


Abstract
The voltage-gated delayed-rectifier-type K+ channel Kv2.1 is expressed in high-density clusters on the soma and proximal dendrites of mammalian central neurons; thus, dynamic regulation of Kv2.1 would be predicted to have an impact on dendritic excitability. Rat brain Kv2.1 polypeptides are phosphorylated extensively, leading to a dramatically increased molecular mass on sodium dodecyl sulfate gels. Phosphoamino acid analysis of Kv2.1 expressed in transfected cells and labeled in vivo with 32P shows that phosphorylation was restricted to serine residues and that a truncation mutant, ΔC318, which lacks the last 318 amino acids in the cytoplasmic carboxyl terminus, was phosphorylated to a much lesser degree than was wild-type Kv2.1. Whole-cell patch-clamp studies showed that the voltage-dependence of activation of ΔC318 was shifted to more negative membrane potentials than Kv2.1 without differences in macroscopic kinetics; however, the differences in the voltage-dependence of activation between Kv2.1 and ΔC318 were eliminated by in vivo intracellular application of alkaline phosphatase, suggesting that these differences were due to differential phosphorylation. Similar analyses of other truncation and point mutants indicated that the phosphorylation sites responsible for the observed differences in voltage-dependent activation lie between amino acids 667 and 853 near the distal end of the Kv2.1 carboxyl terminus. Together, these parallel biochemical and electrophysiological results provide direct evidence that the voltage-dependent activation of the delayed-rectifier K+ channel Kv2.1 can be modulated by direct phosphorylation of the channel protein; such modulation of Kv2.1 could dynamically regulate dendritic excitability.
Voltage-dependent ion channels expressed on the dendrites of principal neurons have been proposed to play a crucial role in regulating dendritic excitability (1). The voltage-dependent K+ channel Kv2.1 (or drk1; Ref. 2) is an abundant K+ channel in mammalian brain, in which it is primarily expressed in high-density clusters on the soma and proximal dendrites of principal neurons (3-7). Because voltage-dependent K+ channels are fundamental components in the control of membrane excitability (8), modulation of the gating, conductance, or kinetics of the Kv2.1 K+ channel would be expected to have an impact on dendritic excitability. Analysis of the modulation of Kv2.1 activity will be important in understanding mechanisms involved in shaping the active electrical properties of dendrites.
Protein phosphorylation may be the most important post-translational mechanism that can modulate ion channel function (9). Studies in squid axon have shown that protein phosphorylation can have dramatic effects on voltage-dependent activation of K+ channels, changing the threshold for recruitment of the delayed-rectifier current and thus its involvement in modulation of electrical activity (10, 11). A number of studies have suggested the involvement of phosphorylation in the modulation of the function of cloned and expressed K+ channels (12); however, for the most part, these studies have involved electrophysiological analyses in the absence of biochemical characterization of channel phosphorylation or biochemical analyses of channel phosphorylation in the absence of parallel functional characterization of the modified channels. Some combined electrophysiological/biochemical studies of channel modulation have been performed (13-20), but these studies have mainly focused on modulation of the amplitude of the expressed currents and have not investigated effects on voltage-dependent activation such as those observed by Bezanilla et al. (10, 11) in squid axons.
In our previous studies (3, 21), immunoblot analysis showed that Kv2.1 in crude rat brain membranes existed as a microheterogeneous polypeptide pool with a molecular mass of 110–130 kDa, which is much larger than its deduced molecular mass of 95.3 kDa (2). Transfected COS-1 cells expressed recombinant Kv2.1 as a 108-kDa protein, which on AP treatment migrated at 97 kDa (22), indicating that in these cells, phosphorylation was the major, if not sole, post-translational modification to Kv2.1. Here, we investigate the phosphorylation of Kv2.1 in rat brain and excitable cell lines. In transfected mammalian cells, AP sensitivity and in vivo 32P labeling combined with peptide mapping and phosphoamino acid analysis are used to characterize the phosphorylation on wild-type and mutant Kv2.1 isoforms. We examined the functional consequences of Kv2.1 phosphorylation using whole-cell patch-clamp analysis and in vivo AP treatments of transfected cells expressing the same Kv2.1 isoforms. The results indicate that the bulk of phosphorylation of Kv2.1 is on serine residues at the distal carboxyl terminus and that differential phosphorylation of the channel dynamically regulates voltage-dependent activation.
Experimental Procedures
Materials.
The expression vector pRBG4 was kindly provided by Dr. R. Kopito (Stanford University, Palo Alto, CA). The cDNAs encoding wild-type Kv2.1 (drk1) and the PKA mutant were the generous gifts of Dr. R. H. Joho (University of Texas Southwestern Medical Center, Dallas, TX) and Dr. J. A. Drewe (Baylor College of Medicine, Houston, TX), respectively. COS-1 cells were purchased from the Microbiology Department Tissue Culture Facility (State University of New York at Stony Brook, NY). DMEM, phosphate-free DMEM, methionine-free DMEM, and penicillin/streptomycin were from GIBCO BRL (Gaithersburg, MD). Newborn calf serum was from Hyclone Laboratories (Logan, UT). [35S]Methionine (Expre35S35S) was from Dupont-New England Nuclear (Boston, MA), and [32P]phosphoric acid (H3PO4 in H2O, 285 Ci/mg) was from ICN (Costa Mesa, CA). Anti-Kv2.1 antibodies KC and pGEX-drk1 were generated as described previously (3). Calf intestinal AP was from Boehringer-Mannheim (Indianapolis, IN). The cDNA encoding the lymphocyte surface antigen, CD8, was kindly provided from Dr. B. Seed (Massachusetts General Hospital, Boston, MA). Anti-CD8 antibody-coated beads were purchased from Dynal (Lake Success, NY). All other reagents were from Sigma Chemical (St. Louis, MO).
Construction of mammalian expression vectors for Kv2.1 and Kv2.1 truncation mutants.
Wild-type Kv2.1 and the cDNA encoding the PKA mutant were subcloned into the mammalian expression vector pRBG4 as described previously (22). Generation of the ΔC318 and ΔC187 truncation mutants in the pRBG4 mammalian expression vector was described previously (7).
Transient transfection of COS-1 cells.
COS-1 cells were plated onto Falcon 3001 dishes at 5% confluence for electrophysiological use and at 10% for biochemical use. Plated cells were grown in DMEM containing 10% calf serum, 100 units/ml of penicillin, and 100 mg/ml of streptomycin at 37° under 5% CO2. Within 24 hr after plating, cells were transfected by the calcium phosphate method (22). For electrophysiological use, cells were cotransfected with cDNA encoding CD8 surface antigen to identify visually transfected cells through the use of anti-CD8 antibody-coated beads (23). The CaPO4/DNA mixture was prepared at a final concentration of 4 μg/ml of K+ channel DNA and 0.8 μg/ml of cDNA encoding CD8 antigen. The media was replaced at 18–24 hr after transfection.
Metabolic labeling, immunoprecipitation, immunoblotting, and SDS-PAGE.
Steady state and pulse-chase labeling with [35S]methionine, immunoprecipitations, fractionation by SDS-PAGE, and fluorography were performed as described previously (22, 24). In vivo 32P labeling was performed on COS-1 cells 24 hr after transfection or on undifferentiated PC12 or L6 cells. Cells were washed with phosphate-free DMEM and labeled for 16 hr in phosphate-free DMEM containing 1 mCi/ml [32P]orthophosphate. The cells were then washed, lysed, and subjected to immunoprecipitation as described (22, 24) except that lysis buffer used for antibody incubation and washes was supplemented with 0.2% SDS and 0.5% deoxycholate. Immunoblotting was performed as described previously (22). Lauryl sulfate (Sigma Chemical) was the SDS source for all SDS-PAGE procedures to accentuate differences between phosphorylated and dephosphorylated forms of Kv2.1 (22, 25).
Phosphoamino acid analysis.
Acid hydrolysis followed by double electrophoresis (26) was used for phosphoamino acid analysis.In vivo 32P-labeled Kv2.1 was isolated by immunoprecipitation and fractionated on SDS-PAGE, and the migration of the 32P-labeled Kv2.1 band identified by autoradiography of the wet gel. The portion of the gel containing the32P-labeled Kv2.1 was excised and hydrolyzed, and the resultant phosphoamino acids were resolved by two-dimensional thin layer electrophoresis on precoated cellulose thin layer chromatography plates (Merck, Darmstadt, Germany) using a pH 1.9 buffer [2.5% formic acid/7.8% acetic acid/89.7% water (v/v/v)] and a pH 3.5 buffer [5% acetic acid/0.5% pyridine/94.5% water (v/v/v)] in the first and second dimensions, respectively. Phosphoamino acid standards were from Sigma Chemical and were visualized by staining with ninhydrin. Detection of 32P-labeled phosphoamino acids was by autoradiography of the thin layer plate.
One-dimensional peptide mapping.
In vivo 32P-labeled wild-type Kv2.1, the PKA mutant, and the ΔC318 truncation mutant were isolated by immunoprecipitation and fractionated on SDS-PAGE, and the migration of the32P-labeled bands were identified by autoradiography of the wet gel. The portion of the gel containing the32P-labeled polypeptide was excised and washed three times in 62.5 mm Tris·HCl, pH 6.8, and 150 mm HCl. The excised gel slice was resuspended in 1200 μl of this same buffer, followed by the addition of 120 μl of acetonitrile containing 225 μg of CNBr and incubation for 2 hr at room temperature. The gel slice was washed twice for 10 min with 500 μl of 125 mm Tris·HCl, pH 6.8, resuspended in 50 μl of reducing SDS sample buffer, and boiled, and the entire sample (gel slice and buffer) was fractionated by SDS-PAGE on a 20% polyacrylamide gel. The 32P-labeled phosphopeptides were detected by autoradiography of the dried gel.
Electrophysiology.
Recordings were made using whole-cell patch-clamp configuration (27) 36 hr after transfection. Before use, cotransfected cells were incubated for 3–5 min with external solution containing anti-CD8 antibody-coated beads diluted 1000-fold to allow CD8-transfected cells to be decorated with beads, which made possible visual identification of transfected cells (23). Electrodes (1–3 MΩ) pulled from borosilicate glass were fire-polished and filled with a pipette solution (see below). Currents were recorded with a patch-clamp amplifier (EPC-7), sampled at 10 kHz on an ITC-16 A/D converter, and filtered at 2 kHz by a digital bessel filter. All currents were capacitance and leak subtracted using the P/n procedure (28). All experiments were carried out at room temperature. The membrane potential was held at −80 mV and depolarized to +50 mV for 200 msec with 10-mV increments. The current (I) was converted into conductance (G) using the following equation: G = I/(V − EK). The Nernst K+equilibrium potential EK was calculated as −84 mV. The normalized conductances were then plotted against the test potential V and fitted to a single Boltzmann equation G = Gmax/{1 + exp[−(V − V½)/k]}. Gmaxis the maximum conductance, V½ is the test potential at which the channel has a half-maximal conductance, andk is the slope parameter that represents the slope of the activation curve. Data were presented as mean ± standard error. Statistical significance was evaluated by paired or nonpairedt test between two groups. If p < 0.05, the value was considered to be statistically significant.
Solutions for electrophysiology.
The pipette solution contained 140 mm KCl, 1 mmCaCl2, 10 mm Na-EGTA, and 10 mm Na-HEPES, pH 7.2. The bath solution contained 140 mm NaCl, 5 mm KCl, 1 mmCaCl2, and 10 mm Na-HEPES, pH 7.2. Calf intestinal AP (1 unit/ml) was diluted 100-fold in the pipette solution (to a final concentration of 10 units/ml). As a control, AP was boiled for 30 min to inactivate its enzymatic activity before dilution with pipette solution. Loss of AP enzymatic activity was confirmed by colorimetric assay (29) using p-nitrophenyl phosphate (not shown).
Results
Kv2.1 is phosphorylated in rat brain and excitable cell lines.
Kv2.1 is expressed in rat brain (2, 3), rat pheochromocytoma PC12 cells (30), and in rat skeletal muscle L6 cells.2 To examine the extent of Kv2.1 phosphorylation in adult rat brain, crude membrane preparations were subjected to AP digestion (Fig.1A). AP digestion caused a shift in molecular mass of the brain Kv2.1 polypeptide from the major 130-kDa form to a band of ∼105 kDa (Fig. 1A). This large shift on incubation with AP was inhibited by the presence of NaF (Fig. 1A). Note that the mobility of Kv2.1 in the −/− lane, which was incubated at 37° in the absence of added AP and NaF, is slightly shifted relative to the +/+ lane (Fig. 1A). This shift is presumably due to the action of endogenous phosphatase activity in the −/− crude brain membrane sample that is inhibited by the presence of NaF in the +/+ sample.

Kv2.1 phosphorylation in brain and excitable cell lines. A, Rat brain membranes (50 mg) were digested in the presence (+) or absence (−) of AP (100 units/ml) overnight at 37°. The digestion was done in the presence (+) or absence (−) of 100 mm NaF. Samples were then fractionated on a 7.5% SDS gel, transferred to nitrocellulose, and probed with anti-Kv2.1 antibody. Numbers on left, mobility of prestained molecular mass standards (in kDa). B, Undifferentiated pheochromocytoma PC12 or skeletal muscle L6 cells were incubated in phosphate-free DMEM containing 1 mCi/ml of [32P]orthophosphate for 16 hr and lysed. Cell lysates were subjected to immunoprecipitation in the presence (+) and absence (−) of anti-Kv2.1 antibody pGEX-drk1. Immunoprecipitation products were analyzed on 7.5% SDS gel and by autoradiography for 12 hr. Numbers on left, mobility of 14C-labeled molecular mass standards (in kDa).
In cultured PC12 and L6 cells, labeling in vivo with [32P]orthophosphate followed by immunoprecipitation and analysis by SDS-PAGE and autoradiography was used to investigate the phosphorylation of endogenous Kv2.1. Fig. 1B shows that in both cell lines, 32P-labeled Kv2.1 can be specifically immunoprecipitated with anti-Kv2.1 antibodies. Thus, endogenous Kv2.1 in rat brain and cultured cell lines contains AP-sensitive phosphorylation that contributes to its molecular mass and that in cultured cells can be labeled in vivo with [32P]orthophosphate.
In vivo32P-labeling and AP sensitivity of wild-type and mutant Kv2.1 show the major sites for phosphorylation are on serine residues near the distal carboxyl terminus.
The predicted molecular mass of the Kv2.1 core polypeptide deduced from the cDNA clone is 95.3 kDa (2). The Kv2.1 polypeptide expressed in COS-1 cells has a mobility on SDS gels corresponding to a molecular mass of ∼108 kDa (22). All of the increase in molecular mass of the Kv2.1 polypeptide in COS-1 cells seems to be due to the covalent addition of phosphates early in biosynthesis, based on sensitivity of the increase in molecular mass to digestion with AP (22). In vivo 32P-labeling was performed to confirm and extend these initial findings. When COS-1 cells transfected with the Kv2.1 cDNA were incubated with [32P]orthophosphate followed by immunoprecipitation and analysis by SDS-PAGE and subsequent autoradiography, a heavy 32P-labeled band of 108 kDa was present in immunoprecipitation products from reactions containing anti-Kv2.1 antibody (Fig. 2).

In vivo phosphorylation of wild-type and mutant Kv2.1 in COS-1 cells. COS-1 cells transfected with wild-type Kv2.1 or the indicated mutants were incubated in phosphate-free DMEM containing 1 mCi/ml of [32P]orthophosphate for 16 hr and lysed. Cell lysates were immunoprecipitated in the presence (+) and absence (−) of anti-Kv2.1 antibody pGEX-drk1. Immunoprecipitated samples were analyzed on 7.5% SDS gel and by autoradiography for 12 hr. Numbers on left, mobility of 14C-labeled molecular mass standards (in kDa). Arrows,32P-labeled wild-type and mutant Kv2.1 polypeptides.
Consistent with previous observations on Kv2.1 isolated from pulse-chase labeled cells (22), treatment of Kv2.1 isolated from steady state 35S-methionine-labeled COS-1 cell lysates with AP resulted in a shift in the molecular mass from 108 to 97 kDa (Fig. 3). The shift in mobility on AP treatment was inhibited by NaF, a potent phosphatase inhibitor, demonstrating the specificity of the phosphatase digestion. Thus, in COS-1 cells, most, if not all, of the post-translational increase in molecular mass of Kv2.1 is due to the rapid, constitutive phosphorylation of the Kv2.1 core polypeptide.

AP treatment of wild-type and mutant Kv2.1 in COS-1 cells. Duplicate cultures of transfected cells used for in vivo 32P-labeling shown in Fig. 1 were incubated in methionine-free DMEM containing 300 mCi/ml of [35S]methionine for 16 hr and then lysed. Cell lysates were immunoprecipitated with anti-Kv2.1 antibody pGEX-drk1, followed by digestion in the presence (+) and absence (−) of 100 units/ml of AP for 16 hr. The digestion was done in the presence (+) or absence (−) of 100 mm NaF. Digests were run on 7.5% SDS gel and detected by fluorography for 16 hr. Numbers on left, mobility of 14C-labeled molecular mass standards (in kDa).
Among all voltage-dependent K+ channels, Kv2.1 (853 amino acids) has the longest cytoplasmic carboxyl-terminal domain (443 amino acids), which is unusually rich in serine (61 residues) but less so in threonine (27 residues) and tyrosine (7 residues). Phosphoamino acid analysis of Kv2.1 labeled with32P in vivo indicated that all of the detectable labeling was present on serine (Fig.4A). To begin to address which of these residues were important in the observed phosphorylation events, site-directed and truncation mutants were analyzed for their extent of phosphorylation by in vivo 32P-labeling and sensitivity to AP digestion.
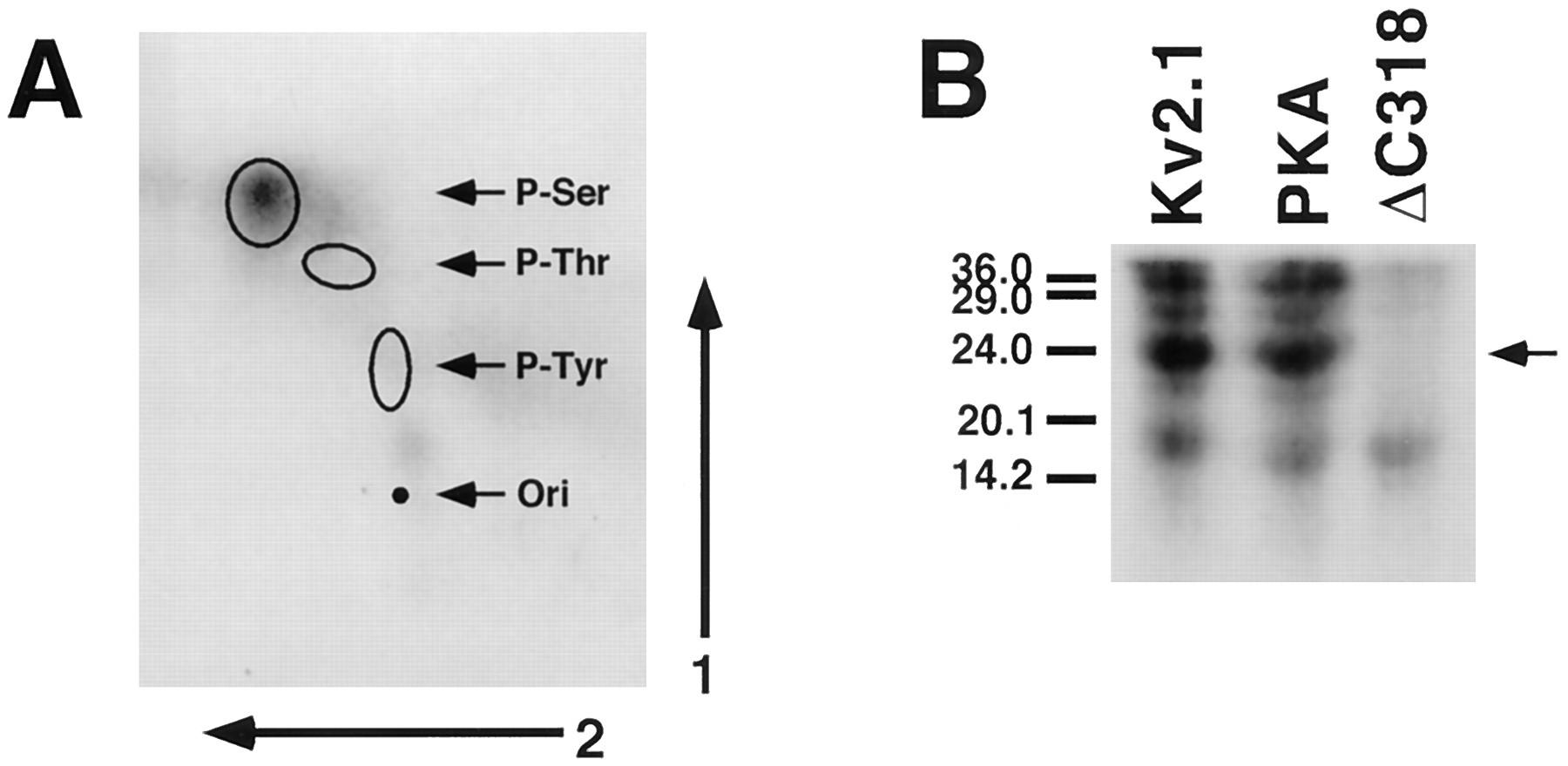
Analysis of in vivo 32P-labeled Kv2.1. A, Phosphoamino acid analysis. Kv2.1 was expressed in COS-1 cells, labeled in vivo with [32P]orthophosphate, and isolated as described in legend to Fig. 1. The labeled Kv2.1 band was excised and hydrolyzed using constantly boiling HCl. Phosphoamino acids were resolved by two-dimensional thin layer electrophoresis and detected by autoradiography of the thin layer plates. Exposure time for autoradiography was 5 days. Locations of ninhydrin-stained phosphoamino acid standards are shown, with the origin marked (ori). B, One-dimensional CNBr-generated peptide maps. Wild-type Kv2.1, the PKA mutant, and the ΔC318 truncation mutant expressed in COS-1 cells and labeled in vivo with [32P]orthophosphate were isolated by immunoprecipitation and fractionation by SDS-PAGE. The excised bands were digested with CNBr, and the resultant phosphopeptides were fractionated by 20% SDS-PAGE and detected by autoradiography. Exposure time for autoradiography was 4 days. Arrow, major phosphopeptide present in wild-type Kv2.1 and PKA mutant and absent in ΔC318.
A double-point mutant was analyzed in which the serine residues at the two consensus PKA phosphorylation sites (Ser440 and Ser492) were replaced by alanines. Expression of this mutant (PKA mutant) in COS-1 cells yielded a protein of a similar molecular mass as wild-type Kv2.1 (108 kDa), which could be similarly labeled in in vivo 32P-labeling experiments (Fig. 2). Like wild-type Kv2.1, the PKA mutant exhibited sensitivity to digestion with AP (Fig.3). The AP-sensitive biosynthetic shift in molecular mass of the PKA mutant occurs (Fig. 5) with the same kinetics (t ½ = 5 min) as for wild-type Kv2.1 (22). Thus, the PKA mutant is indistinguishable from wild-type Kv2.1 in these characteristics, indicating that the phosphorylated residues lie outside the two consensus protein kinase A sites.

AP treatment of pulse-chase labeled mutant Kv2.1 polypeptides. COS-1 cells transfected with the indicated mutant Kv2.1 cDNAs were labeled with a pulse of [35S]methionine for 5 min and then subjected to chase with unlabeled media for the indicated times. Cells were lysed and subjected to immunoprecipitation, and the immunoprecipitation products were digested in the presence (+) or absence (−) of AP. Digests were analyzed on 9% SDS-PAGE and fluorography on preflashed film.
A truncation mutant, ΔC318, was generated (7) that lacks the final 318 amino acids of Kv2.1. This polypeptide, containing amino acids 1–535 of Kv2.1, was expressed in COS-1 cells that were then incubated with [32P]orthophosphate, followed by immunoprecipitation and analysis by SDS-PAGE and autoradiography. Unlike wild-type Kv2.1 and the PKA mutant, very little32P-labeling was seen on the 58-kDa ΔC318 polypeptide (Fig. 2). Duplicate cultures from the same experiments were steady state labeled with [35S]methionine and subjected to immunoprecipitation and fluorography to verify that comparable levels of expression were obtained for wild-type Kv2.1, the PKA mutant, and the ΔC318 truncation mutant (Fig. 3). This suggests that the observed differences in the levels of in vivo 32P-labeling were not simply due to differences in expression levels or immunoprecipitation efficiencies.
Consistent with the lack of in vivo 32P-labeling, the molecular mass of the ΔC318 truncation mutant matched closely the predicted molecular mass deduced from the truncated cDNA (58 kDa), indicating that this mutant was not as extensively post-translationally modified as were wild-type Kv2.1 and the PKA mutant. When the ΔC318 truncation mutant isolated from steady state 35S-methionine-labeled cells was subjected to immunoprecipitation followed by AP digestion, no shift in mobility was observed in the presence or absence of NaF (Fig. 3). In addition, pulse-chase labeling experiments showed that the biosynthetic shift in molecular mass seen for wild-type Kv2.1 (22) and the PKA mutant (Fig. 5) is absent in ΔC318 (Fig. 5). These data indicate that the phosphorylation sites modified to yield the shift in molecular mass on SDS gels seem to reside within the residues deleted in the ΔC318 mutant (amino acids 536–853). Consistent with this, phosphopeptide mapping reveals that the major in vivo 32P-labeled phosphorylated CNBr-generated fragment of ∼23 kDa that is present in wild-type Kv2.1, and in the PKA mutant, is absent in ΔC318 (Fig. 4B). Other, less heavily32P-labeled fragments are present in all three forms of Kv2.1 (Fig. 4B). Together, these data indicate that the bulk of phosphorylation on Kv2.1 in COS-1 cells is on serine residues that lie on the carboxyl-terminal cytoplasmic tail between amino acids 535 and 853.
An additional truncation mutant was generated to localize further the critical sites. The ΔC187 truncation mutant lacks the last 187 amino acids and encodes a polypeptide containing amino acids 1–666 of Kv2.1. Pulse-chase labeling experiments showed that the AP-sensitive biosynthetic shift in molecular mass is absent in ΔC187 (Fig. 5) and ΔC187, like ΔC318, is a poor substrate for in vivo 32P-labeling (not shown). Thus, the residues critical for the biosynthetic shift in molecular mass lie within the last 187 amino acids (667–853) of Kv2.1.
Wild-type and mutant Kv2.1 isoforms exhibit differences in voltage-dependent activation.
Whole-cell patch-clamp analysis was used to characterize the electrophysiological properties of wild-type Kv2.1, the PKA mutant, and the ΔC318 truncation mutant expressed in COS-1 cells. COS-1 cells transfected with the cDNA encoding wild-type Kv2.1 exhibited slowly activating outward currents that displayed little or no inactivation during the 200-msec test pulse. Currents were evoked when the membrane potential was depolarized to >−30 mV (Fig.6A, top). The PKA mutant and ΔC318 exhibited currents with similar macroscopic kinetics as seen for wild-type Kv2.1 (Fig. 6A, middle and bottom); the thresholds for activation were −30 and −40 mV, respectively. These results are generally consistent with previous studies of wild-type Kv2.1 expressed in COS-1 (22) and MDCK (7) cells and of ΔC318 expressed in MDCK cells (7).

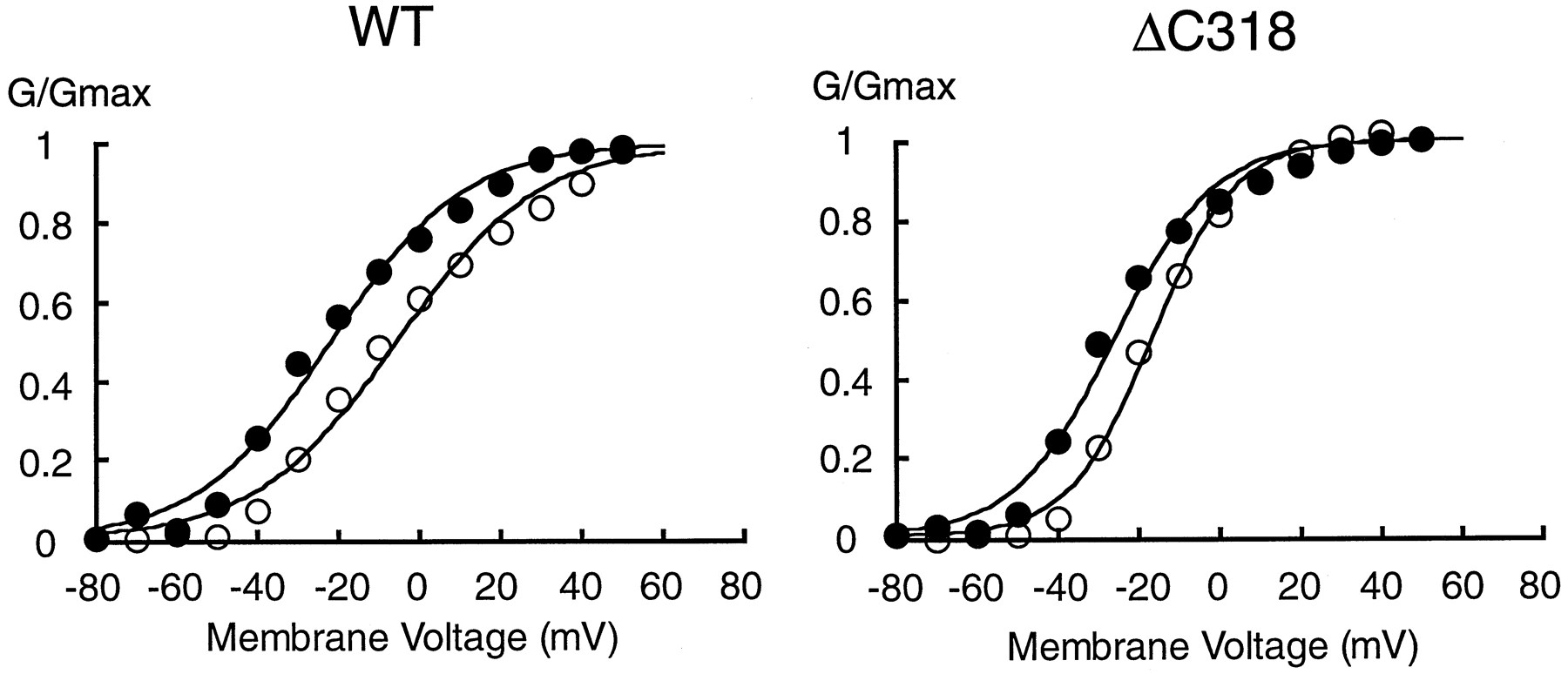
Voltage-dependent activation of wild-type (WT) and mutant Kv2.1 in COS-1 cells. A, Typical membrane currents recorded from COS-1 cells expressing Kv2.1 (top), ΔC318 (middle), and the PKA mutant (bottom). Current traces, recorded by step depolarizations from −40 mV (wild-type and the PKA mutant) or −50 mV (ΔC318) to +50 mV in increments of 10 mV for 200 msec. B, Activation curves for currents from the cells expressing wild-type Kv2.1 (○), ΔC318 (•), and the PKA mutant (▵) shown in A. Normalized conductances were calculated as described in Experimental Procedures. The V½ values for Kv2.1, ΔC318, and the PKA mutants were −3.1, −18.3, and −3.3 mV, respectively. Thek values for Kv2.1, ΔC318, and the PKA mutant were 13.0, 10.7, and 13.2 mV, respectively.
To determine the characteristics of the voltage-dependent activation of the currents shown in Fig. 6A, the normalized conductance was plotted against the membrane voltage (Fig. 6B). Normalized conductance was calculated as detailed in Experimental Procedures. The activation data for wild-type Kv2.1 were well fit by a single Boltzmann distribution, with a membrane potential for half-activation potential (V½) of −3.1 mV and a slope value of 13.0 mV. The activation curve for the PKA mutant was similar to that for Kv2.1 wild-type, with a V½ of −3.3 mV and a slope value of 13.2 mV, respectively. In contrast, the activation curve for ΔC318 was shifted toward more negative potentials compared with that for wild-type Kv2.1 and the PKA mutant, with a V½ value of −18.3 mV, although the slope parameter k was similar (10.7 mV). The activation parameters are summarized in Table 1. There was a significant difference in the V½ value between Kv2.1 and ΔC318, whereas there were no significant differences in the k value between wild-type and mutant Kv2.1 isoforms. There also was no significant difference in the amplitudes of the respective currents recorded at +50 mV (data not shown).
Activation parameters
The voltage-dependence of activation for ΔC187, with a V½ value of −11.2 ± 6.0 mV (mean ± standard deviation, seven experiments) was very similar to that for ΔC318 (Table 1). This is consistent with the finding that ΔC187, like ΔC318, does not undergo a shift in molecular mass during biosynthesis in COS-1 cells (Fig. 4). Other macroscopic activation and inactivation properties were similar between wild-type Kv2.1, the PKA mutant, and the ΔC318 and ΔC187 truncation mutants. Thus, the residues critical for the shift in molecular mass and voltage-dependence of activation lie within the 187 amino acids of Kv2.1 (667–853) that are deleted in this mutant.
In vivo AP treatments show that the voltage-dependence of Kv2.1 activation is dynamically regulated by phosphorylation.
To confirm that the shift in the activation curve for ΔC318 versus the wild-type Kv2.1 and the PKA mutant correlated with the observed differences in phosphorylation state, the effect ofin vivo AP treatment on voltage-dependent activation was studied. Whole-cell patch-clamp recordings were performed under conditions where the pipette solution contained 10 units/ml of calf intestinal AP. Fig. 7 shows the effects of AP treatment on the V½ of activation of wild-type Kv2.1 and the ΔC318 truncation mutant expressed in COS-1 cells. The activation curve for wild-type Kv2.1 was shifted by >20 mV to a more negative potential (−26.1 ± 3.7 mV, mean ± standard deviation, seven experiments) after a 30-min exposure to AP. The AP treatment caused the V½ for ΔC318 to shift by a lesser extent (−13.7 ± 6.0 mV, mean ± standard deviation, eight experiments). After AP treatment, both wild-type Kv2.1 and the ΔC318 truncation mutant had approximately the same V½ value (Table 1). When heat-inactivated AP was used in the pipette, no significant changes were seen in the V½ of either wild-type Kv2.1 or the ΔC318 truncation mutant (data not shown). No differences in the k values for wild-type Kv2.1 or ΔC318 were observed with either treatment (Table 1). Together, these results suggest that the rightward shift in the activation curve of wild-type Kv2.1 relative to the ΔC318 truncation mutant is due to AP-sensitive phosphorylation that yields the observed shift in molecular mass of Kv2.1 during biosynthesis. It should be noted that the ΔC318 truncation mutant can still be further left-shifted in its voltage-dependence of activation by treatment with AP, suggesting that phosphorylated residues exist outside of the carboxyl terminus that contribute to modulation of activation.

Effects of in vivo AP treatment on voltage-dependent activation. Activation curves for currents from the cells expressing wild-type Kv2.1 (WT) or the ΔC318 truncation mutant (ΔC318) before (○) and 30 min after (•) treatment with AP. Normalized conductances were calculated as described in Experimental Procedures. The V½ values before and after AP treatment were −5.6 and −21.9 mV for Kv2.1 and −16.7 and −25.9 mV for ΔC318, respectively. The k values before and after AP treatment were 17.5 and 16.5 for wild-type Kv2.1 and 10.2 and 12.4 for ΔC318, respectively.
Discussion
We have shown previously that Kv2.1 is constitutively phosphorylated early in biosynthesis in COS-1 cells (22). Here, we further analyzed the phosphorylation of Kv2.1 and subsequent effects on the macroscopic properties of Kv2.1 currents. Forms of the Kv2.1 polypeptide (ΔC318 and ΔC187) that were less extensively modified by phosphorylation had activation curves shifted by 10–15 mV to more negative membrane potentials relative to those forms (wild-type and the PKA mutant) that exhibited large AP-sensitive shifts in molecular mass during biosynthesis. It is formally possible that the observed negative shifts in voltage-dependence of the truncation mutants relative to wild-type Kv2.1 were due to a gross conformational change of the channel protein resulting from these rather large truncations in the cytoplasmic carboxyl terminus. However, in our experiments, the V½ values for Kv2.1 and ΔC318 were virtually identical after a 30-min treatment by AP. The fact that the observed differences in voltage-dependent activation disappeared on AP treatment argues that the sole basis for the differences in activation between wild-type Kv2.1 and ΔC318 resided in differences in phosphorylation between the two channel polypeptides. It also indicates that the differences in net amino acid charge between the wild-type Kv2.1 (+6) and ΔC318 (+10) core polypeptides do not contribute to the observed differences in voltage-dependence of activation.
How might differences in phosphorylation state lead to changes in voltage-dependence of activation? It has been demonstrated that the activation curve of the delayed-rectifier K+current in squid axons is shifted toward more positive potentials in the presence of ATP (10, 11). The effects of ATP were shown to be mediated through a change in the charge density on the cytoplasmic surface of the channel protein. Bezanilla et al. (10, 11) proposed that the negative charge of the incorporated phosphate residues (two net negative charges per phosphoamino acid) could have an electrostatic interaction with the voltage sensor of the channel. The negative charges contributed by phosphorylation would affect the voltage sensor such that additional depolarization was necessary to activate the channel, resulting in the rightward shifts in voltage-dependent activation observed on ATP treatment. The shifts in voltage-dependent activation of wild-type and truncated Kv2.1 observed here are consistent with this model. Forms of Kv2.1 that were more extensively phosphorylated (wild-type, PKA mutant) required more positive potentials for activation, whereas forms that were less phosphorylated (ΔC318 and ΔC187) activated at more hyperpolarizing potentials. Removal of these negatively charged phosphates led to leftward shifts in the activation curves, as predicted for the removal of negatively charged phosphates from the region of the voltage sensor. Effects of post-translationally added surface charge on voltage-dependent activation have been documented for Na+ channels (31) and for the Kv1.1 K+ channel (32). However, for these channel proteins, the addition of charged sialic acid residues to extracellular asparagine-linked sugar chains altered activation potentials. As demonstrated previously(22), the Kv2.1 polypeptide was insensitive to digestion with endo-β-N-acetylglucosaminidase H or PNGase F, suggesting that the Kv2.1 expressed in COS-1 cells was not glycosylated.
VanDongen et al. (33) studied the electrophysiological properties of wild-type Kv2.1 and various Kv2.1 cytoplasmic deletion mutants in Xenopus laevis oocytes. They reported that the V½ value for wild-type Kv2.1 was shifted left (−9.2 ± 3.8 mV) in oocytes (33) compared with our results on Kv2.1 expressed in COS-1 cells (2.1 ± 4.2 mV). However, the V½ value for ΔC318 (−14.3 ± 5.6 mV) in oocytes (33) was similar to that obtained in the current study in COS-1 cells (−11.8 ± 2.9 mV). The difference in the extent of phosphorylation of wild-type Kv2.1 in the different expression systems may in fact underlie the observed differences in V½ for Kv2.1. Immunoblot analysis showed that the recombinant Kv2.1 polypeptide expressed in X. laevis oocytes had a molecular mass of 100 kDa, but on the same immunoblot, this cDNA expressed in COS-1 cells had a molecular mass of 108 kDa (22). Because the only identified contribution to the increased molecular mass of Kv2.1 in COS-1 cells is AP-sensitive phosphorylation (22), it can be assumed that the difference in molecular mass between Kv2.1 in oocytes and Kv2.1 in COS-1 cells is that the COS-1 cell form is hyperphosphorylated relative to the oocyte form. This assumption would be consistent with the leftward shift in V½ of wild-type Kv2.1 in oocytes relative to COS-1 cells. The lack of a difference in the V½ between ΔC318 expressed in oocytes and COS-1 cells implies that this truncation mutant may be similarly poorly phosphorylated in both cell types. Recently, we have shown (7) that Kv2.1 expressed in MDCK cells also had a mobility (123 kDa) distinct from that in COS-1 cells and oocytes, presumably due to increased phosphorylation in this cellular background, whereas the ΔC318 truncation mutant expressed in MDCK cells has a molecular mass similar to that predicted for the core polypeptide (61.5 kDa) and present in COS-1 cells. Consistent with our model of increased molecular mass leading to rightward shifts in activation, we found that the calculated V½ value for Kv2.1 in MDCK cells (6.1 ± 1.7 mV, mean ± standard deviation, four experiments) was more positive than that in COS-1 cells or oocytes (33), whereas the calculated V½ value of ΔC318 in MDCK cells (−11.3 ± 2.7 mV, mean ± standard deviation, six experiments) was not significantly different from that in COS-1 cells or oocytes.
The native Kv2.1 polypeptide in rat brain runs on SDS gels as a microheterogeneous population of polypeptides with a molecular mass of 120–130 kDa (3, 21). Interestingly, the molecular mass of Kv2.1 changes during development, with a lower molecular mass species (110 kDa) predominating embryonically and the higher molecular mass species (120–130 kDa) appearing postnatally and persisting as the adult forms (21). The Kv2.1 polypeptide in brain is not glycosylated, as demonstrated by resistance to endoglycosidase digestion and lack of binding to lectins (34). AP digestion of adult rat brain membranes led to shifts in the molecular mass of Kv2.1, which is consistent with phosphorylation being the major determinant of the increased molecular mass of Kv2.1 in adult brain. It is interesting to speculate that the changes in the molecular mass of Kv2.1 during development and the microheterogeneity of Kv2.1 in adult brain are due to differences in phosphorylation state and that such differences could lead to the developmentally regulated expression of Kv2.1 K+channels with distinct functional properties. In addition, the heterogeneity of Kv2.1 phosphorylation in adult brain could underlie functional differences in Kv2.1 currents in different neuronal populations and raises the possibility that dynamic modulation of Kv2.1 phosphorylation could lead to plasticity in neuronal excitability. The comparison of the temporal expression patterns of K+ channel α subunits implied that Kv2.1 may contribute to delayed-rectifier K+ current in rat hippocampus (5). It was predicted by computational simulation that the voltage sensitivity of the K+ conductance in dendrites determines the maximum amplitudes of the voltage transients (35), resulting in control of the neuronal signal intensity. Because Kv2.1 is exclusively expressed in somatodendritic membrane of principal neurons (3-7) and the voltage-dependent activation of Kv2.1 is modulated by phosphorylation, it would be reasonable to speculate that the phosphorylation-induced changes in the activation of Kv2.1 could be involved in the mechanisms of neuronal plasticity associated with neurotransmitter-induced modulatory events, such as long term potentiation, which result in enhanced neuronal kinase activity (36,37).
Phosphoamino acid analyses indicated that Kv2.1 was constitutively phosphorylated in COS-1 cells on serine, and not on threonine or tyrosine residues. Wilson et al. (38) speculated that Kv2.1 was fully phosphorylated in X. laevis oocytes by PKA. The two consensus phosphorylation sites for PKA in Kv2.1 are present on serine (Ser440 and Ser492); however, these sites were not responsible for the constitutive phosphorylation leading to the shifts in molecular mass during biosynthesis and the more depolarized V½ value in COS-1 cells because the PKA mutant was similar to wild-type Kv2.1 in both regards. The ΔC187 truncation mutant lacks 21 serine residues compared with Kv2.1. These residues are expected to contain the sites responsible for differences in the shifts in molecular mass and V½between this mutant and wild-type Kv2.1. Among the serines missing in the ΔC187 truncation mutant are those contained within four consensus sites for phosphorylation by protein kinase C (SXR or SXK; Ref. 39) at Ser725, Ser778, Ser800, and Ser847, and one by calmodulin-dependent protein kinase II (XRXXS; Ref. 39) at Ser852. Future mutational analyses will focus on these 21 serine residues as determinants of voltage-dependent activation of Kv2.1. It should be noted, however, that the ΔC318 and ΔC187 truncation mutants, while lacking the biosynthetic shifts in molecular mass due to phosphorylation, can still be further left-shifted in their voltage-dependence of activation by treatment with AP. This implies that additional phosphorylated residues exist in these truncation mutants that contribute to modulation of activation. The fact that phosphorylation at these sites does not contribute significantly to the levels of in vivo 32P-labeling or the molecular mass of Kv2.1 on SDS gels, yet leads to a large shift in V½, may imply that these represent a small number of sites in close proximity to the voltage sensor. The subtype-specific sequences present in the cytoplasmic tail of K+ channels therefore may be involved not only in determining subcellular localization through interaction with other cellular proteins (7, 40) but also in the dynamic regulation of channel function through phosphorylation.
Acknowledgments
We thank Drs. Paul Brehm, Simon Halegoua, and Robert Haltiwanger for invaluable assistance during this project; Dr. Kenneth J. Rhodes for a critical reading of the manuscript; and Drs. Rolf H. Joho and John A. Drewe for Kv2.1 cDNAs.
Footnotes
- Received June 20, 1997.
- Accepted July 22, 1997.
-
Send reprint requests to: Dr. James S. Trimmer, Department of Biochemistry and Cell Biology, SUNY at Stony Brook, Stony Brook, NY 11794-5215. E-mail:trimmer{at}life.bio.sunysb.edu
-
↵1 Current affiliation: Hormone Research Institute, University of California, San Francisco, CA 94143.
-
↵2 K. L. Lopez and J. S. Trimmer, unpublished observations.
-
This work was supported by a Grant-in-Aid from the American Heart Association, New York State Affiliate, Inc. (H.M.), and by National Institutes of Health Grants NS34375 and NS34383 (J.S.T.). This work was done during the tenure of an Established Investigatorship from the American Heart Association (J.S.T.).
Abbreviations
- AP
- alkaline phosphatase
- DMEM
- Dulbecco’s modified Eagle’s medium
- PKA
- cAMP-dependent protein kinase
- CNBr
- cyanogen bromide
- MDCK
- Madin-Darby canine kidney
- V½
- membrane potential for half-activation potential
- SDS
- sodium dodecyl sulfate
- PAGE
- polyacrylamide electrophoresis gel
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}