


Abstract
Although γ-aminobutyric acid (GABA)A receptor α subunits are important for benzodiazepine (BZD) binding and GABA-current potentiation by BZDs, the presence of a γ subunit is required for high affinity BZD effects. To determine which regions unique to the γ2S subunit confer BZD binding and potentiation, we generated chimeric protein combinations of rat γ2S and α1 subunits using a modified protocol to target crossover events to the amino-terminal extracellular region of the subunits. Several chimeras with full open reading frames were constructed and placed into vectors for either voltage-clamp experiments in Xenopus laevisoocytes or radioligand binding experiments in human embryonic kidney 293 cells. Chimeras (χ) containing at least the amino-terminal 161 amino acids of γ2S bound BZDs with wild-type affinity when coexpressed with α1 and β2 subunits. Further analysis of the γ2S binding site region uncovered two areas, γ2S K41-W82 and γ2S R114-D161, that together are necessary and sufficient for high affinity BZD binding. Surprisingly, although the 161-amino acid residue amino terminus of the γ2S subunit is sufficient for high affinity BZD binding, it is not sufficient for efficient allosteric coupling of the GABA and BZD binding sites, as demonstrated by reduced diazepam potentiation of the GABA-gated current and GABA potentiation of [3H]flunitrazepam binding. Thus, by using γ/α chimeras, we identified two γ2 subunit regions required for BZD binding that are distinct from domain or domains responsible for allosteric coupling of the BZD and GABA binding sites.
GABAAreceptors are the major inhibitory neurotransmitter receptors in the mammalian brain and are members of a ligand-gated ion channel superfamily (Ortells and Lunt, 1995), which includes receptors for acetylcholine, glycine, and serotonin. Molecular cloning studies have identified several different classes and isoforms of GABAA receptor subunits, including 6 α, 4 β, 3 γ,1 δ, and 2 ρ subunit subtypes (Sieghart, 1995). The majority of the GABAA receptors in the brain are likely to consist of α1, β2, and γ2 subunits (Stephenson, 1995). These receptors are pentameric proteins containing an integral chloride-selective channel with specific binding sites for GABA, BZDs, barbiturates, steroids, and picrotoxin (Sieghart, 1995; Smith and Olsen, 1995). BZDs, clinically used for their anxiolytic, muscle-relaxant, sedative, and antiepileptic actions, exert their therapeutic effects by allosterically modulating the activation of the GABAA receptor. Because of their clinical usefulness, a substantial effort has been made to understand the structural determinants within the receptor that underlie BZD binding and allosteric coupling.
Evidence suggests that both the α and γ subunits play critical roles in BZD binding and potentiation. By analogy to the agonist binding site of the nicotinic acetylcholine receptor (Karlin and Akabas, 1995), the BZD binding site of the GABAAreceptor has been modeled with a γ subunit apposed to an α subunit, with adjacent faces of the subunits contributing to the binding site (Smith and Olsen, 1995). Alternatively, any subunit may bind BZD itself but have this ability enhanced by conformational changes conferred by the presence of the γ subunit, which is required for high affinity BZD effects (Pritchett et al., 1989). Regardless, understanding the roles of the α and γ subunits in BZD binding and modulation requires discovery of the specific structural elements involved.
In the α1 subunit, several amino acid residues have been identified that are important for BZD effects. Photoaffinity-labeling (Smith and Olsen, 1995; Duncalfe et al., 1996) and mutagenesis experiments (Wieland et al., 1992; Kleingoor et al., 1993) have identified histidine at position 101 (H101) as forming part of the BZD binding site. Experiments using α1/α3 chimeras point to α1G200 as another potential site for BZD effects (Pritchett and Seeburg, 1991). Other residues in α1 implicated in BZD binding include T162 and V211 (Wieland and Luddens, 1994), Y161 and T206 (Buhr et al., 1996), and Y159 and Y209 (Amin et al., 1997). Taken together, these results suggest that three separate domains of the α1 subunit, near H101, Y159-T162, and G200-V211, are involved in BZD binding.
Less evidence has been gathered regarding the BZD-responsive regions of the γ subunit. Mutagenesis experiments have identified two amino acids (F77 and T142) in the γ2 subunit that may play a role in BZD effects. Mutation of Thr142 to serine (γ2T142S) altered the efficacy of several BZD ligands; both an antagonist (Ro15–1788) and a weak inverse agonist (Ro15–4513) took on the character of partial agonists (Mihic et al., 1994). Mutation of Phe77 to leucine (γ2F77L) enhanced diazepam potentiation of the GABA-mediated Cl− current (Buhr et al., 1996), even though the binding affinity of diazepam was reduced. Substitution of γ2F77 with other amino acids had complex effects on BZD pharmacology (Buhr et al., 1997).
Both γ2F77 and γ2T142 are conserved in the aligned sequence of α1. The α subunit, even though it contains the homologous phenylalanine and threonine residues, cannot substitute for a γ subunit in conferring BZD effects. αβ receptors do not bind BZDs or exhibit BZD-induced potentiation of the GABA-activated Cl− current, whereas αβγ receptors do. Thus, other residues specific to the γ subunit are required for BZD binding and modulation.
To determine which regions unique to the γ2S subunit confer BZD binding and potentiation, we generated chimeric protein combinations of rat γ2S and α1 subunits. Chimeric studies have the potential to target whole domains, which is important if we envision the drug binding site as a pocket formed by the side chains of a variety of amino acids from one or more regions of a subunit. Using this method, we identified two domains of γ2S that are, in conjunction, necessary and sufficient for high affinity BZD binding. In addition, we demonstrated that the γ2S regions responsible for high affinity BZD binding are distinct from the γ2S regions necessary for efficient allosteric coupling of the BZD binding site to the GABA binding site. The construction of chimeric subunits that exhibit wild-type binding but reduced allosteric coupling of GABA and BZD binding sites affords new probes for elucidating the structural components of allosteric modulation.
Materials and Methods
Molecular cloning.
Chimeras (χ) were generated by placing the rat γ2 coding region 5′ to and in register with the rat α1 sequence in pBlueScript SK− (Stratagene, La Jolla, CA). The dual plasmid (pTRCP, Fig.1A) was digested, and the linearized plasmid was recircularized in bacteria by random homologous crossover events (Moore and Blakely, 1994). To create chimeric subunits containing amino-terminal domains of the γ2S subunit and carboxyl-terminal domains of the α1 subunit, we cut the dual-subunit plasmid with a restriction enzyme that cuts only in each coding region of γ2S and α1 (either AflII or BbsI). A fragment consisting mostly of the transmembrane and 3′ coding regions of γ2S was released. The remaining linearized plasmid contained γ2S and α1 sequences with restricted regions of homology. Because appropriate crossovers can occur only in a small area delimited by the chosen restriction enzyme or enzymes, we named this method TRCP. Using this method, dozens of chimeric subunits with crossovers in the 5′ (extracellular) region were generated in XL1-Blue cells, anendA − strain that facilitates plasmid miniprep production. The chimeric open reading frames were subcloned into pGH19 (Liman et al., 1992; Robertson et al., 1996) for expression in oocytes or into pCEP4 (InVitrogen, San Diego, CA) for transient expression in HEK 293 cells. For the TRCP chimeras generated in this study (Fig. 1B), the γ2S and α1 amino acids at which the crossovers occur are γN40/αR28 (χ40), γW82/αK70 (χ82), γW107/αT95 (χ107), γF113/αH101 (χ113), γD161/αA149 (χ161), and γL167/αK155 (χ167). Chimeras χ40, χ82, χ107, and χ113 were generated by AflII digestion, whereas χ161 and χ167 used BbsI digestion.

TRCP. A, Chimeras were generated by placing the γ2S coding region (2.0 kb) 5′ to and in register with the α1 sequence (1.65 kb) in pBlueScript SK− (2.9 kb; Stratagene) to yield pTRCP (6.55 kb). When linearized and introduced into competent coliform cells, the plasmid was recircularized by crossover events in homologous regions within the plasmid construct (see Materials and Methods). By choosing restriction enzymes that cut both γ2S and α1 (e.g., AflII), the transmembrane and 3′ coding region of γ2 was released, and sufficient α1 and γ2 5′ sequence was left to allow for crossover events. Black, γ2S sequence.White, α1 sequence. Gray, crossover areas made available by digestion with AflII. B, TRCP chimeras were screened from four independent trials and contained 5′ γ2S and 3′ α1 sequence, the amount of which was determined by restriction digest mapping and DNA sequencing. The chimeras (χ) generated by TRCP are named for the amino acid of where the crossover transitions occurs and fell into six major groups (χ40, χ82, χ107, χ113, χ161, and χ167). Three additional non-TRCP chimeras were made (see Materials and Methods) and are named for the γ2S segments each contains. For example, χ40/114–161 contains γ2S sequence from Q1 to N40 and from R114 to D161. χ114–161 contains only γ2S sequence from R114 to D161. Black, γ2S sequence. White, α1 sequence. Gray, transmembrane segments M1 through M4.
Chimera χ114–161 (Fig. 1B) was produced by recombinant polymerase chain reaction using an oligonucleotide (5′-CCAGTAAAATCTGGACTCCAGACACTTTCTTCAGGAACTCC-3′) designed to create an αF100/γR114 crossover. Using this 5′ oligonucleotide and a downstream complementary α1 oligonucleotide (5′-CTGGGAGAGAATGACTGTC-3′) with chimera χ161 as template, a 456-base pair polymerase chain reaction fragment with α1 5′ and 3′ flanks and γ2 114–161 sequence was generated and subcloned into wild-type α1 cDNA using BalI and NsiI. The resulting chimera contained α1 sequence except in the region from H101 to D148. This region contained the homologous γ2 region (R114 to D161). Chimeras χ40/114–161 and χ82/114–161 (Fig. 1B) were produced by digesting χ114–161 with MscI and NdeI, which flank the γ2 114–161 sequence, and subcloning the resultant 749-base pair fragment into χ40 and χ82. The resulting chimeras replaced the α1 region from H101 to D148 in both χ40 and χ82 with the homologous γ2S region (R114-D161). All chimeras were verified by restriction digest and double-stranded DNA sequencing using standard techniques (Sambrook et al., 1989).
Transient expression in HEK 293 cells.
Rat α1, β2, γ2S, and chimeric subunit cDNAs were subcloned into the multiple cloning site of a mammalian expression vector (pCEP4; InVitrogen) for transient transfection of HEK 293 cells (American Type Culture Collection CRL 1573). Cells were grown onto 100-mm tissue culture dishes in minimum essential medium with Earle’s salts (Life Technologies, Grand Island, NY) containing 10% fetal bovine serum (Hyclone Laboratories, New Brunswick, NJ) in a 37° incubator under a 5% CO2 atmosphere. Cells were cotransfected at 40–50% confluency with pCEP-α1, pCEP-β2, pCEP-γ2, and/or pCEP-χ using a standard CaHPO4 method (Graham and Eb, 1973). In general, cells were transfected with equal ratios of subunit DNA (5 μg/subunit). Cells were harvested and membrane homogenates prepared 48–72 hr after transfection.
Binding assays.
Cells were scraped from the dishes and pelleted by centrifugation (1000 × g, 10 min, 4°). The cells were washed once and resuspended in a HEPES buffer containing 124 mm NaCl, 2.9 mm KCl, 1.3 mmMgSO4, 1.2 mmKH2PO4, 25.0 mmHEPES, 5.2 mm d-glucose, and 2 mmEDTA; pH 7.4 and homogenized using a Polytron homogenizer (Brinkmann Instruments, Westbury, NY). The homogenates were centrifuged (30,000 × g, 20 min, 4°), and the resulting pellets were resuspended in HEPES buffer. Protein concentrations were determined using a Bradford assay (BioRad, Hercules, CA) with bovine serum albumin as a standard.
For BZD saturation binding experiments, membrane homogenates (100 μg) were incubated at room temperature with seven to nine concentrations of [3H]flunitrazepam (86 Ci/mmol; DuPont-New England Nuclear, Boston, MA) in the absence and presence of 20 μm diazepam or 100 μm flurazepam to determine total and nonspecific binding, respectively (final volume, 250 μl). The unlabeled BZDs, flunitrazepam, diazepam, Ro15–1788, and Ro15–4513 were generously supplied to us by Dr. Sepinwall (Hoffman-La Roche, Nutley, NJ). Flurazepam was obtained from Research Biochemicals (Natick, MA). [3H]Muscimol (15.7 Ci/mmol; DuPont-New England Nuclear) binding experiments were performed similarly; 1 mm GABA or 100 μm muscimol was used to determine nonspecific binding. All points were determined in triplicate. After reaching equilibrium, the incubations were vacuum filtered through glass-fiber filters (Reeves Angels; Whatman, Clifton, NJ) using a cell harvester (model MB-48; Brandel, Montreal, Quebec, Canada) and washed with eight times with 0.25 ml of HEPES buffer. Specific binding was defined as the amount of tritium bound in the absence of displacing ligand minus the amount bound in the presence of displacer. Nonspecific binding was ∼20–30% of total binding atKD concentrations of radioligand. In general, KD andB max were determined by fitting specific binding data to a single site using the equation y =B max*x/(KD + x), where y is specifically bound dpm, andx is the radiolabeled drug concentration (Prism; GraphPAD Softward, San Diego, CA).
Competition experiments with various BZD-site ligands were done under the same general conditions, except seven to nine concentrations of nonradioactive competing ligand were used to displace specifically bound radioligand. Data were fit by using a nonlinear least-squares method to the equation y =B max/[1+ (x/IC50)], where y is the specifically bound dpm, B max is maximal binding, and x is the concentration of displacing drug (Prism). KI was calculated according to the Cheng-Prusoff/Chou equation (Cheng and Prusoff, 1973; Chou, 1974).
To measure GABA potentiation of [3H]flunitrazepam binding (Czajkowski et al., 1989), membrane homogenates were incubated for 60 min at room temperature with 3–5 nm[3H]flunitrazepam in the presence of six different concentrations of GABA (ranging from 100 nm to 10 μm) and then filtered as described. The potentiation was calculated for each GABA concentration as follows: p = (dpmGABA/dpmcontrol) − 1, where dpmGABA is the specific [3H]flunitrazepam bound in the presence of GABA, and dpmcontrol is the specific [3H]flunitrazepam bound in the absence of GABA.
Expression in oocytes.
Capped cRNA coding for the wild-type and chimeric subunits was synthesized by in vitrotranscription from NheI-linearized cDNA template using the mMessage mMachine T7 kit (Ambion, Austin, TX). Oocytes fromXenopus laevis were prepared by incubating small pieces of ovary in collagenase (2 mg/ml) in ND96/Ca2+-free media containing 96 mm NaCl, 2 mm KCl, 1 mm MgCl2, and 5 mm HEPES, pH 7.6, for 40 min at room temperature. The digested ovaries were washed several times in ND96, followed by several washes in recording solution (ND96 with 1.8 mm CaCl2). Individual oocytes were defolliculated manually or en masseby 40-min incubation at room temperature in osmotic shock solution (130 mm K2HPO4, 1 mg/ml bovine serum albumin, pH 6.5 with HCl; Pajor, 1995) followed by several washes in recording solution. Within 1 day, they were injected with 5–50 nl of mRNA (10–200 pg/nl/subunit) mixed in a ratio of 1:1 (α:β, β:γ, or β:χ) or 1:1:10 (α:β:γ or α:β:χ). These ratios were determined to produce maximal assembly of γ- or χ-containing channels (Boileau AJ and Czajkowski C. Improved measurements of GABA-elicited currents and diazepam potentiation in recombinant GABAA receptor channels expressed inXenopus oocytes, manuscript in preparation). Oocytes were stored at 17–19° in recording solution supplemented with 100 μg/ml gentamicin and 100 μg/ml bovine serum albumin and were used for electrophysiological experiments 2–14 days after injection. The total amount of cRNA was scaled to yield maximal GABA-induced currents of ∼3–8 μA for α1β2γ2S and α1β2χ. The β2γ2S and β2χ subunit combinations yielded less current, usually 0.5–3 μA. cRNA concentrations were calculated by UV absorption and corroborated by comparison with RNA standards on 1.5% agarose gels.
Voltage-clamp analysis.
Oocytes under two-electrode voltage-clamp (Vhold = −80 mV) were perfused continuously with ND96/Ca2+ recording solution at a rate of 5 ml/min. In general, drugs and reagents were dissolved in ND96/Ca2+. The stock diazepam solution was made in dimethylsulfoxide. No differences in currents were observed with the vehicle. GABA responses were scaled for run-down or run-up by comparison with a low, nondesensitizing concentration of drug applied just before the drug concentration tested. Diazepam potentiation was recorded at ∼EC7 to EC20for GABA (1 μm GABA for α1β2γ2S and α1β2χ, 40 μm GABA for β2γ2S). Potentiation is defined as [I(GABA + DZ)/IGABA) − 1], where I(GABA + DZ) is the current response in the presence of diazepam, and IGABA is the control GABA current. Standard two-electrode voltage-clamp recording was performed using a GeneClamp 500 (Axon Instruments, Burlingame, CA) interfaced to a computer with an IT-16 A/D device (Instrutech, Great Neck, NY). Electrodes were filled with 3 m KCl and had a resistance of 0.5–1.5 MΩ.
Data acquisition and analysis were performed using AxoData, AxoGraph (Axon Instruments), and Prism (GraphPAD Software, San Diego, CA). All statistical comparisons used Student’s t test for independent samples (Snedecor and Cochran, 1980).
Results
BZD and GABA Binding to αβχ Receptors
To create chimeric subunits containing amino-terminal domains of the γ2S subunit and carboxyl-terminal domains of the α1 subunit, we modified a published method (Moore and Blakely, 1994) to specifically target crossovers to occur in the extracellular amino-terminal domain before M1 (see Materials and Methods; Fig. 1A). Chimeras (χ) used here, named for the γ2S amino acid at which the crossovers occur, are χ40, χ82, χ107, χ113, χ161, and χ167 (Fig. 1B).
To determine whether the chimeric subunits contained appropriate γ2S domains for BZD binding, they were individually expressed with wild-type α1 and β2 subunits in HEK 293 cells to form α1β2χ receptors, and the binding of 100 nm[3H]flunitrazepam was measured. Only two chimeras, χ161 and χ167, which contain the amino-terminal 161 or 167 amino acid residues of the γ2S subunit, exhibited significant levels of specific [3H]flunitrazepam binding (Fig. 2). No significant specific [3H]flunitrazepam binding was detected after expression of single subunits of wild-type or chimeric origin; two-subunit combinations using α1β2, α1γ2S, β2γ2S, or β2χ; or α1β2χ combinations with χ40, χ82, χ107, or χ113.

αβχ161 and αβχ167 receptors bind [3H]flunitrazepam. Chimeric subunits were individually expressed with wild-type α1 and β2 subunits in HEK 293 cells, and the binding of 100 nm [3H]flunitrazepam was measured (see Materials and Methods). Note that only two chimeras, χ161 and χ167, which contain the amino-terminal 161 and 167 amino acid residues of the γ2 sequence respectively, specifically bound [3H]flunitrazepam. Percentages were calculated by normalizing specific [3H]flunitrazepam binding of α1β2γ2S, α1β2, or α1β2χ receptors to α1β2γ2S binding. Results are mean ± standard error. The number of individual experiments are shown in parentheses.
The affinity of α1β2γ2S, α1β2χ161, and α1β2χ167 receptors for [3H]flunitrazepam (BZD agonist), Ro15–1788 (BZD antagonist), and Ro15–4513 (BZD inverse agonist) was measured by radioligand saturation and competition experiments to determine whether χ161- and χ167-containing receptors were altered in their ability to bind different classes of BZDs. Results from saturation binding experiments demonstrated that α1β2χ161 and α1β2χ167 receptors had B max values and equilibrium dissociation constants (KD ) for [3H]flunitrazepam similar to those of α1β2γ2S receptors, with KD values of 13.3, 11.3, and 9.9 nm, respectively (Fig. 3, Table1). Competition binding experiments using Ro15–1788 or Ro15–4513 showed no significant differences from α1β2γ2S in the KI values for these compounds (Table 1).

Saturation binding of [3H]flunitrazepam to membranes prepared from HEK 293 cells expressing α1β2γ2 and α1β2χ161 receptors.K D andB max values for [3H]flunitrazepam were calculated by nonlinear least-squares fit of specifically bound [3H]flunitrazepam (see Materials and Methods). Data shown are from a single experiment repeated multiple times with similar results; points,mean ± standard error of triplicate determinations. Results are summarized in Table 1.
Binding affinities for three different types of BZDs using wild-type and chimeric receptors
The dissociation constants for [3H]muscimol binding (a GABA binding site agonist) to α1β2, α1β2γ2S, and α1β2χ161 receptors also were determined. The affinity andB max values for [3H]muscimol binding to χ161-containing receptors were similar to α1β2γ2S receptors (α1β2χ161:KD = 88.3 ± 5.9 nm, B max = 1.32 ± 0.19 pmol/mg, 3 experiments; α1β2γ2S:KD = 70.0 ± 8.0 nm, B max = 1.21 ± 0.18 pmol/mg, 20 experiments). α1β2 receptors bound [3H]muscimol with a ∼2-fold higher affinity (KD = 46.2 ± 9.0 nm, B max = 1.23 ± 0.14 pmol/mg, 5 experiments).1 The small but significant difference in [3H]muscimol affinity in α1β2γ2S and α1β2χ161 receptors versus α1β2 receptors (p < 0.01) may be diagonistic for the presence of γ2 domains in the pentameric receptor complex.
Allosteric Coupling of the GABA and BZD Binding Sites
Two-electrode voltage-clamp studies.
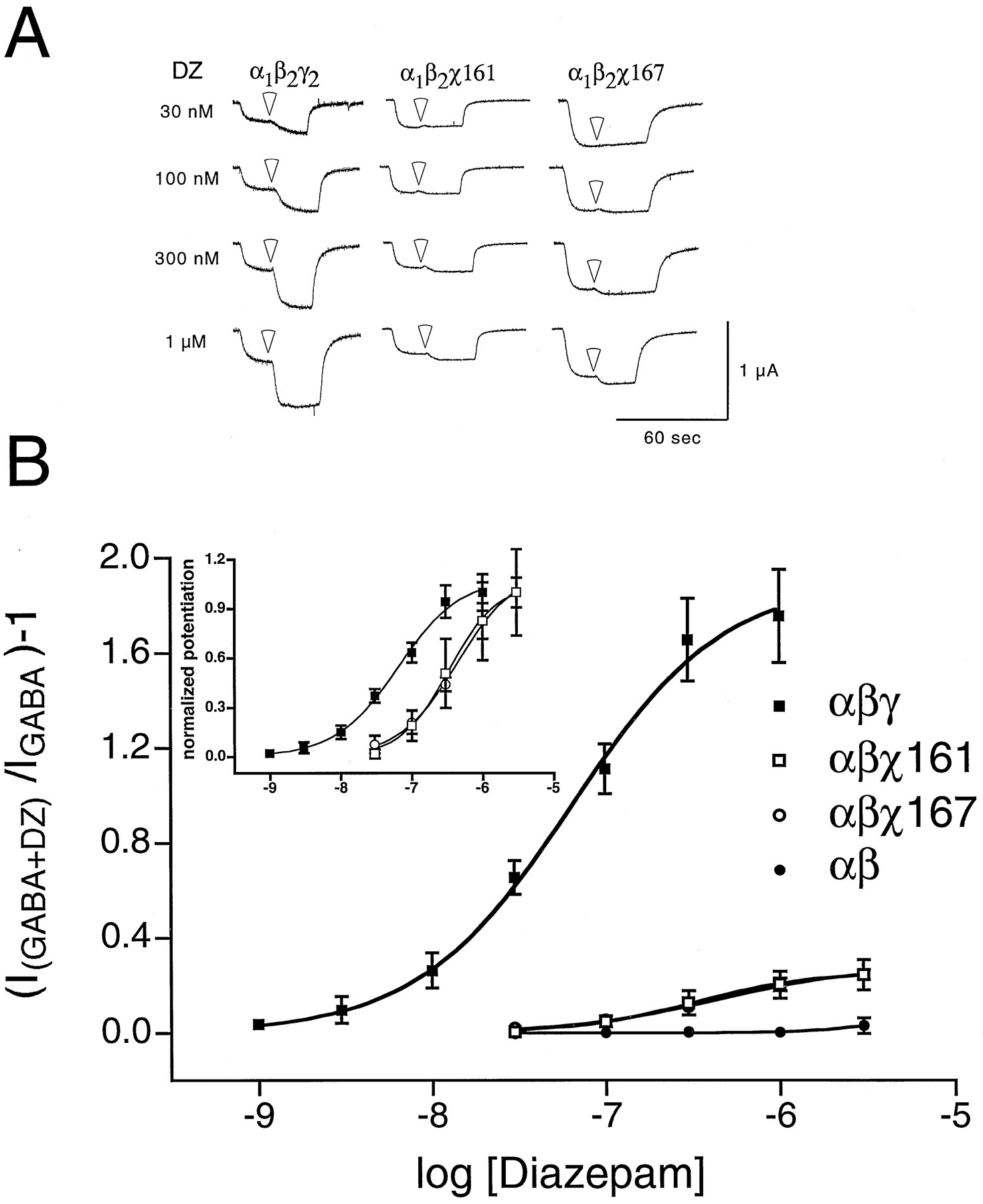
Because robust BZD binding does not necessarily indicate functional coupling of the BZD and GABA binding sites, the chimeras were tested with two-electrode voltage-clamp for the ability of diazepam to potentiate the GABA-mediated Cl− current. χ40, χ82, χ107, and χ113 showed no diazepam potentiation of the GABA response when coexpressed with wild-type α1 and β2 cRNA in X. laevisoocytes, whereas χ161 and χ167 exhibited small but detectable amounts of potentiation. The traces in Fig.4A show diazepam potentiation of GABA-activated currents from oocytes expressing α1β2γ2S, α1β2χ161, and α1β2χ167 GABAAreceptors. Fig. 4B plots the potentiation of GABA-activated currents for α1β2γ2S, α1β2χ161, α1β2χ167, and α1β2 receptors as a function of diazepam concentration. The maximal diazepam potentiation of α1β2χ161 and α1β2χ167 receptors was dramatically lower (∼7-fold) than that for wild-type α1β2γ2S receptors (Table 2). This result was surprising, considering that α1β2χ161 and α1β2χ167 receptors bound BZDs with wild-type affinity (Table 1), and indicates an uncoupling of high affinity BZD binding from BZD potentiation. Although the potentiation of α1β2χ161 and α1β2χ167 receptors was small, it was significant (p < 0.05) at diazepam concentrations above 100 nm compared with α1β2, α1β2χ40, or α1β2χ113 (Table 2). On normalization of the data to maximal potentiation, a ∼6-fold increase in the EC50 for diazepam potentiation was observed in χ161- and χ167-containing receptors compared with wild-type receptors (Fig. 4B, inset; Table 2).

Diazepam potentiates α1β2χ161 and α1β2χ167 receptors. A, Trace recordings from cells injected with chimeric construct α1β2γ2S (left), α1β2χ161 (middle), and α1β2χ167 (right). Cells were voltage-clamped at −80 mV and perfused with ND96 recording solution or ND96 with 1 μm GABA or 1 μmGABA plus diazepam (transition to diazepam-containing solutions:white arrowheads). Far left, diazepam concentrations. Cells were washed with ND96 recording solution for 5–20 min between drug applications. Note that wild-type α1β2γ2S subunits show a large potentiation, whereas chimeras show smaller potentiation even at a high concentration of diazepam (1 μm). B, Oocytes injected with wild-type α1β2γ2S (1:1:10), α1β2 (1:1), and α1β2χ (1:1:10) cRNA mixtures were treated with a range of diazepam concentrations in the presence of GABA and further analyzed. A potentiation response ratio was determined by dividing the peak current for α1β2γ2S (▪), α1β2 (•), α1β2χ161 (□), and α1β2χ167 (○) exposed to 1 μm GABA plus diazepam (DZ) by the response to 1 μm GABA alone. Data were fitted to a curve described by the equation Y = Min + (Max − Min)/{1 + 10[(logEC50 − X) ·n]}, where Max is the maximal potentiation, Min is the potentiation at the lowest drug concentration tested,X is the logarithm of diazepam concentration, EC50 is the half-maximal potentiation response, andn is the Hill coefficient. Data pointsrepresent mean potentiation from four or more cells from two or more batches of oocytes. Error bars, standard deviation. The parameters from the curve fits are presented in Table 2.Inset, a plot of the same data after normalizing to the maximum response for α1β2γ2 (▪), α1β2χ161 (□), and α1β2χ167 (○) receptors displays the shift in EC50value for chimera-containing receptors.
Summary of voltage-clamp results
Because a change in GABA EC50 value could potentially explain the decrease in BZD potentiation observed, GABA dose responses were measured. Current amplitudes at 1 μm(test concentration) and 10 mm GABA (maximal concentration) for all six α1β2χ combinations (data not shown) and GABA dose-response curves for α1β2χ161 and α1β2χ167 were similar to those for wild-type α1β2γ2S receptors (Fig.5, Table 2). These data indicate that the decrease in diazepam potentiation measured for α1β2χ161 and α1β2χ167 receptors was not caused by an alteration in GABA-mediated activation. Interestingly, the GABA EC50 values for α1β2γ2S, α1β2χ161, and α1β2χ167 receptors were statistically different than that for α1β2 receptors (p < 0.001, Table 2). The small change in GABA potency in the triple subunit combinations compared with α1β2 receptors may be indicative of the presence of the γ2S subunit or domains (see Discussion) and suggests that after injection of α1β2χ subunit combinations into X. laevisoocytes, a majority of the expressed receptors contain a chimeric subunit.

GABA dose response for chimeras is similar to that of wild-type α1β2γ2 receptors. Oocytes were injected with α1β2γ2 (1:1:10, ▪), α1β2 (1:1, •), β2γ2 (1:1, ▴), and α1β2χ161 (1:1:10, □) cRNA to determine whether reduced diazepam potentiation of chimeras was due to any shift in GABA dose-response curves. Data were fitted to a curve described by the equation Y = Min + (Max − Min)/{1 + 10[(logEC50 − X) ·n]}, where Max is the maximal response, Min is the response at the lowest drug concentration tested, Xis the logarithm of GABA concentration, EC50 is the half-maximal response, and n is the Hill coefficient. Dose response for both α1β2χ161 and α1β2χ167 (not shown, for clarity) are most similar to that of wild-type α1β2γ2.Data points, mean peak current from four or more cells from two or more batches of oocytes; error bars, standard deviation. Parameters determined from the curve fits are presented in Table 2.
Equilibrium binding studies.
To gain further insight into whether the decrease in the allosteric coupling of the GABA and BZD binding sites was due to an intrinsic property of the chimera-containing receptors, the ability of GABA to potentiate [3H]flunitrazepam binding to membrane homogenates prepared from HEK 293 cells expressing α1β2γ2S, α1β2χ161, and α1β2χ167 receptors was measured. In this experimental paradigm, only the receptor populations containing a γ2S or chimeric subunit were monitored because α1β2 receptors do not bind BZDs. Fig. 6 plots the potentiation of specific [3H]flunitrazepam binding of α1β2γ2S and α1β2χ161 receptors as a function of GABA concentration. The GABA-mediated potentiation of [3H]flunitrazepam binding in α1β2χ161 receptors was nearly abolished at concentrations of GABA up to 100 μm. Similar results were seen for α1β2χ167 receptors. In contrast, GABA potentiated [3H]flunitrazepam binding of α1β2γ2S receptors with an EC50 value of 1.20 ± 0.15 μm and a maximal potentiation of 1.25 ± 0.05 (Fig.6). These results suggest that the BZD and GABA binding sites are uncoupled in α1β2χ161 and α1β2χ167 receptors and that the uncoupling is due to a property of the chimera-containing receptors. In addition, these results corroborate the markedly reduced diazepam potentiation observed electrophysiologically.

GABA potentiation of [3H]flunitrazepam binding on wild-type and chimeric receptors. GABA potentiation of 2.5 nm[3H]flunitrazepam binding was measured in membrane homogenates prepared from HEK 293 cells expressing α1β2γ2 (▪) and α1β2χ161 (□) receptors (see Materials and Methods). Potentiation was calculated by dividing specific dpm in the presence of GABA by specific dpm in the absence of GABA, and the resulting data were fit to a single-site sigmoidal dose-response curve (see Materials and Methods; Fig. 4). Data points, mean potentiation of binding from eight experiments with α1β2γ2 and six experiments with α1β2χ161. Error bars, standard error.
Further Localization of the BZD Binding Site
By comparing the γ/α crossover positions (Fig. 1B) in chimeras that bound BZDs with high affinity (χ161, χ167) with those that did not (χ40, χ82, χ107, and χ113), a region of 48 amino acid residues (R114-D161) of the γ2S subunit that is essential for BZD binding can be identified. This determination requires that α1β2χ receptor combinations using χ40, χ82, χ107, or χ113 subunits were assembled and expressed efficiently. To address this question, the chimeric subunits were individually expressed with β2 subunits inX. laevis oocytes and the ability of GABA to activate a Cl−-specific current was tested. Because the dual subunit combinations α1β2 and β2γ2S form functional GABA-gated receptors when expressed in X. laevis oocytes (Table 2; Sigel et al., 1990) and β2 subunits expressed alone cannot, expression of β2χ combinations directly tests the capability of the chimeras to assemble into functional receptors. We observed GABA-mediated Cl− currents using all six β2χ subunit combinations (data not shown). β2χ40, β2χ82, β2χ161, and β2χ167 had maximal GABA current amplitudes similar to β2γ2S (3≥ μA). The maximal GABA currents of β2χ107 and β2χ113 receptors were ∼5-fold smaller. Interestingly, although diazepam potentiated the GABA response in β2γ2S receptors (EC50 = 24 ± 2 nm), diazepam did not potentiate the GABA current of any of the β2χ receptors (see Discussion). Nevertheless, these results demonstrate that the chimeric subunits can be assembled into functional β2χ receptors. If the chimeric subunits assemble into functional α1β2χ receptors in a similar manner, a region of 48 amino acids delimited by χ113 to χ161 in γ2S is required for BZD binding.
To determine whether this region is not only necessary but also sufficient for BZD binding, a chimeric subunit (χ114–161, Fig. 1B) was constructed that replaced the region from H101 to D148 in the α1 subunit with the homologous γ2S region (R114-D161). This chimera, when expressed with wild-type α1 and β2 subunits, did not specifically bind [3H]flunitrazepam, [3H]Ro15–1788, or [3H]Ro15–4513 at concentrations up to 200 nm (data not shown). To determine whether χ114–161 could assemble into a functional receptor, it was expressed with wild-type β2 subunits, and the binding of [3H]muscimol was measured. The χ114–161β2 receptor specifically bound [3H]muscimol with aKD value of 108 ± 30 nm and a B max value of 0.6 ± 0.4 pmol/mg (four experiments). Membrane homogenates prepared from HEK 293 cells expressing β2 alone did not specifically bind [3H]muscimol. These data suggest that the lack of BZD binding by α1β2χ114–161 receptors cannot be explained by an impairment in the assembly or expression of the χ114–161 subunit. Therefore, although the R114-D161 region of γ2S may be necessary for BZD binding, it clearly is not sufficient.
Because χ114–161 did not bind BZDs, two γ/α/γ/α chimeras were constructed (χ40/114–161 and χ82/114–161; Fig. 1B) that replaced in both χ40 and χ82 the α1 region from H101 to D148 with the homologous γ2S region (R114-D161). These chimeras were expressed with wild-type α1 and β2 subunits in HEK 293 cells and the binding of [3H]flunitrazepam was measured. The α1β2χ40/114–161 receptors did not specifically bind [3H]flunitrazepam or [3H]Ro15–4513, whereas α1β2χ82/114–161 receptors bound [3H]flunitrazepam in a similar fashion to α1β2γ2S receptors with aKD of 17.8 ± 5.4 nm and a B max of 0.36 ± 0.06 pmol/mg (six experiments) (Fig.7). α1β2χ82/114–161 receptors showed no significant differences from α1β2γ2S receptors in theKI values for Ro15–1788 (KI = 12.7 ± 3.1 nm, three experiments) or Ro15–4513 (KI = 23.0 ± 9.4 nm, four experiments). Thus, only two regions of the γ2S subunit, Q1-W82 and R114-D161, are required for high affinity BZD binding. Amino acid sequence comparison of α1β2χ40/114–161 receptors, which do not bind BZDs, and α1β2χ82/114–161 receptors suggests that high affinity BZD binding requires only the γ2S domains K41-W82 and R114-D161.

αβχ82/114–161 receptors bind [3H]flunitrazepam. Chimeric subunits were individually expressed with wild-type α1 and β2 subunits in HEK 293 cells and the binding of 100 nm [3H]flunitrazepam was measured (see Materials and Methods). Percentages were calculated by normalizing specific [3H]flunitrazepam binding of α1β2χ receptors to α1β2γ2S binding. Results are presented as mean ± standard error. The number of individual experiments is shown in parentheses. ▨, α1β2γ2S receptors; ▪, α1β2χ40/114–161 receptors; □, α1β2χ82/114–161 receptors.
Discussion
TRCP.
The use of TRCP was successful. By choosing available restriction sites, we specifically targeted DNA sequence crossovers to the amino-terminal regions of the α1 and γ2S subunits (see Materials and Methods). Moreover, by engineering a sequence with silent mutations to provide new restriction enzyme sites, one could choose any region to target for crossover events. Thus, TRCP should prove useful for any multisubunit protein.
Despite relatively low amino acid identity (Shivers et al., 1989), the intersubunit chimeras described in this study (γ2S/α1) formed functional channels. Intersubunit chimeras can furnish different structural/functional information from that furnished by subunit subtype chimeras (e.g., α1/α3), such as determining areas that are unique to each subunit and regions that are interchangeable between subunits. This was particularly useful because we were interested in identifying structural determinants of BZD binding and potentiation that were unique to the γ subunit.
Identification of γ2S BZD binding region.
The results demonstrate that a chimeric subunit with γ2S sequence in the first 161 amino acid residues and α1 sequence in the remainder can efficiently substitute for a wild-type γ2S subunit in assembling functional cell surface GABAA receptors that bind BZDs with high affinity. This proves that the γ2S determinants for binding BZD agonists (diazepam and flunitrazepam), inverse agonists (Ro15–4513), and antagonists (Ro15–1788) lie in the major extracellular portion of the subunit from the amino terminus through the cysteine-cysteine loop. Because electrophysiological experiments with β2χ combinations indicate that all of the chimeric subunits are expressible, we assume the chimeras also are expressed in α1β2χ receptors. Amino acid sequence comparison of α1β2χ113 receptors, which do not exhibit BZD binding, with α1β2χ161 receptors leads to the conclusion that high affinity BZD binding requires γ2S domain or domains between amino acid residues 113 and 161. However, a chimera that contains only these γ2S residues (the α/γ/α chimera, χ114–161), does not exhibit BZD binding, demonstrating that this 48-amino acid span is not sufficient for high affinity BZD binding. In other words, elements of γ2S upstream from position 114 must be involved in the formation of the binding pocket.
A chimera that contains the amino-terminal 82 amino acid residues from γ2S in addition to residues 114–161 (the γ/α/γ/α chimera, χ82/114–161) bound BZDs when expressed with α1 and β2 subunits. This result demonstrates that the γ2 subunit determinants for expression of high affinity BZD binding are contained within two domains of the γ2 subunit, Q1-W82 and R114-D161. Furthermore, if χ40/114–161, which does not bind BZDs, is expressed, the results indicate that BZD binding requires the γ2S domains K41-W82 and R114-D161. Although direct evidence that χ40/114–161 is expressed is not shown, it is reasonable to assume that it is because both χ40 and χ114–161 are expressed.
It has been postulated that the BZD and GABA binding sites are conserved structures, with a BZD binding site at a γ/α subunit interface and a GABA binding site at an α/β interface. Many of the γ/α interface residues that have been identified as being important for BZD binding (γ2F77, α1Y159, α1T206, and α1Y209) are homologous to the α/β interface residues that are important for binding GABA (α1F64, β2Y157, β2T202, and β2Y205). In the aligned sequences of the subunits, these residues are conserved. However, because the molecular structures of GABA and BZDs are quite distinct, it seems likely that other nonconserved residues will be required to impart pharmacological specificity to these sites.
Our results confirm the potential roles for γ2F77 and/or γ2T142 in BZD effects. These residues, which have previously been implicated in BZD effects (Mihic et al., 1994; Buhr et al.,1996, 1997), occur within the two γ2 regions identified in this study. However, although these residues may be important for BZD binding, both are conserved between the α1 and γ2 subunits. Thus, other γ2 residues are needed to impart a unique γ2 flavor and pharmacologically distinguish the BZD binding site pocket from the GABA binding site pocket. In this study, we have localized the specific γ2 determinants for BZD binding to K41-W82 and R114-D161.
A recent study (Buhr and Sigel, 1997) using γ3/γ2 chimeras has identified one amino acid residue (γ2 M130) and perhaps a second (γ2 M57) that seem to be important for controlling the affinity of zolpidem, an imidazopyridine. These residues occur within the two γ2 regions identified in this study. Mutations of these residues, however, have little or no effect on binding of classic BZDs. Thus, although subunit subtype chimeras are useful in examining the subtle pharmacological differences between γ subunits, these types of chimeras are not as helpful for identifying residues that are absolutely necessary for BZD binding. In our study, the novel use of γ2/α1 intersubunit chimeras allowed us to identify two distinct regions unique to the γ subunit that are required for the high affinity binding of a variety of different types of BZD ligands.
Galzi and Changeux (1994) proposed a four-loop model for ligand binding. Perhaps the three potential BZD binding regions of α1 (H101, Y159-T162, and G200-V211) correspond to three of these loops. If the model holds true, our results suggest that the fourth loop, presumed to be a part of an adjacent subunit, is contributed by residues in two regions of the γ2 subunit (K41-W82 and R114-D161). Moreover, if the GABA-binding region of α is homologous to the BZD-binding region of γ, then our finding that more than one region in the γ subunit is needed for BZD binding suggests that there may be other regions in the α subunit in addition to F64 that are involved in the formation of a GABA-binding site.
Chimeric uncoupling of BZD binding and potentiation.
Despite high affinity BZD binding with α1β2χ161 or α1β2χ167, there are significant differences in the behavior of the resultant channels with regard to BZD allosteric coupling. By using two different assays (electrophysiological and equilibrium radioligand binding) and two different expression systems (X. laevis oocytes and HEK 293 cells), we have shown that the allosteric interactions between the GABA and BZD binding sites of α1β2χ161 and α1β2χ167 receptors are markedly decreased compared with wild-type α1β2γ2S receptors (Figs. 4 and 6). Neither the decrease in diazepam potentiation of the GABA response nor the near abolishment in GABA potentiation of BZD binding is caused by a reduction in GABA sensitivity (Table 2) or muscimol binding affinity for α1β2χ161 or α1β2χ167 receptors. In addition, several pieces of evidence indicate that it is unlikely that the reduced potentiation is due to impaired assembly or expression of the χ161 and χ167 subunits: (1)B max for BZD binding of α1β2χ161 and α1β2χ167 receptors expressed in HEK 293 cells (Table 1) is similar to α1β2γ2S receptors and suggests that these chimera-containing receptors are efficiently expressed, (2) α1β2χ161 and α1β2χ167 GABA EC50 andKD values for muscimol binding are more similar to those of α1β2γ2S than to those of α1β2 receptors (Fig. 5, Table 2), suggesting full expression of a γ2 element, and (3) β2χ161 and β2χ167 receptors expressed inX. laevis oocytes have maximal GABA-activated currents, similar to those of β2γ2S receptors.
Taken together, these results indicate that the decreased coupling of BZD binding and potentiation is an intrinsic property of the χ161- and χ167-containing receptors and demonstrate that transduction of high affinity BZD binding (binding indistinguishable from wild-type) to full agonist potentiation must require additional and/or different γ2 subunit regions than those contained in the amino-terminal 167 amino acids of the γ2S subunit. Furthermore, these results suggest that a γ element is responsible for the ability of GABA to potentiate BZD binding. Experiments are under way to delineate residues, downstream from χ167, that are determinant for the allosteric interactions. In addition, these findings underscore the fact that a measured alteration in allosteric coupling does not necessarily imply any change in ligand binding.
Also of interest is the lack of measurable diazepam potentiation of the GABA response with any β2χ subunit combination expressed inX. laevis oocytes. β2γ2S receptors display both GABA activation and BZD potentiation of the Cl−current (Table 2), yet β2χ161 and β2χ167 receptors seem to be devoid of diazepam response despite the fact that when they are coexpressed with α1, they exhibit BZD binding (Table 1) and diazepam potentiation (Fig. 4). We postulate that the rightward shift in the GABA dose-response curve for β2γ2S receptors (Fig. 5; Sigelet al., 1990) represents a diminished capacity for γ2 GABA-binding regions to substitute for homologous α1 regions in the formation of a GABA-binding pocket at the interface with a β subunit. Recent work indicates a similar inability of γ2 to substitute for α1 in BZD actions (Amin et al., 1997), even though β2γ2S receptors display BZD sensitivity akin to that of α1β2γ2S receptors (Table 2; Sigel et al., 1990; Imet al., 1993). If inefficient allosteric coupling in the chimeras is superimposed on an already low GABA binding affinity contributed by the γ2 regions, this might make potentiation of the β2χ receptors difficult to measure. Alternatively, residues carboxyl-terminal to γ2L167 might be needed for formation of fully functional GABA or BZD binding pockets in β2χ receptors. Finally, it is possible that our chimeras will not bind BZDs in the absence of an α subunit. Whether β2χ receptors have altered BZD binding or altered allosteric coupling of the GABA and BZD binding pockets requires further investigation.
Previous studies have shown that chronic exposure to GABAA receptor modulators, including BZDs, can cause a decrease in coupling between binding sites, as measured by potentiation of [3H]flunitrazepam binding (Hu and Ticku, 1994; Friedman et al.,1996). This observation has been postulated to represent one mechanism underlying drug tolerance. In α1β2χ161 and α1β2χ167 receptors, the GABA and BZD binding sites are already uncoupled; therefore, these chimeras can be used to glean structural information about regions of the γ2S and α1 subunits involved in allosteric interactions. Mutation of these chimeric subunits such that coupling is enhanced will lead to a better understanding of allosteric cross-talk between binding sites and may help identify the structural elements underlying allosteric modulation. Studies using whole-cell and outside-out patch-clamp on chimeras expressed in HEK 293 cells also will be of interest, to determine the kinetics underlying these alterations in coupling. The chimeric subunits described in this report may be especially informative in this regard.
In summary, we demonstrated that γ/α chimeras can be expressed and form functional GABAA receptors in the presence of wild-type subunits. We identified two domains of the γ2 subunit, K41-W82 and R114-D161, that together are required for high affinity BZD binding. In these regions, several amino acid residues are not conserved between the α1 and γ2 subunits and are likely candidates for BZD binding site residues. Notably, diazepam potentiation of the GABA response and GABA potentiation of BZD binding are reduced in chimeras containing these regions due to impaired allosteric coupling. Thus, by using intersubunit chimeras, the molecular dissection of multiple domains associated with ligand-binding sites has been described and distinguished from areas that function in allosteric coupling.
Acknowledgments
We thank Polo Chen for technical assistance, Dr. Nicholas Cozzi for help in developing the transient transfection and radioligand binding protocols used in this study, Dr. David Wagner for critical reading of the manuscript, and Drs. Eugenia Jones, Robert Pearce, and Gail Robertson for helpful discussions.
Footnotes
- Received September 2, 1997.
- Accepted October 24, 1997.
-
Send reprint requests to: Dr. Cynthia Czajkowski, Department of Neurophysiology, University of Wisconsin, 1300 University Avenue, Room 197A MSC, Madison, WI 53706. E-mail:czajkowski{at}neurophys.wisc.edu
-
↵1 The affinities for [3H]muscimol binding measured are 2–5-fold lower than values reported by others in the field due to different assay conditions. In our binding assays, incubations with radioligand are carried out at room temperature (as opposed to on ice or 4°) to more closely mimic our electrophysiological recording conditions.
-
This work was supported in part by a grant to the University of Wisconsin Medical School under the Howard Hughes Medical Institute Research Program for Medical Schools and by grants to C.C. from The Epilepsy Foundation of America, National Institutes of Health–National Institute of Neurological Disorders and Stroke, and the March of Dimes Birth Defects Foundation. C.C. is a recipient of the Burroughs Wellcome Fund New Investigator Award in the Basic Pharmacological Sciences. Portions of this work have been reported previously in abstract form (Boileau et al., 1996).
Abbreviations
- BZD
- benzodiazepine
- GABA
- γ-aminobutyric acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HEK
- human embryonic kidney
- TRCP
- targeted random chimera production
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}