


Abstract
The whole-cell patch-clamp technique was used to examine the effects of retigabine, a novel anticonvulsant drug, on the electroresponsive properties of individual neurons as well as on neurotransmission between monosynaptically connected pairs of cultured mouse cortical neurons. Consistent with its known action on potassium channels, retigabine significantly hyperpolarized the resting membrane potentials of the neurons, decreased input resistance, and decreased the number of action potentials generated by direct current injection. In addition, retigabine potentiated inhibitory postsynaptic currents (IPSCs) mediated by activation of γ-aminobutyric acidA(GABAA) receptors. IPSC peak amplitude, 90-to-10% decay time, weighted decay time constant, slow decay time constant, and, consequently, the total charge transfer were all significantly enhanced by retigabine in a dose-dependent manner. This effect was limited to IPSCs; retigabine had no significant effect on excitatory postsynaptic currents (EPSCs) mediated by activation of non–N-methyl-d-aspartate ionotropic glutamate receptors. A form of short-term presynaptic plasticity, paired-pulse depression, was not altered by retigabine, suggesting that its effect on IPSCs is primarily postsynaptic. Consistent with the hypothesis that retigabine increases inhibitory neurotransmission via a direct action on the GABAA receptor, the peak amplitudes, 90-to-10% decay times, and total charge transfer of spontaneous miniature IPSCs were also significantly increased. Therefore, retigabine potently reduces excitability in neural circuits via a synergistic combination of mechanisms.
The novel anticonvulsant drug retigabine [D-23129;N-(2-amino-4-(4-fluorobenzylamino)phenyl)carbamic acid ethyl ester] has been found to effectively reduce or block seizure activity in a wide variety of animal models of epilepsy (Dailey et al., 1995;Rostock et al., 1996; Tober et al., 1996). Retigabine is structurally different and has a higher protective index than many of the commonly prescribed anticonvulsants (Rostock et al., 1996). Of particular interest is the recent finding that a primary mechanism of action of retigabine is the enhanced activation of heteromeric potassium channels composed of the KCNQ2 and KCNQ3 subunits (Main et al., 2000; Rundfeldt and Netzer, 2000b; Wickenden et al., 2000). It has been demonstrated that the channels formed by the KCNQ2/Q3 subunits underlie a neuronal potassium current commonly referred to as the M current (Wang et al., 1998; Shapiro et al., 2000). The M current is critical in determining resting membrane potential and neuronal excitability in many brain regions because of its sustained activation at potentials below the threshold for action potential generation (Marrion, 1997). Consistent with its ability to augment M channel currents, retigabine has been found to effectively hyperpolarize and reduce action potential generation in projection neurons located in layers II and III of the entorhinal cortex (Hetka et al., 1999).
Previous work suggests that the mechanism of action of retigabine is not restricted to potassium channels. Experiments in the hippocampal slice preparation suggest that retigabine can increase synthesis of the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (Kapetanovic et al., 1995). In addition, work by Rundfeldt and Netzer (2000a)demonstrated that, in cultured rat cortical neurons, retigabine potentiates Cl− currents induced by subsaturating concentrations of exogenously applied GABA. However, these findings contrast with those in the entorhinal cortex brain slice preparation, where retigabine was found to have no effect on any parameters of GABAA receptor-mediated IPSCs and inhibitory postsynaptic potentials (Hetka et al., 1999). One obstacle faced by Hetka et al. (1999) was that to reverse the effects of retigabine on resting membrane potential and action potential generation, the drug could only be superfused into brain slices for 2 min. Thus, the concentration of retigabine at the synapse was most probably lower than that of the superfusion media; consequently, it is not yet known whether retigabine, at concentrations comparable with effective dose plasma levels [estimated to be between 3 and 10 μM, (Tober et al., 1996)], modifies synaptic transmission. The present set of experiments was therefore conducted in a simple in vitro model system to circumvent these obstacles and directly determine the effects of retigabine on excitatory and inhibitory synaptic transmission.
The whole-cell patch-clamp technique was used to record from monosynaptically connected pairs of cultured cortical neurons. Cultured neurons provide rapid and reversible access of the compound to the synapses. In addition, this experimental paradigm allows us to directly distinguish presynaptic and postsynaptic effects of drugs (Wilcox and Dichter, 1994), because it provides the resolution required to analyze spontaneous miniature postsynaptic currents. We report here that retigabine effectively reduces excitability by hyperpolarizing neurons and by decreasing the input resistance at membrane potentials near action potential threshold, thus decreasing the number of action potentials that can be elicited by direct current injection. Moreover, we have found that retigabine dose dependently and reversibly potentiates IPSCs mediated by activation of GABAAreceptors. Thus, retigabine has multiple actions that serve to dampen excitability in neural circuits; these actions may underlie the potent anticonvulsant profile of this compound.
Materials and Methods
Tissue Culture.
Fetal mouse cortical neurons were maintained in primary culture using standard techniques, and all animals were treated in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Briefly, embryonic Swiss-Webster mice were removed from anesthetized mice (Charles River, Wilmington, MA) on gestational day 18 and the brains were quickly removed (Skeen et al., 1994). Cortical hemispheres were dissected, gently chopped into small pieces, and incubated for 2 min in Dulbecco's modified Eagle's medium (Sigma, St. Louis, MO) containing 0.25% trypsin. Horse serum (2.0 ml) (Invitrogen, Carlsbad, CA) was added for 30 s and the mixture was then placed into a 15-ml centrifuge tube and spun for 3 min at 1800 rpm. Most of the supernatant was discarded and the brains were triturated 10 to 14 times and spun down once more for 2 min at 1500 rpm. The pellet was reconstituted in Dulbecco's modified Eagle's medium supplemented with 10% horse serum, 3% glucose, and 2% l-glutamine. After trituration, cells were filtered and plated on 12-mm coverslips (Carolina Biological Supply Co., Burlington, NC) coated with poly(l-lysine) (Peninsula Labs, Belmont, CA) and maintained in a humidified incubator at 37°C in 5% CO2. Cultures were treated for 24 h with Ara-C when glial cells became about 70% confluent. In some cases, 24 h after plating, the media was replaced with a high-potassium media, supplemented with 20 mM KCl, to enhance survival (Wilcox et al., 1994). Three times per week, the culture media was replaced with either the standard or high-potassium media.
Electrophysiological Recordings.
As described previously, whole-cell patch-clamp recordings (Hamill et al., 1981) were obtained from monosynaptically connected pairs of neurons maintained in culture 2 to 4 weeks (Wilcox et al., 1994; Wilcox and Dichter, 1994; Cummings et al., 1996). Glass capillaries (World Precision Instruments, Inc.) were pulled to 3 to 6 MΩ resistance using a micropipette electrode puller (Sutter Instrument Co.). For all data acquisition, Axopatch 200 Series amplifiers and pClamp 8.0 software were used (Axon Instruments, Union City, CA). Signals were filtered at 5 kHz with the exception of mIPSC currents, which were filtered at 1 kHz. Data were acquired at 10 kHz for offline analysis using CLAMPFIT 8.0, Axograph, and/or the Mini Analysis Program (Synaptosoft, Decatur, GA).
Recording Solutions.
For cell-pair and single-cell recording other than mIPSCs, the HEPES-buffered saline (HBS) extracellular recording solution contained 142 mM NaCl, 1.5 mM KCl, 10 mM HEPES, 1 mM MgCl2, 3 mM CaCl2, 10 mM glucose, and 20 mM sucrose. To determine the effect of retigabine on input resistance, tetrodotoxin (TTX; 500 nM) and CdCl2 (200 μM) were included in the bath. The pH was maintained at 7.34–7.36 and the osmolality ranged from 315 to 318 mOsm. The internal electrode solution contained 130 mM potassium gluconate, 10 mM KCl, 10 mM HEPES, 1 mM EGTA, 0.1 mM CaCl2, and 10 mM glucose. The pH and osmolality of the internal solution were maintained at 7.28 and 290 mOsm, respectively.
For mIPSC recording, the HBS external solution contained 142 mM NaCl, 5 mM KCl, 10 mM HEPES, 2 mM CaCl2, 2 mM MgCl2, and 10 mM glucose. The pH and osmolarity were the same as the above external solution. To block EPSCs and Na+ currents, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (10 μM) and TTX (500 nM) were added, respectively. The internal electrode solution contained 140 mM KCl, 1 mM EGTA, 10 mM HEPES, 0.1 mM CaCl2, and 10 mM glucose. The pH and osmolarity were maintained at 7.28 and 305 to 308 mOsM, respectively.
Retigabine was supplied by ASTA Medica AG (Frankfurt, Germany). Stock solutions of 0.05 M were made in 50% dimethyl sulfoxide, frozen, and thawed as needed. The working concentration of dimethyl sulfoxide was ≤0.01%. Stocks of 0.01 M linopirdine were made in 10% HCl, frozen, and thawed as needed. Linopirdine and all other buffer chemicals were supplied by Sigma.
Data Analysis.
When determining the effects of retigabine on electroresponsive properties of single cortical neurons, the whole-cell current-clamp mode was used. Membrane potentials were maintained at −65 mV by direct current as needed. Currents ranging from −200 pA to +325 pA, in increments of 25 pA, were injected into single neurons. Recordings were terminated if, in control HBS solution, current injections of ≥50 pA did not produce two or more action potentials. The effects of retigabine on action potential firing, input resistance, and resting membrane potential were analyzed.
Neuron pairs were considered monosynaptically connected when evoked postsynaptic currents (PSCs) followed presynaptic action potentials with uniform and immediate latency times and were not interspersed with spontaneous activity (Wilcox and Dichter, 1994). A high external divalent cation concentration was used in the external solution (3.0 mM CaCl2 and 1.0 mM MgCl2) to reduce the probability of spontaneous action potentials and activation of any polysynaptic pathways (Wilcox et al., 1994). Because a low chloride containing internal solution was used, reversal potentials could be used to distinguish IPSCs from EPSCs. At −40 mV, IPSCs were outward, whereas EPSCs were inward; in addition, IPSCs and EPSCs were also distinguishable based on the duration of the PSC decay (Wilcox et al., 1994). As previously demonstrated for rat hippocampal cultures (Wilcox et al., 1994), IPSCs could be completely and reversibly blocked with bicuculline (10 μM), a selective GABAAreceptor antagonist, and EPSCs could be completely and reversibly blocked with the non-NMDA receptor antagonist CNQX (10 μM) (data not shown).
Using Clampfit 8.0 (Axon Instruments), the evoked PSC and mIPSC peak amplitude, 90-to-10% decay time, decay time constant (τ), total charge transfer, and paired-pulse ratio were analyzed. EPSCs were adequately fit with first-order exponential equations in both control and retigabine solutions; IPSCs could be fit with first-order equations in control solution, but second-order equations were almost always required in the presence of retigabine. Decay phases of the currents were fit with a double exponential equation of the form: I(t) = If × exp(−t/τf) + Is × exp(−t/τs), where If is the amplitude of the fast component, Is is the amplitude of the slow component, and τf and τs are the fast and slow time constants, respectively. Weighted time constants are calculated using an equation of the form: τw = [If / (If + Is)] × τf + [Is / (If + Is)] × τs (Rumbaugh and Vicini, 1999). Spontaneous mIPSC peak amplitude and total charge transfer were plotted on relative cumulative frequency histograms, and the Kolmogorov-Smirnov test statistic was used to determine the significance of retigabine's effects on mIPSCs. All values are presented as mean ± S.E.M.
Results
Effects of Retigabine on the Electroresponsive Properties of Neurons.
Initial experiments examined the effects of retigabine on the electroresponsive properties of cultured cortical neurons. Retigabine (1, 10, and 50 μM) significantly attenuated the ability of prolonged current injection to elicit action potentials (Fig.1A). During retigabine administration, and especially at the highest concentrations, current amplitudes that had previously elicited action potentials in control solution often failed to generate action potentials (Fig. 1B). This effect was completely and rapidly reversible. Interestingly, after washing the drug out, it was common for the number of elicited action potentials to surpass that of the initial control solution. The number of action potentials elicited in both control and drug solutions varied from cell to cell, but retigabine consistently reduced the number of action potentials elicited. In addition, retigabine significantly hyperpolarized the resting membrane potential of all cells dose dependently (Fig. 2.). From a membrane potential of −65 mV, neurons were consistently hyperpolarized by 2.4 ± 0.6 mV in 1 μM retigabine (n = 5), 5.4 ± 1.0 mV in 10 μM retigabine (n = 8); and by 7.2 ± 1.0 mV in 50 μM retigabine (n = 16). This hyperpolarization does not account entirely for action potential attenuation, because a steady-state current was injected to return the cells to the same membrane potential as during control (−65 mV). Even with this steady-state current injection, it was still necessary to inject larger current amplitudes to elicit action potentials in the presence of drug. To determine whether the hyperpolarization induced by retigabine was caused by enhanced activation of the KCNQ2/3 channel, the KCNQ2/3 antagonist linopirdine (10 μM) was coperfused with retigabine (10 μM) (Rundfeldt and Netzer, 2000b; Wickenden et al., 2000). Under these conditions, linopirdine completely blocked the effect of retigabine on resting membrane potential (Fig. 2).

Retigabine (RGB) decreases the number of action potentials elicited by identical current injections. A, the effect of retigabine on action potentials elicited by a 300-ms current injection. Note that upon drug washout, current injections elicited more action potentials than in control. B, shows the dose-dependent decrease in the number of action potentials elicited by various positive current injections in the presence of retigabine. In the presence of retigabine, more positive current is required to elicit action potentials. Data shown are from one representative cell.

Retigabine hyperpolarizes neurons from a resting membrane potential of −65 mV (1 μM, n = 5; 10 μM, n = 16; 50 μM, n = 16). All concentrations of retigabine produced a significant change in membrane potential (*, p < 0.05; Student's pairedt test.). The hyperpolarization induced by 10 μM retigabine was blocked by the KCNQ2/3 antagonist linopirdine (10 μM,n = 8). Linopirdine (LPD) (10 μM) alone was found to significantly depolarize the resting membrane potential (n = 8).
During retigabine administration, the input resistance of the neurons decreased during depolarizing current injection but was unaffected by hyperpolarizing current injection (Fig.3). Current-voltage relationships in either control conditions or during retigabine administration were evaluated under conditions in which sodium-dependent action potentials were blocked by TTX (500 nM), and voltage-gated calcium channels were blocked by CdCl2 (200 μM). In all cells examined (n = 4), retigabine (10 μM) significantly reduced the change in voltage in response to a given amplitude current pulse at all currents ≥ +100 pA. Retigabine decreased input resistance in the depolarized range, which is reflected as a decreased slope.

Retigabine decreases input resistance in response to depolarizing current. A, current pulses ranging from −150 pA to 325 pA, in increments of 25 pA, were injected into current-clamped cells and the resulting changes in membrane potential were measured. TTX (500 nM) and CdCl2 (200 μM) were included in the external solution to block voltage-dependent sodium channels and voltage-dependent calcium channels, respectively. B, in the presence of retigabine (10 μM), the change in voltage/current relationship from a representative cell shows outward rectification, and thus a decrease in input resistance, in response to depolarizing current injections. Vm was maintained at −65 mV for both conditions with steady state current injection as necessary. Statistically significant reductions in ΔV occurred for all current injections ≥100 pA for all cells examined (n = 4).
Effects of Retigabine on IPSCs and EPSCs.
While recording from pairs of monosynaptically connected neurons, presynaptic cells were recorded in current-clamp mode so that action potentials could be evoked, and postsynaptic cells were voltage-clamped at −70 or −80 mV while PSCs were elicited and recorded. All analyzed pair data were acquired from 25 pairs of monosynaptically connected neurons: four excitatory pairs and 21 inhibitory.
In monosynaptically connected neurons, presynaptic action potentials resulted in either evoked EPSCs or IPSCs and failures of neurotransmission were seldom observed. Peak amplitudes of IPSCs were significantly increased by 10 μM and 50 μM retigabine (n = 5 and 9, respectively) with no significant effect at 1 μM (n = 4) (Fig.4B). IPSC peak amplitudes were 122.0 ± 5.0% of control in 10 μM, and 150 ± 9.0% of control in 50 μM. At all concentrations tested, retigabine significantly increased the 90-to-10% decay times (see Table 1), whereas 10-to-90% rise times were unaffected (data not shown).

Effects of retigabine on monosynaptically evoked IPSCs. A, sample traces in control conditions versus retigabine show that 10 and 50 μM retigabine enhance evoked IPSC peak amplitudes and 90-to-10% decay times. Each trace is an average of 10 episodes, and the bottom trace is from the presynaptic cell. B, retigabine significantly potentiates IPSC peak amplitudes at 10 and 50 μM retigabine (*, p < 0.05, Student's pairedt test); C, total charge transfer of IPSCs is significantly increased by retigabine at 10 and 50 μM retigabine. For 1, 10, and 50 μM IPSC data, n = 4, 5, and 9, respectively (*, p < 0.05, Student's pairedt test; †, the effect of 1 μM retigabine approached statistical significance at 95% confidence, with p< 0.085.) D, for IPSC decay phases that were adequately fit with second-order exponential equations, retigabine significantly enhanced weighted τ values (*, p < 0.05, Student's paired t test). Refer to Table 1 for further comparison of weighted τ values in control and retigabine.
Effects of retigabine on the IPSC decay kinetics
By integrating the area of the current trace, the total charge transfer across the postsynaptic membrane was calculated. Retigabine significantly and selectively increased the IPSC total charge transfer at 10 and 50 μM. The IPSC total charge transfer in 10 μM retigabine was 167.6 ± 23.1% of control (n = 5) and was increased in 50 μM retigabine to 486.7 ± 69.2% of control (n = 9) (Fig. 4C). Consistent with the finding that the decay time constant was significantly enhanced in 1 μM retigabine, although peak amplitude was unaffected, the change in total charge transfer at this concentration approached statistical significance atp < 0.085.
To analyze the effect of retigabine on IPSC kinetics in more depth, the decay phases were fit with second-order exponential equations to determine slow and fast τ values. Weighted τ values were then calculated from the data as described previously (Rumbaugh and Vicini, 1999). All IPSC kinetic data are represented in Table 1. Kinetic data in control conditions were pooled. To compare IPSCs across conditions, only decays that could be adequately fit by second-order equations were analyzed. Retigabine at 10 and 50 μM significantly enhanced the percentage of the decay phase of the IPSC that was attributed to the slow τ, and the overall weighted τ values were also significantly increased (Fig. 4D; Table 1). In addition, the fast τ component was significantly decreased in 50 μM retigabine (Table 1), although this is most probably not caused by any direct action on factors contributing to the fast component of decay, but instead by the dramatic enhancement of the percentage of the decay that the slow τ encompassed after administration of 50 μM retigabine.
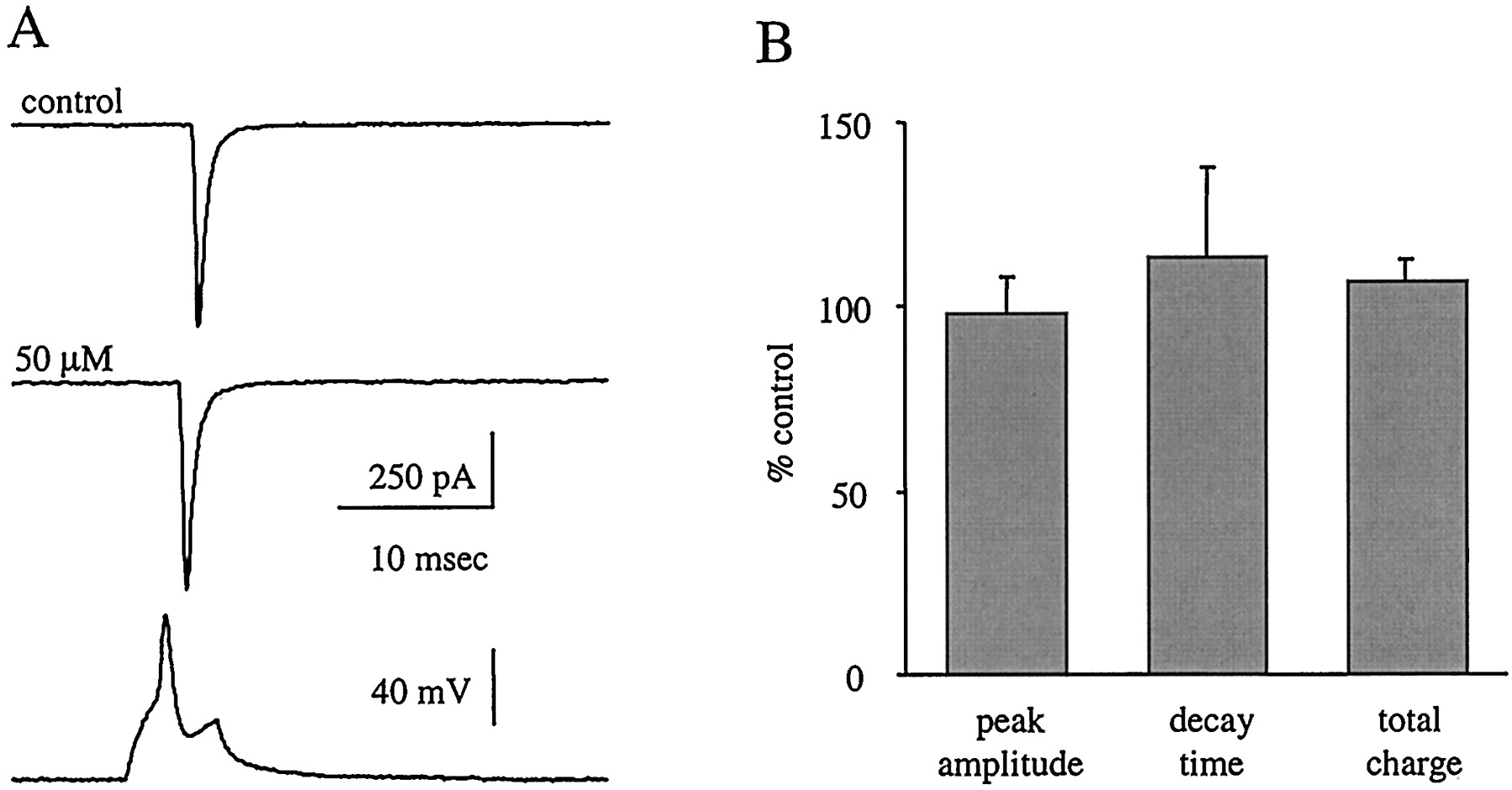
In contrast to the effects on IPSCs, all measured parameters of EPSCs, including rise time, peak amplitude, 90-to-10% decay time, decay time constant, and total charge transfer, were unaffected by retigabine (50 μM, n = 4) (Fig. 5, A and B). First-order exponential equations adequately fit EPSCs in control and retigabine conditions.

Retigabine has no effect on monosynaptically evoked non-NMDA mediated EPSCs. A, sample EPSC traces in control and 50 μM retigabine. Each trace is an average of 10 episodes from a single paired recording, and the bottom trace is from the presynaptic cell. B, retigabine (50 μM) has no effect on peak amplitude, decay time, or total charge transfer of EPSCs (n = 4).
Effects of Retigabine on Short-Term Plasticity.
Evoking two PSCs with a short interstimulus interval of less than 4 s often leads to a decreased amplitude of the second PSC (Deisz and Prince, 1989; Mott et al., 1993; Wilcox and Dichter, 1994; Cummings et al., 1996); this presynaptic phenomenon is referred to as paired-pulse depression (PPD). It is thought that changes in the degree of PPD are caused by presynaptic mechanisms related to the release of neurotransmitter (del Castillo and Katz, 1954; Otis and Mody, 1992;Wilcox and Dichter, 1994). PPD elicited with interstimulus intervals of 300 ms at both excitatory (n = 4) and inhibitory synapses (n = 16) was not effected by retigabine at any concentration (Fig. 6, B and C). This suggests that the observed effects of retigabine on IPSC amplitude and decay time constant are caused primarily by a direct postsynaptic action on the GABAA receptor rather than a presynaptic alteration of neurotransmitter release.
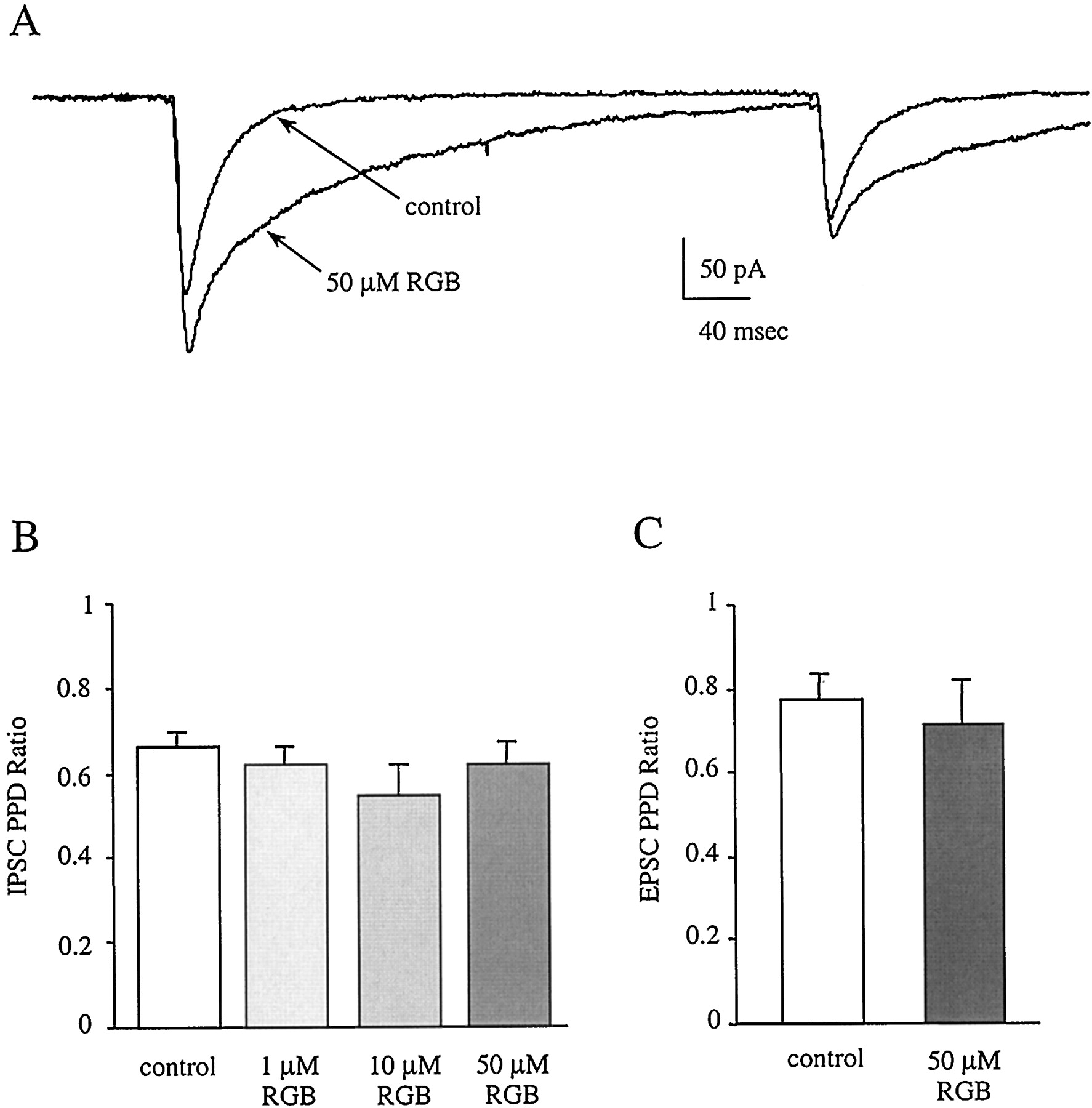
The effect of retigabine on paired-pulse depression at inhibitory and excitatory synapses. A, IPSC sample traces from the postsynaptic cell in control and 50 μM retigabine demonstrate that PPD is unchanged. Each trace is an average of 10 episodes. Although the amplitudes of both the first and second IPSCs are increased by retigabine, the ratio of the second to the first amplitude remains the same. Neither IPSC (B) nor EPSC (C) PPD is altered by retigabine at any concentration tested (IPSCs, n = 5, 5, and 10; EPSCs, n = 4.)
Effects of Retigabine on mIPSCs.
To test the hypothesis that the effects of retigabine on IPSCs were mediated by a direct action on postsynaptic GABAA receptors, we analyzed mIPSCs in the presence of retigabine (50 μM). Single cells were voltage-clamped at −70 mV in the presence of TTX and CNQX to block Na+ channels and excitatory non-NMDA receptors, respectively. The effects of retigabine (50 μM) on peak amplitude, 90-to-10% decay time, and total charge of mIPSCs were examined (n = 5 cells). Cumulative probability histograms were generated from the mIPSC data obtained, and the Kolmogorov-Smirnov test statistic was used to determine the significance of the effects of retigabine. Although retigabine significantly potentiated mIPSC peak amplitude in only three of the five cells recorded, (Fig.7C), both the 90-to-10% decay times and total charge transfer were substantially and significantly increased in all five cells (Fig. 7, D and E). The 10-to-90% rise time was also significantly increased in two of five cells (data not shown). The effects of retigabine on mIPSCs closely paralleled those on evoked IPSCs, confirming that its effect on IPSCs is caused primarily by a postsynaptic action at GABAA receptors.

Effects of retigabine on spontaneous miniature IPSCs. Traces from a representative cell in control solution (A) and in the presence of 50 μM retigabine (B). Retigabine markedly increases mIPSC peak amplitude and 90-to-10% decay times. Notice that in the presence of drug, there is a marked increase in baseline noise. C, cumulative frequency histogram for a representative cell demonstrating that 50 μM retigabine significantly increases the probability that mIPSC peak amplitudes will be larger than in control conditions (*,p < 0.05 for three of five cells; Kolmogorov-Smirnov test statistic). D, cumulative frequency histogram demonstrating the 50 μM retigabine increases 90-to-10% decay time. (*, p < 0.05 in 5 of 5 cells) E, total charge transfer is also potentiated by 50 μM retigabine. (*,p < 0.05) in 5 of 5 cells). All histograms were generated from data obtained from a single representative cell.
It was noted that retigabine, especially at 50 μM, dramatically enhanced the baseline noise of the recordings (Fig. 7, A and B, and Fig. 8, A and B). To determine whether this effect was due to a direct activation of K+channels or a direct action on GABAA receptors, retigabine (50 μM) was coapplied with bicuculline (10 μM), a competitive antagonist of the GABAA receptor (n = 3). It was hypothesized that if activation of K+ channels were responsible for the increased noise, it would persist in the presence of bicuculline. During coapplication of retigabine and bicuculline, the baseline noise did not increase as it did in retigabine alone (Fig. 8). This suggests that the increased baseline noise during retigabine application was the result of enhanced current flow through GABAA receptors and not activation of K+ channels (Leao et al., 2000).

The baseline noise is increased in retigabine but can be blocked by coapplication of bicuculline (10 μM). A and B, sample traces show enhanced noise in the presence of retigabine. C, during coapplication of retigabine and bicuculline, the mIPSCs and the increased noise are both blocked.
Discussion
The experiments presented here investigated the effects of the novel anticonvulsant retigabine on cortical neurons maintained in culture. Retigabine was found to modulate several electroresponsive properties of all neurons tested and to significantly enhance inhibitory neurotransmission. Specifically, in all neurons examined, retigabine hyperpolarized the resting membrane potential, reduced input resistance at membrane potentials depolarized from resting membrane potential, decreased action potential generation, and significantly enhanced IPSCs through a direct action on postsynaptic GABAA receptors. Therefore, retigabine can dampen excitability in neural circuits not only by inhibiting action potential generation in many types of neurons but also by enhancing inhibition at GABAergic synapses.
The direct actions of retigabine on individual neurons that we describe in our culture system are similar to what has been described in the entorhinal cortex brain slice preparation and in rat sympathetic neurons in culture (Hetka et al., 1999; Tatulian et al., 2001). Retigabine was found to decrease the input resistance of neurons at potentials depolarized from the resting membrane potential, thereby making it necessary to inject more depolarizing current to reach threshold for action potential generation. This outward rectification effectively reduced the number of action potentials that could be elicited via direct current injection. This action has been attributed to the ability of retigabine to enhance flow through the M-channel, a potassium channel that is thought to be a heteromultimer composed of KCNQ2/Q3 potassium channel subunits (Hetka et al., 1999; Main et al., 2000; Rundfeldt and Netzer, 2000b; Wickenden et al., 2000; Tatulian et al., 2001). This is of particular interest with regard to the treatment of epilepsy because it has been demonstrated that compounds that reduce current flow through the M-channel are potent convulsants (Rogawski, 2000). In addition, loss of function mutations in the KCNQ2/Q3 channel underlie the epilepsy syndrome known as benign familial neonatal convulsions (Biervert et al., 1998; Charlier et al., 1998; Singh et al., 1998). Therefore, mechanisms that enhance current flow through this channel, such as that which occurs in the case of retigabine, are likely to reduce excitability and should prove useful in the treatment of seizure disorders.
Our experiments also demonstrate that retigabine, at physiologically relevant concentrations, can significantly increase GABAA receptor-mediated IPSCs via a direct action on these receptors. The experiments on IPSCs, spontaneous mIPSCs, and baseline noise support the hypothesis that retigabine, like diethyl-lactam, is most effective under conditions in which the synapse is not saturated by GABA (Hill et al., 1998; Perrais and Ropert, 1999;Hajos et al., 2000; Leao et al., 2000). Thus, the most robust effect of retigabine on IPSCs occurs during the decay phase of the IPSC, when concentrations of GABA in the synapse are diminishing. Single-channel experiments should be performed to determine the precise mechanism of action of retigabine on the GABAA receptor-gated ion channel complex.
Previous efforts to study the effects of retigabine on GABAA receptors have produced conflicting reports. Although in cultured rat cortical neurons, retigabine concentrations of 10 μM and above resulted in potentiation of currents evoked by the exogenous application of GABA (Rundfeldt and Netzer, 2000a), 100 μM retigabine had no significant effect on the peak amplitudes of evoked IPSCs in rat entorhinal cortex brain slice preparations (Hetka et al., 1999). It is not currently understood why the brain slice experiments did not demonstrate any effects of retigabine on IPSCs. However, this discrepancy could be attributed to a variety of differences in the experimental protocols, such as an insufficient concentration of retigabine at the synapses in the brain slice experiments, the temperature at which experiments were performed (Perrais and Ropert, 1999), and possible differences in the subunit composition of postsynaptic GABAA receptors in culture versus the brain slice.
The culture preparation used in these experiments is a useful tool for determining the effects of novel compounds on synaptic transmission in a simple neural circuit. Using this experimental paradigm, we have determined that retigabine, at concentrations comparable with therapeutically effective plasma concentrations, effectively decreases excitability in neural circuits by hyperpolarizing neurons, impeding action potential generation, and greatly enhancing current flow through GABAA receptors by prolonging IPSCs. Furthermore, these unique synergistic actions of retigabine provide a novel approach for the therapeutic management of seizure disorders.
Acknowledgments
We thank Drs. Harold Wolf and H. Steve White for encouragement and support. In addition, we thank Cynthia Levinthal and Tim Pruess for tissue culture preparation and maintenance and David Daberkow with help on initial miniature IPSC experiments.
Footnotes
- Received June 27, 2001.
- Accepted January 15, 2002.
-
This work was supported by National Institutes of Health contract N01-NS42311 and an ASPET Summer Fellowship to M.M.K.
Abbreviations
- GABA
- γ-aminobutyric acid
- IPSC
- inhibitory postsynaptic current
- TTX
- tetrodotoxin
- EPSC
- excitatory postsynaptic currents
- mIPSC
- miniature inhibitory postsynaptic current
- HBS
- HEPES-buffered saline
- CNQX
- 6-cyano-7-nitroquinoxaline-2,3-dione
- PSC
- postsynaptic current
- PPD
- paired-pulse depression
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}