


Abstract
Toxins isolated from scorpion, snake, and spider venoms are valuable tools to probe the physiologic function and structure of ion channels. In this study, we have isolated three new toxins (heteropodatoxins) from the venom of a spider, Heteropoda venatoria. These toxins are structurally similar peptides of 29 to 32 amino acids and share sequence homology with hanatoxins isolated from the venom of a Chilean tarantula. The heteropodatoxins prolonged the action-potential duration of isolated rat ventricular myocytes, suggesting that the peptides block K+ currents. The effect of toxins on cardiac K+ currents were studied using voltage clamp techniques. The toxins blocked the transient outward K+ current but not other K+ currents in isolated rat cardiac myocytes. The mechanism of block was studied further using Kv4.2, a cloned channel believed to underlie transient outward K+ current in rat myocytes. The toxins blocked Kv4.2 current expressed in Xenopus laevis oocytes in a voltage-dependent manner, with less block at more positive potentials. In addition, the toxins slowed the time course of current activation and inactivation and shifted the voltage dependence of current inactivation to more positive potentials. The heteropodatoxins represent new pharmacologic probes to study the role of Kv4.2 channels in cardiac and neural tissue.
Voltage-sensitive K+ channels modulate excitability and synaptic transmission of neurons and repolarization of cardiac myocytes. A large number of K+ channels have been cloned recently, indicating the great diversity of these channels compared with other types of voltage-sensitive channels. The study of their physiologic roles has been aided greatly by the discovery of specific blockers. For example, charybdotoxin isolated from venom of the scorpion, Leiurus quinquestriatus, was used to define the function of large conductance Ca2+-activated K+ channels (1), and the structural determinants of function in Shaker-like K+ channels (2-4). Potassium channel toxins also have been used to distinguish the role of a particular channel type to net current resulting from the overlap of many different types of channels (5, 6) and between different subtypes of a single class of K+ channels (7).
Transient outward K+ (Ito) channels are characterized by rapid activation and inactivation. These K+ channels are important in modulation of action-potential configuration and neuronal firing patterns. In the heart, activation of Ito channels initiates rapid repolarization of the action potential and is important in regional- and rate-dependent differences in action-potential duration (8). Ito channels are blocked by 4-aminopyridine (9), but this compound also blocks other types of K+ channels (10). Specific blockers of Itochannels have not been described.
Two different Ito channels have been cloned from cardiac tissue. Kv1.4 (Shaker subfamily) and Kv4.2 (Shalsubfamily) were cloned from heart cDNA libraries, their mRNAs were detected in Northern blots of heart tissue, and have properties similar to voltage-dependent cardiac Ito (Ito1) when studied in heterologous expression systems (11-13). However, Kv4.2, but not Kv1.4, was detectable in isolated rat myocytes using immunohistochemical techniques (14). These results suggest that Kv4.2 subunits form functional Ito1 channels in rat heart muscle and that Kv1.4 forms Ito channels in other tissues (e.g., neural or smooth muscle) within the heart. Specific blockers of one or both of these channel types would be useful in confirming that Kv4.2 subunits form Ito1 channels in cardiac myocytes.
In this study, we describe three peptide toxins isolated from the venom of a spider, Heteropoda venatoria, that prolong cardiac action potentials and block transient outward K+ current in rat ventricular myocytes. These peptides (heteropodatoxins) were found to block cloned Kv4.2 (but not Kv1.4) K+ channels expressed in X. laevis oocytes, consistent with the hypothesis that Kv4.2 forms Ito1 channels in rat heart. The heteropodatoxins will be useful for probing the physiologic role of Kv4.2 K+ channels and defining the structural basis of Kv4.2 channel function.
Materials and Methods
Isolation/purification of active components from whole venom.
H. venatoria Linneaus (Araneae, Heteropodidae) were collected from Malaysia and maintained in the vivarium at NPS Pharmaceuticals (Salt Lake City, UT). Whole venom was obtained by electrical stimulation of the cephalothorax of live spiders after anesthetization with CO2. Whole venom was stored at −80° until used for fractionation. Aliquots of venom (75–120 μl) were diluted to 1 ml with 0.1% aqueous TFA (solution A). The diluted venom was fractionated on a Vydac C-18 reversed-phase HPLC column (10 × 250 mm) equilibrated in 80% A/20% B (0.1% TFA in CH3CN). After 3 min, the gradient was changed to 24% B over 1 min; at 5 min, a linear gradient from 24 to 35% B over 44 min was begun. The flow rate was 3.5 ml/min and the UV absorbance of the effluent was monitored at 220 nm (Fig. 2A). Fractions were collected and like fractions from the eight chromatographic runs were combined, concentrated by lyophilization, and stored at −80°.

Reversed-phase HPLC chromatogram of H. venatoria venom and sequence of HpTxs. A, Venom (120 μl) was dissolved in 0.1% TFA (aq) and applied to a Vydac C-18 column as described in Materials and Methods. UV absorbance of the effluent was monitored at 220 nm. Fractions were collected as noted on the chromatogram. B, Sequences of HpTxs and comparison to hanatoxin2 (22).
Further purification of fractions 5, 6, and 7 was performed using a HEMA-IEC BIO SB cation-exchange column (10 μm, 4.6 × 150 cm). The lyophilized material was dissolved in 50 mm sodium acetate, pH 4.0, and loaded onto the column. The following gradients were used for elution of the toxins. For reversed-phase fraction 5, which contains HpTx1, the column was developed with a 45-min linear gradient from 0.1 to 1 m NaCl in 50 mm sodium acetate, pH 4.0. HpTx1 eluted between 27 and 34 min. For reversed-phase fraction 6, which contains HpTx2, the column was developed with a 50-min linear gradient from 0 to 0.5 m NaCl in 50 mm sodium acetate. HpTx2 eluted between 27 and 29 min. For reversed-phase fraction 7, which contains HpTx3, the column was developed with a 4-min linear gradient from 0 to 0.3 mNaCl, followed by a 35-min linear gradient from 0.3 to 1.0m NaCl in 50 mm sodium acetate, pH 4.0. HpTx3 eluted between 30 and 33 min. For each of these chromatographies, the flow rate was 1 ml/min and the effluent was monitored at 280 nm.
The cation-exchange fractions containing HpTx1, HpTx2, and HpTx3 were desalted on a semipreparative Vydac C18 reversed-phase column (10 × 250 mm, 300 Å) equilibrated in 0.1% aqueous TFA (solution A). After loading a cation-exchange fraction onto the column, the column was developed with a 10-min linear gradient from 0 to 25% B (0.1% TFA in acetonitrile), followed by a 20-min linear gradient from 25 to 35% B. The flow rate was 3.5 ml/min and the absorbance of the effluent was monitored at 220 nm. HpTx1 eluted between 24 and 25.5 min, HpTx2eluted between 26 and 28 min, and HpTx3 eluted between 27 and 29 min. The lyophilized fractions were submitted for amino-terminal sequence and mass spectral analysis.
Mass determination and peptide sequencing.
A triple-quadrupole mass spectrometer with an ion-spray interface (SCIEX API III system; Perkin-Elmer, Thornhill, Ontario) was used for mass determination (15). The sample was dissolved in 10% acetic acid and the sample solution (0.2 mg/ml) was delivered at a flow rate of 1.0 μl/min to a sprayer by a syringe infusion pump. The molecular mass of the sample was determined with the first quadrupole, which was calibrated with the ammonium adduct ions of polypropylene glycols.
PTC amino acid analysis was performed on 1–10 nmol of each peptide (in triplicate) with a Waters Pico-Tag system (Waters, Milford, MA). Amino-terminal sequencing was carried out on a pulse-liquid sequenator (Applied Biosystems, Foster City, CA) on both native andS-pyridylethylated derivatives.S-pyridylethylated peptides were generated in situ according to the method of Kruft (16).
Isolation of cardiac myocytes.
Adult rats of either sex (200–300 g) were anesthetized with sodium pentobarbital (200 mg/kg, intraperitoneal), then killed by cervical dislocation. The heart was excised rapidly and the aorta was cannulated to an apparatus for retrograde perfusion with oxygenated solutions at 35–36°. The method used to isolate single ventricular myocytes was the same as described previously (17), except that 150 U/ml of collagenase type II (Worthington Biochemicals, Freehold, NJ) and 0.5 units/ml protease type XIV (Sigma Chemical, St. Louis, MO) were used. Myocytes were stored at room temperature in standard extracellular solution before use in voltage clamp experiments.
Whole-cell voltage clamp data acquisition and analysis.
The suction microelectrode voltage clamp technique described by Giles and Shibata (18) was used to record currents in single rat ventricular myocytes. Pipettes were fabricated from 1-mm outer-diameter square-bore borosilicate glass (Frederick and Dimmock, Millville, NJ) and had resistances of 3–6 MΩ when filled with an intracellular solution that consisted of 0.5 m potassium gluconate and 25 mm KCl, pH 7.3. Series resistance was compensated by at least 80%. The limitations (high series resistance, loading of cells with K+ during prolonged recordings) and advantages (no immediate alteration of intracellular contents, ease of seal formation) of using these electrodes and filling solutions has been described previously (19). Command voltage pulses were generated using pClamp software (Axon Instruments, Burlingame, CA), a TL-1 interface (Axon Instruments) connected to a 486DX/50 MHz desktop computer, and an Axoclamp 1D amplifier (Axon Instruments). Currents were low-pass filtered with a cutoff frequency (−3 db) of 1 or 2 kHz.
Rat ventricular myocytes were bathed in a nominally Ca2+-free solution at room temperature. The standard extracellular solution contained 132 mm NaCl, 4 mm KCl, 1 mm CoCl2, 1.2 mm MgCl2, 5 mm HEPES, and 5 mm glucose. The pH was adjusted to 7.2 with NaOH. Currents were elicited with stepped pulses from a holding potential of −60 mV. Each test pulse was preceded by a 20-msec pulse to −30 mV to inactivate INa. To determine the current-voltage relationship for Ito1, pulses were applied every 8 sec in 10-mV increments to test potentials ranging from −40 to +40 mV. This rate of pulsing was sufficiently slow to allow complete recovery of Ito1 from inactivation at −60 mV between successive test pulses. The magnitude of Ito1 current was defined as time-dependent current during the 500-msec test pulse. The sustained current (Isus) remaining at the end of the pulse represents a small leak component and a much larger outward K+ current (20). The voltage dependence of Ito1 activation was determined from the amplitude of tail currents after 20-msec test pulses. Data was fit with a Boltzmann relation: Ito/Ito-max = 1/(1 + exp[(V1/2 − Vt)/k]), to estimate the average half-point (V1/2) and slope factor (k) for this relationship.
Isolation of oocytes and injection of RNA.
X. laevis frogs were anesthetized by immersion in 0.2% tricaine for 15–30 min. Ovarian lobes were digested with 2 mg/ml Type 1A collagenase (Sigma Chemical) in Ca2+-free ND96 solution for 1.5 hr to remove follicle cells. Stage IV and V oocytes (21) were injected with Kv4.2 or Kv1.4 cRNA (0.05 mg/ml, 50 nl) and then cultured in Barth’s solution supplemented with 50 μg/ml gentamycin and 1 mm pyruvate at 18°. Barth’s solution contained 88 mm NaCl, 1 mm KCl, 0.4 mmCaCl2, 0.33 mmCa(NO3)2, 1 mm MgSO4, 2.4 mm NaHCO3, 10 mm HEPES, pH 7.4.
Plasmids containing Kv1.4 and Kv4.2 cDNA were kindly provided by Dr. Mike Tamkun (12, 13). Templates for cRNA synthesis from channel DNA were prepared as described by Po et al. (13).
Two-microelectrode voltage clamp of oocytes.
Oocytes were bathed in ND96 solution. This solution contained 96 mmNaCl, 2 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2, 5 mm HEPES, pH 7.6. Currents were recorded at room temperature (21–23°) using standard two-microelectrode voltage clamp techniques. Glass microelectrodes were filled with 3 m KCl and their tips were broken to obtain tip resistances of 1–3 mΩ for the voltage-recording electrode and 0.6–1 mΩ for the current-passing electrode. Oocytes were voltage clamped with an Axoclamp 2A amplifier (Axon Instruments). Voltage commands were generated using pCLAMP software (version 5.5; Axon Instruments), a 486DX2 personal computer, and a Digidata 1200 D/A interface (Axon Instruments). Current signals were sampled digitally at a rate equal to 2–4 times the low-pass cutoff frequency (−3 db) of an 8-pole Bessel filter. The oocyte membrane potential was maintained at −70 mV between test pulses, applied at a rate of 1–3 pulses/min.
The time course of Kv4.2 inactivation was fit with a single exponential relationship: I(t) = A0 + A1e(−t/τ), using a Chebyshev noniterative fitting technique (pCLAMP). The voltage dependence of Kv4.2 inactivation was determined using 8-sec conditioning test pulses applied once every 30 sec. Each conditioning pulse was applied to a potential ranging from −120 to 0 mV and was followed by a pulse to +40 or +75 mV to monitor the extent of channel inactivation.
Data analyses, including exponential fitting of current traces, were performed using pCLAMP. Fits of appropriate data to a Boltzmann function or Hill equation were performed using Kaleidagraph (Synergy Software, Reading, PA). Data are expressed as the mean ± standard error.
Results
Effects of whole venom on cardiac myocytes.
We determined the effects of whole venom (1:2000 dilution) on K+ current in isolated rat ventricular myocytes. As shown in Fig. 1, the venom decreased the amplitude of Ito1 in a voltage-dependent manner, with relief of block at the most positive test potentials. Ito1 was completely blocked at test potentials between −30 and +10 mV, 53% decreased at +30 mV, and only 2% decreased at +60 mV.
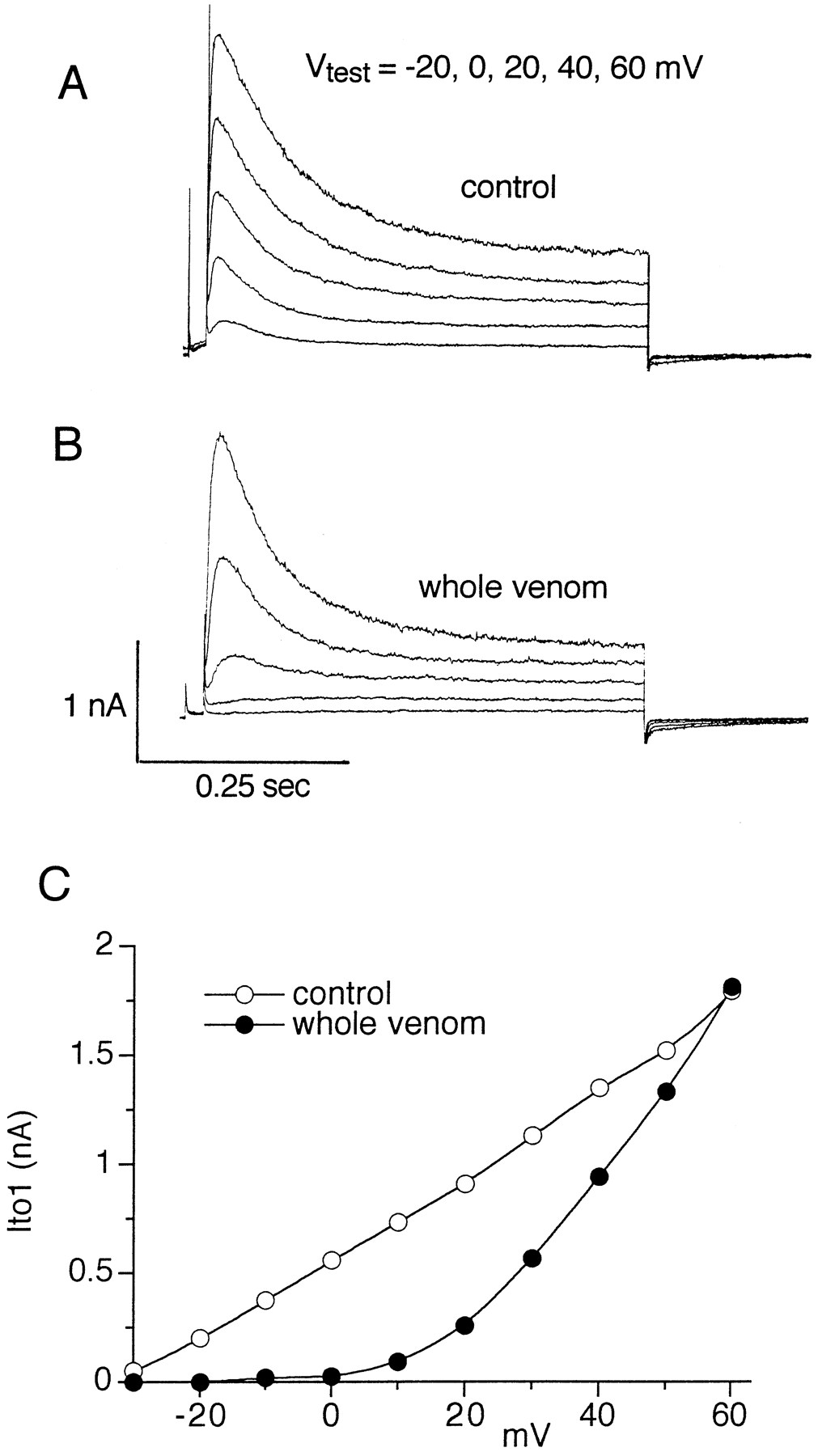
H. venatoria venom causes voltage-dependent block of Ito1 in isolated rat ventricular myocyte. Cell was exposed to 1:2000 dilution of whole venom. A and B, Currents were elicited from a holding potential of −50 mV. A brief prepulse to −30 mV was followed by 0.5-sec pulses to the indicated test potentials. C, Block of Ito1 by venom was decreased as the test potential was increased.
Isolation, purification, and amino acid sequence of active components from whole venom.
Fractionation of H. venatoria whole venom on a Vydac C-18 reversed-phase HPLC column revealed several prominent peaks as detected by UV absorbance at 220 nm (Fig. 2). Eight fractions, as indicated in Fig. 2A, were collected, lyophilized, and stored at −80°. Each fraction was diluted with 1 ml of H2O and tested at a 1:100 dilution of this stock solution for effects on Ito1 in isolated myocytes. Peaks 2 and 5–8 completely blocked Ito1 measured at −10 mV. Three of these fractions (5-7) were chosen for further characterization. Peak 5 eluted from the reversed-phase column between 27 and 30 min, peak 6 eluted between 30 and 33.5 min, and peak 7 eluted between 33.5 and 37 min. These fractions were further purified by cation-exchange and reversed-phase chromatography as described in Materials and Methods.
Mass spectral analysis of the HpTx was obtained using a SCIEX API III ion-spray mass spectrometer. The observed mass of HpTx1, HpTx2, and HpTx3 were 3910.57, 3412.72, and 3599.38, respectively. The peptides all had amides on their carboxyl termini. The amino acid sequences of the purified toxins, HpTx1, HpTx2, and HpTx3, were determined by amino-terminal sequencing and are shown in Fig. 2B. Amino acid identity was 39–41% among the HpTxs. HpTx3 is also 39% identical to hanatoxin2, a peptide toxin isolated from a Chilean tarantula, Grammostola spatulata (22).
HpTx2 prolongs action potential duration and blocks transient outward current in rat cardiac myocytes.
Isolated rat ventricular myocytes were current clamped and stimulated with suprathreshold current pulses (2-msec duration) at a frequency of 0.2 Hz to elicit action potentials. Action potential duration was measured at 90% repolarization, which was prolonged immediately after addition of HpTx2 to the cell chamber, and reached a steady state within 1 min thereafter. HpTx2 (30 nm) lengthened 90% repolarization by 34 ± 5% (n = 5; Fig. 3).

Prolongation of action potential in an isolated rat ventricular myocyte by 30 nm HpTx2.
HpTx2 blocked Ito1 but had no effect on the inward rectifier K+ current (IK1), l-type Ca2+current, or the maintained outward current (Isus). The block of Ito was voltage-dependent; less block was apparent at more positive membrane potentials. For example, 1 μmHpTx2 completely blocked Ito1 at a test potential of −10 mV (Fig. 4A) but decreased peak Ito1 by less than 50% at a test potential of +30 mV (Fig.4, B and C). HpTx2 had no effect on Isus, a very rapidly activating delayed rectifier K+ current (20) (Fig. 4D). The voltage-dependent block of Ito by HpTx2 results in a shift in the voltage dependence of current activation. In the example shown in Fig. 5A, the half-point for Ito activation was −13 mV in control and 6 mV after treatment of the cell with 30 nmHpTx2. The slope factor for the activation curve was increased from 5 mV in control to 12 mV after toxin.

Voltage-dependent block of Ito1 in isolated rat ventricular myocyte by HpTx2. A and B, Current recorded at a test potential of −10 mV was completely blocked by 1 μm HpTx2, but current recorded at +30 mV was only blocked by ∼50%. C, Voltage-dependent block of Ito1by 1.0 μm HpTx2. D, HpTx2 had no effect on Isus.

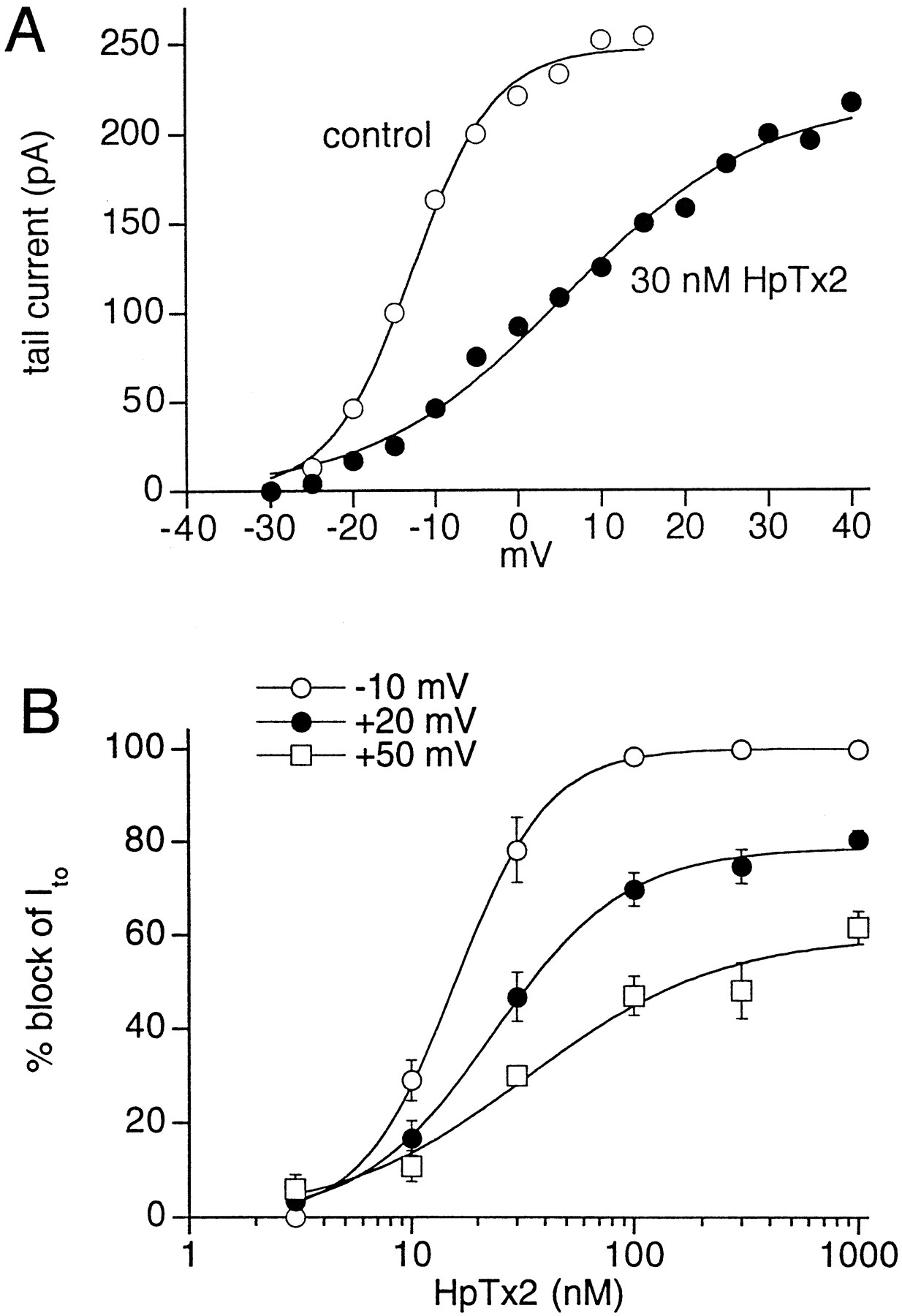
Voltage-dependent effects of HpTx2 on Ito in isolated rat ventricular myocyte. A, HpTx2 shifts the voltage dependence of Ito1activation. Curves represent best fits of the data to a Boltzmann function; control: V1/2 = −13 mV, slope = 5 mV; 30 nm HpTx2: V1/2 = 6 mV, slope = 12 mV. B, Voltage- and concentration-dependent block of Ito1 by HpTx2 in isolated rat ventricular myocytes. Block was determined at test potentials of −10, +20, and +50 mV (n = 5). Each curve represents a fit of the data to a Hill equation.
The concentration-dependent block of Ito1 by HpTx2 was determined at three different test potentials (Fig. 5B). The concentration required for 50% block (IC50) at a test potential of −10 mV was 15.8 ± 0.6 nm, with complete block at about 100 nm. The Hill coefficient for the concentration-effect curve was 2.1. At +20 and +50 mV, block was incomplete. Therefore, the IC50 value and the concentration required for half-maximal block (IC50-max) were determined. The IC50 values were 35 and 138 nm at +20 and +50 mV, respectively. The IC50-max value was 24 ± 1.5 nm (Hill coefficient = 1.5) at +20 mV and 34 ± 11.5 nm(Hill coefficient = 1.0) at +50 mV. The decrease of Ito block at more positive test potentials suggests that depolarization causes dissociation of the toxin from its receptor.
Preliminary experiments have shown that HpTx2 (0.2–1 μm) did not have any effect on delayed rectifier K+ currents recorded from rat neurons (cerebellar granule, Purkinje, sympathetic ganglion cells), GH3 pituitary cells, or rabbit osteoclasts (data not shown).
HpTxs block Kv4.2 channels.
Two types of transient outward K+ channels have been cloned from rat heart cDNA libraries, Kv1.4 and Kv4.2 (11, 12). We tested the effects of the toxins on these two cloned K+ channels expressed in X. laevisoocytes. HpTx2 and HpTx3, at a concentration of 2 μm, had no effect on Kv1.4 (not shown). All three HpTxs blocked Kv4.2 in a concentration-dependent manner. For example, at a test potential of −5 mV, HpTx3 decreased Kv4.2 84% at 400 nm, 72% at 200 nm, and 58% at 67 nm (n = 4). The voltage dependence of block of Kv4.2 was determined using a concentration of toxin that decreased current by 50% at a test potential near 0 mV (100 nm for HpTx1 and HpTx2, and 67 nm for HpTx3). The current-voltage relationships for Kv4.2 determined before and after exposure to each HpTx are shown in Fig.6. The voltage dependence of block was linear for 100 nm HpTx1 and 67 nmHpTx3 (Fig. 7A) but was curvilinear for 100 nm HpTx2 (Fig. 7B). The data for HpTx2 was well described with the sum of a linear component and a sigmoidal component fit with a Boltzmann function. From these data, the voltage at which Kv4.2 was blocked by 50% was determined to be +4 mV for all three toxins.

Effect of HpTxs on the current-voltage relationships of Kv4.2 expressed in X. laevis oocytes. A, Block of Kv4.2 by HpTxs was voltage dependent (n= 4 for each toxin). B, Examples of Kv4.2 currents recorded at test potentials of −5 and +55 mV, before and after exposure of different oocytes to each HpTx at the concentration indicated in A.

Voltage-dependent block of Kv4.2 expressed inX. laevis oocytes by HpTxs. The relationship between fractional block of current and test potential was determined with 100 nm HpTx1 and HpTx2 and with 67 nm HpTx3 (n = 4 for each toxin). The data for HpTx1 were best fit with the linear function: fractional block (Bf) = −0.00262 mV + 0.510. The data for HpTx3 were also best fit with a linear function: Bf = −0.00895 mV + 0.533. The data for HpTx2were best fit with the sum of a Boltzmann and a linear function: Bf = 0.367/[1 + exp((Vt + 3.29)/7.85)] − 0.0065 mV + 0.422.
The toxins increased the time required for currents to reach peak amplitude (Fig. 8A) and decreased the rate of current inactivation (Fig. 8B). The effect of HpTx3 on the voltage-dependence of current inactivation was determined. For these experiments, the holding potential was −90 or −100 mV. An 8-sec conditioning prepulse was applied to potentials ranging from −120 to 0 mV. After each conditioning prepulse, a test pulse was applied to +40 mV in one group of oocytes. In another set of oocytes, a test potential to +75 mV was used to assess current inactivation. When the test pulse was applied to +40 mV, the steady-state inactivation curves were fit with a Boltzmann function and had an average half-point (V1/2) of −67.6 ± 0.9 mV and a slope factor of 6.4 ± 0.2 mV (n = 4) under control conditions. In the presence of 100 nm HpTx3, the V1/2 was −59.5 ± 2.6 mV and the slope factor was 9.6 ± 0.8 mV. Using this pulse protocol, the inactivation curves overlapped; the test current in the presence of toxin was smaller when the conditioning pulse was applied to a voltage lower than −55 mV. However, when the conditioning pulse was applied to voltages higher than −55 mV, the test current was larger in the presence of HpTx3 than in control (Fig. 9A). When a test pulse to +75 mV was used to assess the extent of channel inactivation, control data was still best fit with a Boltzmann function. The average V1/2 was −68.3 ± 0.8 mV, and the slope factor was 8.4 ± 1.4 mV (n = 4). After exposure to 100 nm HpTx3, the inactivation curve had a similar maximum value (due to relief of block) but was shifted to a more positive potential (Fig. 9B). The voltage dependence of inactivation in the presence of toxin did not have a simple sigmoidal shape and was not well described with a single Boltzmann function. However, the data were best fit with a sum of two Boltzmann functions. For these fits, the values for V1/2 and the slope for one of the Boltzmann functions was fixed at the values measured from the matching control experiment. The second Boltzmann component had an average V1/2 of −39.8 ± 2.7 mV and a slope factor of 7.3 ± 0.3 mV (n = 4). The relative amplitude of the second component was 0.69 ± 0.03. These results suggest that approximately 70% of the channels were bound by HpTx3 and inactivated with an altered voltage dependence.

Effect of HpTxs on the rate of activation and inactivation of Kv4.2 expressed in X. laevis oocytes. A, Time to peak outward current is slowed by HpTxs (n= 4). B, The time constant of current inactivation is slowed by HpTxs (n = 4).

HpTx3 shifts the voltage dependence of Kv4.2 inactivation. A, Inactivation assessed with a test pulse to +40 mV. Smooth curves are best fits of the data to a Boltzmann function. In control, V1/2 was −65.5 mV and the slope factor was 6.4 mV. In the presence of HpTx3, V1/2 was −59.6 mV and the slope factor was 9.1 mV. B, Inactivation assessed with a test pulse to +75 mV in another oocyte. In control, V1/2was −67.0 mV and the slope factor was 12.5 mV. In the presence of HpTx3, the data were fit with the sum of two Boltzmann functions. The V1/2 and slope factor for one component were fixed at the values determined in control; the other component had a V1/2 of −34.2 mV and a slope factor of 6.5 mV.
Discussion
Our discovery of K+ channel blockers from the venom ofH. venatoria is the latest of several recent reports demonstrating that spider venom is a rich source of ion channel modulators. Toxins have been isolated that block voltage-dependent Ca2+ and K+ channels and glutamate receptor-operated channels. The ω-agatoxins (ω-Aga-1A–IVA) are peptide toxins isolated from the funnel web spider, Agelenopsis aperta, that block N-, P-, and L-type Ca2+ channels (23, 24). Related toxins isolated from the Chilean tarantula, G. spatulata (ω-grammotoxin) and Hololena curta(Hololena toxin), also block Ca2+ channels (25, 26). Acylpolyamine toxins isolated from the venom of orb weaver spiders (Argiope sp.) and A. aperta block glutamate receptor-operated channels (27). Most recently, peptides (hanatoxins) were isolated from the venom of G. spatulata that block the Kv2.1 K+ channel expressed in X. laevis oocytes (22). It is likely that spider venoms will yield many more interesting and useful ion channel modulators.
We found that the voltage dependence of Kv4.2 block varied among the three different HpTxs. Block was less voltage dependent for HpTx1 than for HpTx2 and HpTx3. The block of Kv4.2 by HpTx1 and HpTx3 was a linear function of voltage, whereas block by HpTx2 was a curvilinear function of voltage. The mechanisms of channel block by these toxins is not known. The strong voltage dependence of block is consistent with relief of block upon depolarization. This could result either from dissociation of the toxin from a binding site in or near the pore caused by outward flow of K+ and/or from a modulation of intrinsic channel-gating behavior.
It is often difficult to correlate channel activity recorded from isolated cells with specific channel types cloned from the the same tissue. Relatively specific pharmacologic probes can help resolve this difficulty. For example, it was suggested that Kv1.4 K+channels may contribute to Ito1 in cardiac ventricular myocytes (13). However, despite some similarities, some biophysical properties of these channels are significantly different. The rate of activation and inactivation and the sensitivity to block by 4-aminopyridine were similar for human Kv1.4 and human Ito1, but the recovery from inactivation was slower, and the half-point of activation and inactivation were more negative for Kv1.4 than for Ito1 in isolated myocytes (13). Our finding that HpTxs block Kv4.2, but not Kv1.4, suggests that Ito1in rat myocytes is formed from Kv4.2 and not from Kv1.4 channel subunits.
Hanatoxin1 and hanatoxin2 share significant sequence homology with the HpTxs (Fig. 2B). The hanatoxins were reported to block Kv2.1 with an IC50 value of 42 nm. Hanatoxin1 (500 nm) also blocked Kv4.2 by 73% at a test potential of 0 mV. Block of Kv4.2 by hanatoxin was characterized by a slowed time to peak and a slowed rate of inactivation, similar to the pattern of block of Kv4.2 by the HpTxs. Although the voltage dependence of Kv4.2 block was not investigated, considering these characteristics of block and similarity in sequence to the HpTxs, it is likely that the hanatoxins will also block Kv4.2 in a voltage-dependent manner. Future experiments are needed to determine whether HpTxs also block Kv2.1.
In situ hybridization of rat brain has shown that Kv4.2 channels are strongly expressed in the cerebellum and hippocampus, especially in the dentate gyrus and the pyramidal cells of the CA3 and CA1 region (28). The level of Kv4.2 mRNA in the excitatory dentate granule cell layer was down-regulated after seizure activity induced with a single dose of pentylenetetrazol in rats. Repression of Kv4.2 channel gene expression may enhance synaptic activity and may be involved in phenomenon such as kindling, a long-lasting epileptic state centered in the hippocampus (28). The HpTxs probably will be useful pharmacologic tools to determine the role of Kv4.2 channel activity in normal and pathological neural activities.
In summary, we have isolated several related peptide toxins from venom of H. venatoria that block Ito1 in isolated cardiac myocytes and Kv4.2 channels expressed in X. laevisoocytes. The heteropodatoxins represent a new class of modulators that will be useful in defining the physiologic role of Kv4.2 channels in a variety of cell types and will aid in structure function studies of these channels.
Acknowledgments
We thank C. Levinthal for collection of spiders, R. Roeloffs for collection of venom, and J. Busby and J. Garrett for synthesis of cRNA. Mass spectral analysis was performed by Y. Konishi at the Biotechnology Research Institute (Montreal, Quebec, Canada).
Footnotes
-
Send reprint requests to: Michael Sanguinetti, Ph.D., Division of Cardiology, University of Utah, Bldg. 533, Room 4220, Salt Lake City, UT 84112.
-
↵1 Current affiliation: Division of Cardiology, Eccles Program in Human Molecular Biology and Genetics, University of Utah, Salt Lake City, UT 84112.
- Abbreviations:
- HpTx
- heteropodatoxin
- Ito1
- voltage-dependent transient outward potassium current
- TFA
- trifluoroacetic acid
- HPLC
- high performance liquid chromatography
- PTC
- phenylthiocarbamyl
- Received September 26, 1996.
- Accepted November 14, 1996.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}