
- tumor necrosis factor
- TIR
- Toll/interleukin-1 receptor
- MyD88
- myeloid differentiation primary response gene 88
- TIRAP
- Toll/interleukin-1 receptor domain-containing adaptor protein
- TRIF
- Toll/interleukin-1 receptor domain-containing adaptor protein inducing interferon-β
- TRAM
- Toll/interleukin-1 receptor domain-containing adaptor protein inducing interferon-β-related adaptor molecule
- IRAK
- interleukin-1 receptor-associated kinase
- HEK
- human embryonic kidney
- TAK-242 (resatorvid)
- ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate
- NF-κB
- nuclear factor-κB
- ISRE
- interferon-sensitive response element
- Ab
- antibody
- mAb
- monoclonal antibody
- PAGE
- polyacrylamide gel electrophoresis
- HA
- hemagglutinin
- FCS
- fetal calf serum
- DMEM
- Dulbecco's modified Eagle's medium
- TK
- thymidine kinase
- BAY 11-7082
- 3-[(4-methylphenyl)sulfonyl]-(2E)-propenenitrile
- SB203580
- 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole
- U0126
- 1,4-diamino-2,3-dicyano-1,4-bis(methylthio)butadiene
- SP600125
- 1,9-pyrazoloanthrone.

- Received August 9, 2010.
- Accepted September 29, 2010.
- Copyright © 2011 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}