


Abstract
G protein-coupled receptor kinase (GRK) 2 plays a crucial role in regulating the extent of desensitization and resensitization of G protein-coupled receptors (GPCRs). We have shown that the expression level of GRK2 in lymphocytes decreases during inflammatory diseases such as arthritis. Reactive oxygen species play an important role in a variety of inflammatory conditions, including arthritis. We demonstrate herein that oxidative stress, induced by exposure of lymphocytes to H2O2, results in a 50% reduction in GRK2 protein levels and GRK activity with no changes in mRNA expression. Treatment of lymphocytes with the tyrosine kinase inhibitor genistein partially reverses the effect of H2O2 on GRK2 levels, although we did not detect direct tyrosine phosphorylation of GRK2. Inhibition of the nonproteasomal protease calpain by calpeptin can prevent the H2O2-induced GRK2 decrease. In vitro experiments confirm that GRK2 is partially digested bym-calpain in a calcium-dependent way. Functionally, H2O2-induced decrease in GRK2 levels is associated with an ∼70% decrease in agonist-induced β2-adrenergic receptor sequestration. We describe oxidative stress as a novel mechanism for regulation of the intracellular level of GRK2 during inflammatory processes. Moreover, our data demonstrate that oxidative stress may change the functioning of GPCRs via calpain-dependent regulation of GRK2 levels.
G protein-coupled receptor kinase (GRK) 2 is the most ubiquitous member of the GRK family. GRKs rapidly phosphorylate G protein-coupled receptors (GPCRs) upon agonist stimulation and facilitate the binding of arrestins to the phosphorylated receptors, leading to uncoupling of the receptor from the G protein (Pitcher et al., 1998). This process, known as homologous receptor desensitization, is the loss of receptor responsiveness upon prolonged stimulation. GRKs and arrestins are also thought to play a central role in agonist-induced GPCR internalization, dephosphorylation, and re-expression (Ferguson, 2001). Thus, GRKs contribute to the extent of desensitization and resensitization of GPCRs.
GRK2 is expressed in many different cell types, and its level of expression is particularly high in cells of the immune system (Oppermann et al., 1999). GPCRs that are known substrates of GRK2 include the β2-adrenergic receptor (β2-AR), the chemokine receptors CCR2b and CCR5, the platelet activating factor receptor, and the neurokinin-1 receptor for substance P (Pitcher et al., 1998). The extent of agonist-induced desensitization and sequestration of GPCRs depends on the intracellular availability of GRKs and β-arrestins (Menard et al., 1997). Changes in the cellular levels or in the activity of GRK2 have been shown to influence desensitization and internalization of β2-AR, chemokine receptors, and opioid receptors (Aramori et al., 1997; Menard et al., 1997; Li et al., 2000).
Interestingly, changes in GRK2 levels have been reported in a number of disease states. In human heart failure, GRK2 levels in myocardial tissue are increased (Ungerer et al., 1994). Increased levels of GRK2 in peripheral blood mononuclear cells (PBMCs) have also been reported in humans with hypertension (Gros et al., 1997). In contrast, our recent data have demonstrated that in humans with rheumatoid arthritis, GRK2 levels in PBMCs are reduced (Lombardi et al., 1999). Moreover, induction in rats of adjuvant arthritis results in marked down-regulation of GRK2 protein in splenocytes and mesenteric lymph node cells at the peak of the disease. After remission of the disease, GRK levels normalize in these cells (Lombardi et al., 2001).
Little is known about mechanisms involved in regulation of intracellular GRK2 levels. In a heterologous system, activation of the β2-AR results in enhanced degradation of the GRK2 protein via the proteasome pathway (Penela et al., 1998). In peripheral blood lymphocytes, mitogenic stimulation induces an increase in GRK2 protein via increased mRNA expression (De Blasi et al., 1995). In contrast, the cytokines IL-6 and IFN-γ reduce intracellular levels of GRK2 protein in these cells (Lombardi et al., 1999). In line with the latter results, we have shown that the activity of the GRK2 promoter is reduced in cardiovascular cells by IFN-γ and also by TNF-α or IL-1β (Ramos-Ruiz et al., 2000)
Reactive oxygen species (ROS) have been implicated in the pathogenesis of arthritis and other autoimmune diseases (Droge, 2002). Moreover, production of ROS has been detected in a variety of cells stimulated with cytokines, peptide growth factors, and agonists of GPCRs (Thannickal and Fanburg, 2000). During inflammatory processes, lymphocytes are exposed to H2O2 and other ROS that are derived from activated macrophages and neutrophils as a first line of defense against invading pathogens. If not produced in too high levels, ROS can act as second messengers and regulate cellular functions, e.g., during immune and inflammatory processes (Remacle et al., 1995). Exposure of T lymphocytes to H2O2 or other donors of ROS results in a rapid tyrosine phosphorylation of a large number of proteins (Schieven et al., 1993). Moreover, H2O2 induces a rapid and transient increase in intracellular calcium concentration (Thannickal and Fanburg, 2000). Further downstream, ROS regulate transcription factors, including nuclear factor κB (NF-κB) (Schreck et al., 1991). H2O2-induced activation of NF-κB is dependent on degradation of IκB, the natural inhibitor of NF-κB (Schreck et al., 1991). This process involves two different pathways of protein degradation: rapid degradation of IκB via the proteasome pathway and a slower process involving calpain-dependent degradation of IκB (Schoonbroodt et al., 2000).
The aim of our present study was to investigate the effect of H2O2 on protein expression levels of GRK2 in T lymphocytes. Our data demonstrate that H2O2 induces a decrease in GRK2 protein level, via a mechanism involving tyrosine phosphorylation and the protease calpain. Consistently, H2O2 treatment promotes a decrease in agonist-mediated β2-AR sequestration. Our results put forward a novel mechanism by which the intracellular level of GRK2 can be modulated.
Experimental Procedures
Materials.
Human Jurkat T cells were obtained from American Type Culture Collection (Manassas, VA). RPMI 1640 medium, penicillin/streptomycin, glutamine, and fetal bovine serum were obtained from Invitrogen (Carlsbad, CA). Ficoll-Hypaque and protein A-Sepharose were from Amersham Biosciences AB (Uppsala, Sweden). Hydrogen peroxide (30%, w/v) and Na3VO4 were obtained from Merck (Darmstadt, Germany). Leupeptin, pepstatin, benzamidine, catalase, PD98059, wortmannin, and 1-5(isoquinoline sulfonyl)-2-methylpiperazine (H7) were purchased from Sigma-Aldrich (St. Louis, MO). PP2 was obtained from Alexis Corporation (Laüfelfingen, Switzerland). Lactacystin and m-calpain were from Calbiochem (La Jolla, CA). Calpeptin, genistein, and 5,6-dichloro-1-β-ribofuranosyl-benzimidazole (DRB) were from BIOMOL Research Laboratories (Plymouth Meeting, PA). CGP12177 was purchased from Tocris Cookson (Bristol, UK). [γ-33P]ATP,125I-cyanopindolol (I-CYP), and peroxidase-conjugated donkey anti-rabbit IgG were purchased from Amersham Biosciences UK, Ltd. (Little Chalfont, Buckinghamshire, UK). Peroxidase-conjugated sheep anti-mouse IgG and 4-(2-aminoethyl)benzenesulfonyl fluoride were from Roche Applied Science (Mannheim, Germany). GRK2 polyclonal rabbit antibody (Sc-562) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). β-Arrestin1 mouse monoclonal antibody and anti-phosphotyrosine PY20 antibody were obtained from Transduction Laboratories (Lexington, KY). Protein concentration was determined using protein assay reagent (Bio-Rad, Hercules, CA) using bovine serum albumin as standard.
Cell Culture and Treatment.
Human PBMCs were prepared by Ficoll-Hypaque gradient centrifugation from healthy blood donors, and T lymphocytes were isolated by rosetting with 2-amino-ethyl-isothiuronium-treated sheep red blood cells (Heijnen et al., 1979). T lymphocytes were cultured overnight in culture medium (RPMI 1640 supplemented with 10% fetal bovine serum, 2 mM glutamine, and antibiotics) to remove erythrocyte ghosts. CD4+ and CD8+ T cells and CD19+ B cells were purified by magnetic cell sorting using MACS beads and specific monoclonal antibodies (Miltenyi Biotec, Nordrhein-Westfalen, Germany). Neutrophils were isolated by incubating the erythrocyte pellet after the Ficoll gradient with 0.15 volume of 5% dextran for 30 min at 37°C. The supernatant, containing neutrophils, was gently removed and washed twice with RPMI 1640 medium. Jurkat T cells were maintained in culture medium. For experimental treatments, cells were incubated at a density of 1 × 106 cells/ml in culture medium in 24-well culture plates (Falcon, Cowley, UK), with the reagents indicated at 37°C. Catalase was added contemporaneously with H2O2. All others inhibitors were added 1 h before H2O2 treatment and maintained during incubation. At the end of the incubation period, cells were collected on ice, washed twice with ice-cold PBS, and tested for viability by trypan blue exclusion. Alternatively, 106 T lymphocytes were incubated with 4 μg/ml Con A and 0.2 × 106 neutrophils for 4 h, in presence or absence of SOD and catalase, both at a concentration of 40 μg/ml. After incubation, cells were collected and T lymphocytes were isolated by Ficoll density gradient centrifugation, washed twice with ice-cold PBS, and tested for viability by trypan blue exclusion. In all samples viability was higher than 90%.
Preparation of Cell Lysates and Western Blotting.
Cells (2–3 × 106) were lysed in ice-cold lysis buffer [20 mM HEPES pH 7.5, 1% Triton X-100, 150 mM NaCl, 10 mM EDTA, 2 mM 4-(2-aminoethyl)benzenesulfonyl fluoride, 20 μg/ml leupeptin, 200 μg/ml benzamidine, and 10 μg/ml pepstatin] for 30 min at 4°C. The lysates were clarified by centrifugation at 13,000g for 15 min at 4°C. Proteins (15–20 μg) were separated by 10% SDS-PAGE and analyzed for GRK2 and β-arrestin1 expression by immunoblot as described previously (Lombardi et al., 1999, 2001). Tyrosine phosphorylation was detected using a 1:1,000 dilution of the anti-phosphotyrosine mouse monoclonal antibody PY20 followed by incubation with peroxidase-conjugated sheep anti-mouse IgG (Roche Applied Science) at 1:10,000 dilution. Immunoreactivity was detected by enhanced chemiluminescence (Amersham Biosciences UK, Ltd.). Immunoprecipitated GRK2 was detected in the same blots by incubating stripped filters with the GRK2 Sc-562 antibody. Band density was determined using a GS-700 imaging densitometer and analyzed by Molecular Analyst software, version 1.5 (Bio-Rad).
Kinase Activity.
GRK enzymatic activity was assessed using light-dependent phosphorylation of rhodopsin by cytosolic and membrane fractions as described previously (Lombardi et al., 1999). Samples were separated by electrophoresis on 10% SDS-PAGE. Phosphorylated rhodopsin was visualized by autoradiography. Bands corresponding to rhodopsin (∼38 kDa) were cut from the dried gel and quantified via liquid scintillation spectroscopy.
Northern Blot Analysis.
Total RNA was isolated using RNAzol-B (Campro-Scientific, Veenendaal, The Netherlands). Ten micrograms of RNA/lane was separated on a 1% agarose-formaldehyde gel and transferred to a Hybond-N+ membrane (Amersham Biosciences UK, Ltd.). Northern blot analysis was performed as described previously (Lombardi et al., 1999). β-Actin mRNA expression was determined on the same membrane, after stripping with 0.5% SDS, using a random primed cDNA probe (1.8-kilobase human β-actin cDNA; CLONTECH, Palo Alto, CA).
Immunoprecipitation.
After incubation in the presence or absence of H2O2, cells (3 × 106) were pelleted and lysed in 200 μl of ice-cold radioimmunoprecipitation assay buffer (20 mM HEPES pH 7.5, 1% Triton X-100, 0.5% sodium deoxycholate, 10 mM EDTA, 1 mM Na3VO4, and a cocktail of protease inhibitors as mentioned above) for 30 min at 4°C. The lysates were clarified by centrifugation at 13,000g for 15 min at 4°C, and an aliquot (20 μl) was used to assess H2O2 -induced tyrosine phosphorylation of total protein. Before GRK2 immunoprecipitation, the supernatants were precleared by 1-h incubation with 1 μg of rabbit IgG (Southern Biotechnology, Birmingham, AL). GRK2 was then immunoprecipitated by overnight incubation at 4°C with 2 μg of GRK2 rabbit polyclonal antibody (Sc-562) followed by addition of 20 μl of (50%) slurry of protein A-Sepharose for 1 h. The immune complexes were washed five times with ice-cold PBS, and the proteins were extracted by boiling for 5 min in 30 μl of Laemmli sample buffer. After resolution by 10% SDS-PAGE, the proteins were transferred on nitrocellulose membranes and subjected to immunoblotting as indicated above.
In Vitro Proteolysis Assay.
Recombinant GRK2 (0.5 μg), purified from baculovirus-infected Sf9 cells as described previously (Penela et al., 1998), was incubated with 1 unit ofm-calpain in proteolysis buffer (20 mM Tris-HCl pH 7.4 and 10 mM β-mercaptoethanol) in the presence or absence of 20 mM CaCl2 at 30°C for 45 min. Samples were resolved on 7.5% SDS-PAGE and transferred to nitrocellulose. Blots were developed with several anti-GRK2 polyclonal antibodies, raised against recombinant GRK2 (Ab 9, dilution 1:1000; a gift from Dr. J. L. Benovic, Jefferson University, Philadelphia, PA), against fusion proteins containing amino acids 50 to 145 (Ab FP1, dilution 1:1000) or 436 to 689 of bovine GRK2 (Ab FP2, dilution 1:600) (Penela et al., 1998), or the anti-peptide antibodies Ab 927 (dilution 1:1000, raised against amino acids 663–679) and Sc-562, raised against amino acids 675 to 689 of GRK2 (Santa Cruz Biotechnology). None of these antibodies cross-react with calpain.
β2-AR Sequestration Assay.
T lymphocytes were resuspended at a density of 106cells/ml and incubated with or without 400 μM H2O2 for 4 h. Cells were then exposed to 1 μM (−)-isoproterenol and 100 μM ascorbate or medium for 10 min at 37°C and washed twice with ice-cold PBS. Subsequently, cells were resuspended in binding buffer (PBS and 0.5% bovine serum albumin) at a density of 2.5 × 106 cells/ml. To determine the amount of internalized receptors a binding assay was performed. Total binding was determined by using 175 pM of 125I-CYP alone. The number of internalized receptors was determined by using 175 pM of125I-CYP plus 1 μM of the hydrophilic antagonist CGP12177, and nonspecific binding was determined by using 175 pM of 125I-CYP plus 1 μM (−)-propranolol (Ferguson et al., 1998). β2-AR sequestration was calculated as the ratio of (specific receptor binding of125I-CYP in the presence of CGP12177)/(specific receptor binding of 125I-CYP in the absence of CGP12177).
Statistical Analysis.
Data are expressed as a mean value ± S.E. All results were confirmed in at least two separate experiments. Specific measurements were compared using Student'st test or one-way analysis of variance followed by Bonferroni's analysis. Two-tailed p < 0.05 was considered statistically significant.
Results
H2O2 Induces a Decrease in GRK2 Protein Levels in T Lymphocytes.
Exposure of cells to H2O2 is frequently used as a model of oxidative stress (Schoonbroodt et al., 2000; Thannickal and Fanburg, 2000; Takeyama et al., 2000). For example, it has been shown that 6-h exposure of cells to 400 μM H2O2 can up-regulate mucin synthesis to a similar extent as ROS produced by activated neutrophils (Takeyama et al., 2000). In our experiments, we exposed human T lymphocytes to increasing concentrations from 100 to 400 μM of H2O2 for 4 h and determined the level of GRK2 in total cell lysates by Western blotting. Exposure of T lymphocytes to 100 to 400 μM H2O2 leads to a significant dose-dependent decrease in GRK2 protein levels in these cells (∼50% at 400 μM; p < 0.01 versus control; Fig.1A). The effect of 400 μM H2O2 becomes apparent at 2 h of incubation (∼20% decrease of GRK2 levels) and reaches a maximum after 4 h of incubation. The effect of H2O2 on GRK2 protein levels is similar after 6 h of incubation (data not shown). Comparable kinetics of GRK2 down-regulation was observed in Jurkat T cells incubated with H2O2 (data not shown).

H2O2 induces a decrease in GRK2 expression in lymphocytes. A,H2O2 induces a dose-dependent decrease of GRK2 expression levels in T lymphocytes. Cells were treated for 4 h with 100 to 400 μM H2O2, and GRK2 expression levels were assessed in total cell lysate resolved by 10% SDS-PAGE and analyzed by Western blotting. Each data point represents the mean ± S.E. from three independent experiments. ∗, p< 0.05; ∗∗, p < 0.01 versus control. Inset, representative Western blot depicting immunodetectable GRK2 in 20 μg of total cell lysate. B, effect of catalase on H2O2-induced decrease of GRK2 in T lymphocytes. Addition of 40 μg/ml catalase completely prevents the effect of 400 μM H2O2. Each data point represents the mean ± S.E. from three independent experiments. ∗∗,p < 0.01 versus control. No effect of H2O2 is observed on expression levels of β-arrestin1 assessed on the same Western blots. Inset, representative Western blot depicting immunodetectable GRK2 and β-arrestin1 in 20 μg of total cell lysate. C, assessment of GRK activity in cytosolic and membrane fractions from T lymphocytes treated for 4 h with 400 μM H2O2 as detailed under Experimental Procedures. Data are expressed as percentage of control values (considered as 100%). Each data point represents the mean ± S.E. from two independent experiments. D, GRK2 mRNA expression assessed in T lymphocytes incubated in the presence or absence of 400 μM H2O2 for 4 h. Each lane contains 10 μg of total RNA. β-Actin mRNA expression was determined on the same membrane after stripping.
The enzyme catalase rapidly converts H2O2 into H2O and O2. As is shown in Fig. 1B, addition of 40 μg/ml catalase to cultures of T lymphocytes treated with H2O2completely abolishes the effect of H2O2 on GRK2. The decrease in GRK2 levels is not the result of cytotoxic effects of H2O2, because recovery and viability were similar in control medium and H2O2-containing cultures (>90%). Moreover, the effect of H2O2 on GRK2 levels in lymphocytes does not result from general protein degradation, because recovery of protein is not significantly different between treated and control samples (data not shown). In addition, exposure of T lymphocytes to H2O2 does not change the level of β-arrestin1, a GRK2 cofactor, as assessed by incubating the same blots with β-arrestin1 antibody (Fig. 1B).
To investigate whether the H2O2-induced decrease in GRK2 expression is associated with a decrease in GRK activity, T lymphocytes were incubated with H2O2 and cytosolic and membrane fractions were prepared. GRK activity in T lymphocyte cytosolic and membrane fractions was determined in vitro by agonist (light)-dependent phosphorylation of rhodopsin. Exposure of T lymphocytes to 400 μM H2O2 for 4 h reduces GRK activity in both cytosolic and membrane fractions by ∼50 and ∼54%, respectively (Fig. 1C).
To analyze whether H2O2influences GRK2 mRNA levels, T lymphocytes were incubated with H2O2 for 4 h, and RNA was subjected to Northern blot analysis. H2O2 does not significantly influence the expression of mRNA encoding GRK2 (∼3.8-kilobase transcript) in these cells (H2O2-treated cells, 106 ± 4% of control; Fig. 1D), suggesting that the effect of H2O2 is at the post-transcriptional level.
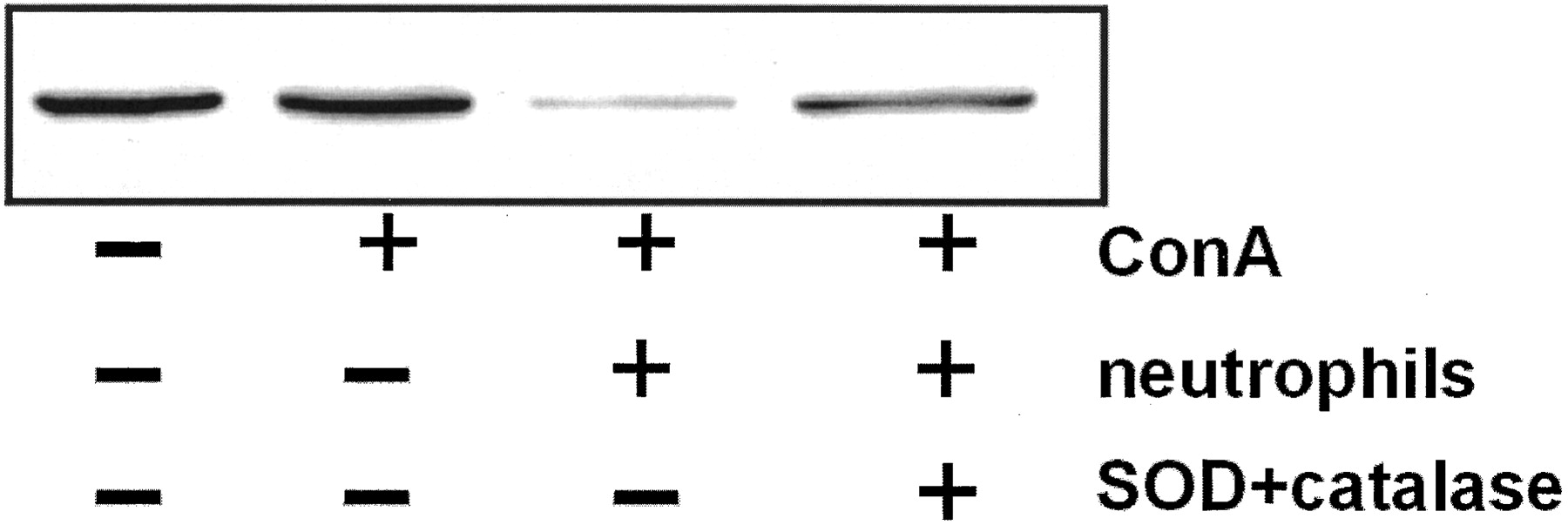
To further address the physiological relevance of the H2O2-induced decrease in GRK2 expression in T lymphocytes, we examined the effect of activated neutrophils on GRK2 expression in T cells. We incubated T cells for 4 h in the presence of 20% neutrophils and Con A and determined GRK2 levels in the T cells. As is shown in Fig.2 culture of T cells with Con A alone for 4 h does not alter GRK2 expression. However, addition of 20% neutrophils to the cultures of T cells with Con A results in an ∼80% reduction in GRK2 protein expression in T cells. Importantly, the effect of the neutrophils can be reversed to a large extent by addition of SOD and catalase, which suggests the involvement of neutrophil-derived reactive oxygen species in this process.

Activated neutrophils can decrease the GRK2 level in T lymphocytes. T lymphocytes (106) were incubated with 4 μg/ml Con A and 0.2 × 106 neutrophils for 4 h, in presence or absence of SOD and catalase, both at a concentration of 40 μg/ml. Cell lysate (30 μg) was analyzed by Western blot. A representative Western blot of two independent experiments is shown.
Cell Type Specificity.
To investigate whether the effect of H2O2 on GRK2 levels is limited to a specific subset of lymphocytes, we analyzed the effect of H2O2 on GRK2 protein levels in isolated fractions of CD4+ T lymphocytes, CD8+ T lymphocytes, and CD19+ B lymphocytes. Each subset was incubated for 4 h with 400 μM H2O2 in the presence or absence of 40 μg/ml catalase, and GRK2 levels were analyzed in total cell lysates. The data in Fig. 3clearly demonstrate that H2O2 induces a decrease in GRK2 protein levels in all subsets of lymphocytes. Moreover, the effect of H2O2 was catalase-reversible, and no changes were observed in β-arrestin1 expression levels in any of the subsets of lymphocytes tested (data not shown).

Cell type specificity. GRK 2 levels in CD4+ T lymphocytes, CD8+ T lymphocytes, and C19+ B lymphocytes after treatment with 400 μM H2O2 for 4 h in the presence or absence of 40 μg/ml catalase. Total cell lysates (20 μg) were analyzed by Western blotting. Gels representative of two independent experiments are shown.
Tyrosine Kinases Are Involved in H2O2-Induced Down-Regulation of GRK2 Levels in Human Lymphocytes.
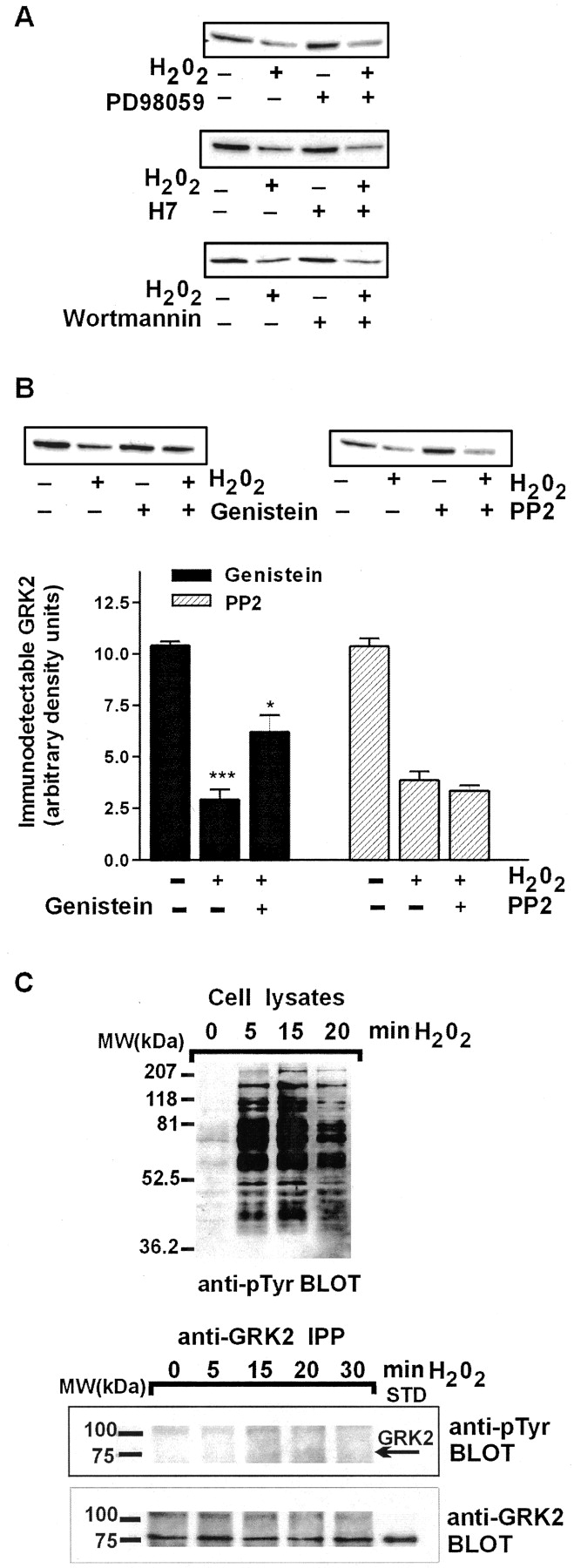
In T lymphocytes, H2O2 is known to stimulate a number of signaling events, including activation of tyrosine phosphorylation (Schieven et al., 1993), protein kinase C (Abe et al., 1998), PI3-kinase (Qin et al., 2000), and MAP kinases (Guyton et al., 1996). It has been shown that GRK2 can be phosphorylated by MAP kinase (Elorza et al., 2000) and PKC (Chuang et al., 1995) as well as tyrosine kinases (Sarnago et al., 1999). To investigate the possible role of these kinases in the H2O2-induced decrease in lymphocyte GRK2 levels, we analyzed the effect of specific kinase inhibitors. Inhibition of MAP kinase by 50 μM PD98059 (Elorza et al., 2000), of PKC by 20 μM H7 (Chuang et al., 1995), and of PI3-kinase by 100 nM wortmannin (Qin et al., 2000), respectively, does not have any effect on the H2O2-induced decrease in cellular GRK2 content (Fig.4A). However, administration of the tyrosine kinase inhibitor genistein does have a significant effect in this system (Fig. 4B, inset). Preincubation of cells with 100 μM genistein for 1 h before addition of H2O2 partially reverses the effect of H2O2 on GRK2 levels (p < 0.05 versus H2O2 alone). We recently demonstrated in COS-7 cells that the c-Src member of the tyrosine kinase family is capable of phosphorylating GRK2 (Sarnago et al., 1999). Therefore, we tested whether PP2, a specific inhibitor of c-Src, could prevent H2O2-induced down-regulation of GRK2 protein in T lymphocytes. However, as shown in Fig. 4B (right), PP2 has no effect on the H2O2-induced decrease in GRK2 protein levels in T lymphocytes.

Tyrosine kinase inhibitor genistein partially reverses the effect of H2O2 on GRK2 expression levels. A, GRK2 expression in T lymphocytes incubated with 400 μM H2O2 for 4 h with or without 1-h preincubation with 50 μM PD98059, 20 μM H7, or 100 nM wortmannin, respectively. Total cell lysates (20 μg) were analyzed by Western blotting. Results are representative of three independent experiments. B, effect of the tyrosine kinase inhibitor genistein on the H2O2-induced decrease of GRK2 protein. T lymphocytes were preincubated for 1 h with 100 μM genistein or 5 μM PP2 (a specific Src inhibitor) and subsequently incubated for 4 h with 400 μM H2O2. ∗∗∗,p < 0.001 versus control; ∗,p < 0.05 versus H2O2-treated cells. Each data point represents the mean ± S.E. from three independent experiments. Insets, representative Western blots depicting immunodetectable GRK2 in 20 μg of total cell lysate are shown. C, time course of tyrosine phosphorylation induced by 400 μM H2O2 of whole cell lysates from T lymphocytes (top) resolved by 10% SDS-PAGE and analyzed with a monoclonal anti-phosphotyrosine (anti-pTyr) antibody. To assess direct tyrosine phosphorylation on GRK2, radioimmunoprecipitation assay buffer lysates from 3 × 106cells were immunoprecipitated (IPP) with an anti-GRK2 polyclonal antibody as described under Experimental Procedures. Immunoprecipitates were resolved by 10% SDS-PAGE and analyzed with anti-pTyr antibody (bottom). After stripping, the presence of GRK2 was assessed in the same gel using the specific GRK2 antibody. The arrow indicates the migration of GRK2 as assessed by the position of the recombinant GRK2 resolved in the same gel (GRK2, STD). Gels representative of two independent experiments are shown.
To investigate whether GRK2 becomes phosphorylated on tyrosine residues after exposure to H2O2, GRK2 was immunoprecipitated from H2O2-stimulated T lymphocytes and analyzed with an anti-phosphotyrosine antibody. In agreement with previous reports, exposure of T cells to H2O2 results in rapid tyrosine phosphorylation of a large number of proteins as assessed in total lysates (Fig. 4C, top) (Carballo et al., 1999). However, H2O2 does not induce detectable tyrosine phosphorylation of GRK2 in a time course of 5 to 30 min (Fig. 4C, bottom).
Lack of Involvement of the Proteasome Pathway.
We have shown previously that transfected (Penela et al., 1998) or endogenous (Penela et al., 2001) GRK2 can be rapidly degraded via the proteasome pathway. We also showed that agonist activation of β-adrenergic receptors results in increased GRK2 degradation via this proteolytic pathway (Penela et al., 1998, 2001). To investigate a putative role for proteasome-dependent H2O2-induced down-regulation of GRK2, T lymphocytes were preincubated with the proteasome inhibitor lactacystin and stimulated with H2O2. However, lactacystin (in concentrations up to 30 μM) did not inhibit the H2O2-induced decrease in cellular GRK2 levels (Fig. 5A), suggesting that the decrease is not due to degradation through the proteasome pathway. Moreover, inhibition of lysosomal protein degradation by 100 μg/ml leupeptin does not prevent H2O2-induced decreases in GRK2 protein level either (Fig. 5A).

Calpain is involved in the H2O2-induced decrease in GRK2. A, GRK2 expression in T lymphocytes preincubated with 30 μM lactacystin or 100 μg/ml leupeptin, respectively, for 1 h before treatment with 400 μM H2O2 for 4 h. Total cell lysates (20 μg) were resolved by 10% SDS-PAGE and analyzed by Western blotting. Representative gels of three independent experiments are shown. B, effect of the calpain inhibitor calpeptin. T cells were preincubated for 1 h with 100 μM calpeptin and incubated with 400 μM H2O2 for 4 h. Total cell lysates (20 μg) were resolved by 10% SDS-PAGE and analyzed by Western blotting. Each data point represents the mean ± S.E. of three independent experiments. ∗∗, p < 0.01 versus H2O2-treated cells. Inset, representative Western blot depicting immunodetectable GRK2 in 20 μg of total cell lysate. C, effect of the casein kinase II inhibitor DRB on H2O2-induced down-regulation of GRK2 in T lymphocytes. Cells were pretreated with 0.2 mM DRB before H2O2 treatment. Total cell lysates (20 μg) were resolved by 10% SDS-PAGE and analyzed by Western blotting. Each data point represents the mean ± S.E. from three independent experiments. ∗, p < 0.05 versus H2O2-treated cells. Inset, representative Western blot depicting rGRK2 standard (S) and immunodetectable GRK2 in 20 μg of total cell lysate.
Calpain Is Involved in GRK2 Decrease after Treatment with H2O2.
It is known that H2O2 can decrease the level of IκB via a nonproteasomal pathway involving the protease calpain (Schoonbroodt et al., 2000). Moreover, the calpain-mediated decrease of IκB induced by H2O2 is a slower process than the proteasome-dependent proteolysis. In view of the relatively slow kinetics of H2O2-induced decreases of GRK2 levels in human T lymphocytes, we examined the effect of the calpain inhibitor calpeptin in our system. The data in Fig. 5B clearly demonstrate that calpeptin can almost completely prevent the H2O2-induced decrease in cellular GRK2 level (p < 0.01 versus H2O2 alone). In addition, the H2O2-induced decrease in GRK2 levels in Jurkat T cells could also be reversed by calpeptin treatment (data not shown).
It is known that calpain-dependent degradation of IκB can be prevented by inhibition of casein kinase II (Schoonbroodt et al., 2000). Therefore, we investigated whether casein kinase II is also involved in the H2O2-induced decrease of GRK2 levels. T lymphocytes were pretreated for 1 h with 0.2 mM DRB that can specifically inhibit casein kinase II without inducing any cytotoxic effect, and cultured for 4 h with 400 μM H2O2. Figure 5C shows that DRB can indeed partially prevent the H2O2-induced decrease in GRK2 (p < 0.05 versus H2O2 alone).
In Vitro Digestion of GRK2 by m-Calpain.
To confirm that calpain can directly digest GRK2, in vitro experiments were performed using recombinant GRK2 (rGRK2) and purifiedm-calpain. As shown in Fig. 6, rGRK2 (∼80 kDa) is partially digested by m-calpain in a calcium-dependent way, into a protein fragment with an estimated weight of ∼76 kDa. This fragment is detected by anti-GRK2 antibodies raised against different N-terminal and C-terminal regions of the kinase (AbFP1, AbFP2) and is still recognized by an antibody raised against the peptide 663-679 (Ab 927), although with reduced intensity. However, an antibody raised against the C-terminal 675-689 peptide (Sc-562) no longer recognizes the 76-kDa GRK2 fragment. These data indicate that the fragment removed by m-calpain comprises the 10 carboxyl-terminal amino acids of GRK2.

Proteolysis of purified recombinant GRK2 bym-calpain. Recombinant GRK2 (∼80-kDa band) was incubated with or without m-calpain in the presence or absence of 20 mM CaCl2 at 30°C for 45 min. Samples were resolved on 7.5% SDS-PAGE and transferred to nitrocellulose. Blots were developed as detailed under Experimental Procedureswith several anti-GRK2 polyclonal antibodies: Ab 9, raised against recombinant GRK2 (dilution 1:1000); Ab FP1 (dilution 1:1000, raised against residues 50–145); Ab FP2 (dilution 1:600, against residues 436–689); Ab 927 (dilution 1:1000, raised against amino acids 663–679); and Sc-562, a commercial antibody raised against amino acids 675–689 of GRK2. A degradation product of ∼76 kDa is detected. Results are representative of three independent experiments.
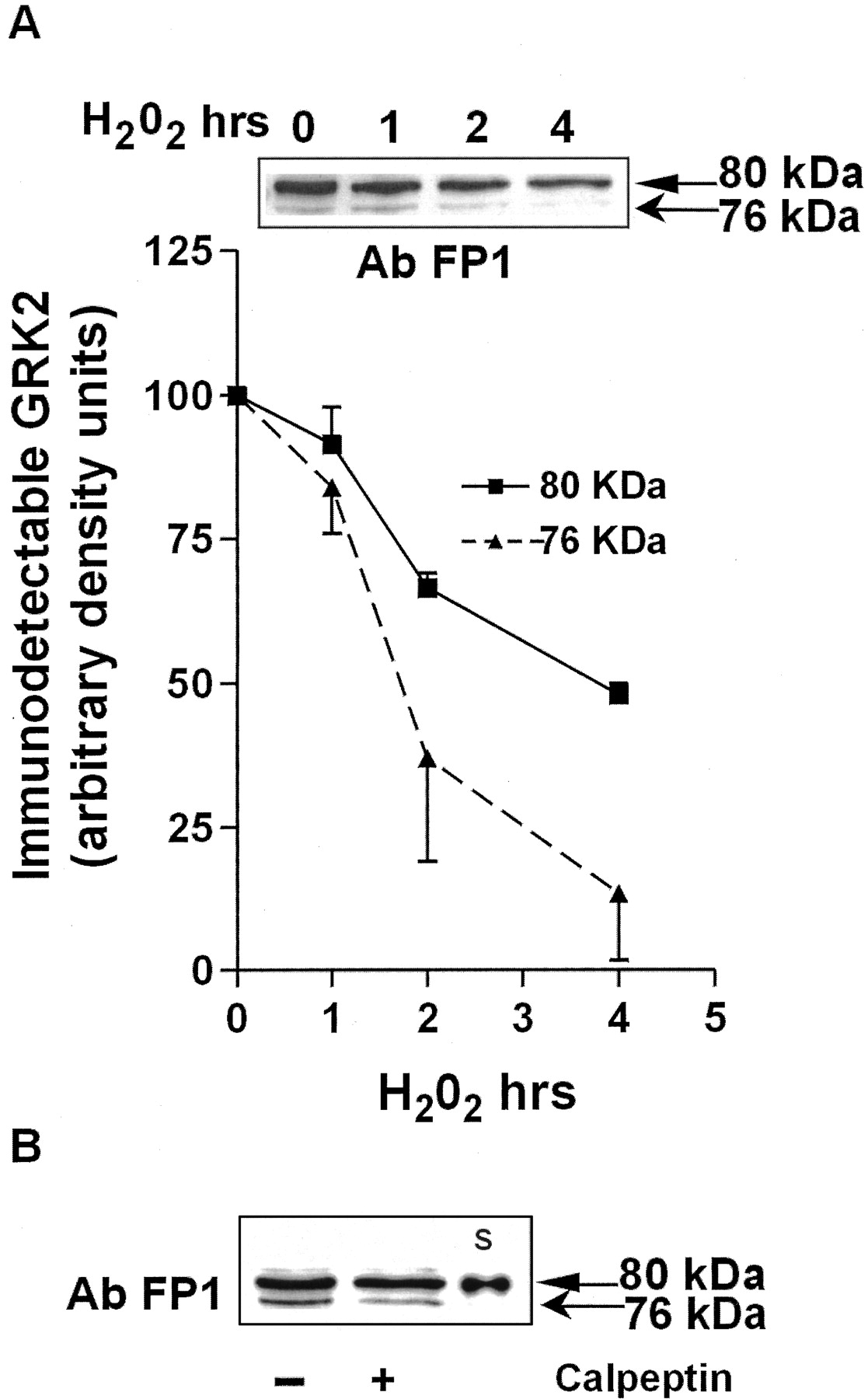
Subsequently, we tested whether the 76-kDa fragment of GRK2 is detected “in vivo” and whether its level is modulated after exposure of T lymphocytes to H2O2. We therefore reprobed the samples used for assessment of the time course of GRK2 degradation, with the AbFP1 antibody, that recognizes the 76-kDa fragment of GRK2 generated by m-calpain in vitro (Fig. 6). Indeed, a similar 76-kDa fragment is detectable in lysates of T lymphocytes, and it is more rapidly degraded than the 80-kDa protein after exposure of cells to H2O2. After 4 h of stimulation with H2O2 the level of the 80-kDa protein is ∼50% of control expression levels, whereas the immunodetectable 76-kDa fragment is reduced to ∼13% of control expression levels (Fig. 7A). In addition, treatment of cells with calpeptin in the absence of H2O2 inhibits the formation of the 76-kDa fragment by ∼50% (Fig. 7B). These data suggest that the 76-kDa band present in control samples results from basal calpain activity.

In vivo detection of an ∼76-kDa GRK2 fragment and its modulation by H2O2 treatment. A, cell lysates from T lymphocytes (n = 2) treated with 400 μM H2O2 for 1 to 4 h were resolved on 7.5% SDS-PAGE and transferred to nitrocellulose. Blots were developed with Ab FP1 (dilution 1:1000) that recognizes both the GRK2 80-kDa band and the ∼76-kDa degradation product (inset). Results are representative of two independent experiments. B, T lymphocytes were preincubated for 1 h in the presence or absence of 100 μM calpeptin and total cell lysates (20 μg/lane) were resolved on 7.5% SDS-PAGE. Blots were developed with Ab FP1 as described above. A representative Western blot that includes rGRK2 standard (S) is shown.
H2O2 Treatment Induces a Decrease in β2-AR Sequestration.
GRK2 is involved in agonist-induced receptor sequestration (Ferguson et al., 1998). To investigate potential functional consequences of H2O2-induced decreases in GRK2, we determined the effect of H2O2 treatment on agonist-induced sequestration of the β2-AR. In line with data in the literature, initial experiments showed that isoproterenol-induced β2-AR sequestration reached a maximum after 5 to 10 min (data not shown). Therefore, we used an incubation period of 10 min with isoproterenol in subsequent experiments. A 10-min incubation with isoproterenol does not change the total number of β2-ARs (data not shown).
The data in Fig. 8 show that exposure of T lymphocytes to H2O2 (400 μM, 4 h) does not significantly alter the percentage of sequestered β2-ARs in the absence of β2-AR agonist. However, pretreatment of T lymphocytes with H2O2results in an ∼70% reduction in isoproterenol-induced receptor sequestration (p < 0.05).

Effect of H2O2 on β2-AR sequestration. T lymphocytes were incubated for 4 h with or without 400 μM H2O2, and β2 -AR internalization in response to 10-min incubation with 1 μM (−)-isoproterenol was analyzed by125I-CYP binding in the presence or absence of the hydrophilic ligand CGP12177 (see Experimental Procedures) and are represented as percentage of total β2-AR. Results are the means ± S.E. of six independent experiments performed in duplicate. ∗,p < 0.05 versus untreated cells stimulated with isoproterenol.
Discussion
We show herein for the first time that exposure of lymphocytes to oxidative stress results in a decrease in cellular GRK2 protein levels. Oxygen radicals that are produced by activated macrophages and neutrophils can alter the activity of lymphocytes. Exposure of lymphocytes to oxygen radicals results in increased intracellular calcium level (Thannickal and Fanburg, 2000), rapid tyrosine phosphorylation of a variety of proteins (Schieven et al., 1993), and activation of transcription factors such as NF-κB (Schreck et al., 1991).
Various immune and other stimuli are known to regulate cellular GRK2 protein levels, although with marked differences in kinetics. For example, mitogenic stimulation of lymphocytes induces increased GRK2 protein expression. This process requires at least 24 to 48 h and is associated with increased synthesis of mRNA encoding GRK2 (De Blasi et al., 1995). More recently, we have shown that 24 h in vitro exposure of lymphocytes to the cytokines IFN-γ or IL-6 leads to reduced intracellular GRK2 protein levels (Lombardi et al., 1999). In line with these findings, the cytokines IFN-γ, TNF-α, and IL-1β have been shown to decrease the activity of the GRK2 promoter in cardiovascular cells (Ramos-Ruiz et al., 2000). Triggering of GPCRs can also modulate GRK2 expression. The GRK2 promoter is activated after stimulation of α1-adrenergic receptors in transfected aortic smooth muscle cells (Ramos-Ruiz et al., 2000). In addition, stimulation of β-adrenergic or chemokine receptors results in increased degradation of coexpressed or endogenous GRK2 (Penela et al., 1998, 2001).
The effect of oxidative stress on GRK2 protein levels in lymphocytes can be detected after 2 h and becomes more pronounced after 4 h of incubation. The half-life of mRNA for GRK2 in lymphoid cells has been shown to be more than 7 h (Parruti et al., 1993). In our experiments, we did not observe any effect of H2O2 on GRK2 mRNA expression during the 4-h incubation period. Together, these results strongly suggest that the effect of H2O2 on GRK2 protein levels is located at the post-transcriptional level.
We show herein that the H2O2-induced decreases in GRK2 protein level can be prevented almost completely by addition of the calpain inhibitor calpeptin, whereas the presence of leupeptin, an inhibitor of protein degradation in lysosomes, or of the proteasome inhibitor lactacystin does not inhibit the effect of H2O2. In addition, in other cell types it has been shown that exposure to H2O2 results in activation of calpain (Ishihara et al., 2000). These data strongly suggest that the H2O2-induced decrease in GRK2 in lymphocytes is mediated via the calpain proteolytic pathway. Interestingly, a similar H2O2-induced decrease in GRK2 levels is observed in smooth muscle vascular cells, and this effect is also blocked in the presence of calpain inhibitors (P. Penela and F. Mayor, Jr., unpublished observations), thus suggesting that this is a general mechanism for GRK2 modulation. Calpains are a family of calcium-dependent enzymes that are involved in the cleavage of several cellular substrates, including transcription factors, cytoskeletal elements, and signaling molecules. A PEST region, rich in proline (P), glutamic acid (E), serine (S), and threonine (T), and/or the presence of calmodulin binding domains are considered to be typical features of proteins that are substrates for calpains (Rechsteiner and Rogers, 1996). Using the PEST-FIND algorithm, we found a putative PEST region (aa 591–615, score +1.57) in GRK2. Moreover, GRK2 contains two calmodulin-binding domains located in the NH2-terminal (aa-1–88) and COOH-terminal domain (aa 593–689), respectively (Levay et al., 1998).
Our in vitro proteolysis experiments confirm that purified calpain is capable of promoting a partial degradation of recombinant GRK2 (∼80 kDa) in a calcium-dependent way. This process leads to the appearance of an ∼76-kDa fragment of the kinase lacking at least the last 10 COOH-terminal amino acids. A similar ∼76-kDa fragment is also detectable in untreated cells and its level decreases by ∼50% after exposure of cells for 4 h to the calpain inhibitor calpeptin. These data indicate that in unstimulated cells a certain degree of constitutive GRK2 turnover may be mediated by calpain and that in these cells the formation of the 76-kDa fragment is a slow process. However, because the degradation the 76-kDa fragment in presence of H2O2 is faster than the degradation of the 80-kDa protein (Fig. 7A), we suggest that the 76-kDa fragment probably reflects an initial degradation step that will be rapidly followed by additional proteolytic processing in vivo. We do not know via which mechanism the 76-kDa fragment could be processed further. It may well be possible, however, that the cleavage of the 10 C-terminal amino acids of GRK2 disrupts inter- or intramolecular interactions in the GRK2 protein that could favor exposure of other domains of GRK2 to kinases and proteolytic enzymes activated by H2O2.
Recently, it has been demonstrated (Schoonbroodt et al., 2000) that oxidative stress also results in calpain-dependent degradation of IκBα that could be prevented by an inhibitor of casein kinase. Moreover, the same study showed that phosphorylation of serine and threonine residues within the PEST sequence of IκB is required for degradation of the protein to occur. The putative PEST region of GRK2, which is located within the pleckstrin-homology (PH) domain of GRK2, also contains a casein phosphorylation site (aa 602–605). Interestingly, we show herein that the casein kinase inhibitor DRB can partially prevent the H2O2-induced decrease in GRK2 expression. NMR structural analysis of the PH domain of GRK2 domain has shown that the threonine at position 602 is the last residue of the fourth β-strand of the PH domain (β-4) (Fushman et al., 1998). It is likely that its R group (i.e., OH) is surface exposed and thus accessible to kinases. It is tempting to speculate that oxidative stress activates casein kinase II, resulting in phosphorylation of S and T residues in the PEST sequence. Phosphorylation of residues in the PEST sequence by casein kinase and maybe also by other kinases could promote a conformational change that could enhance calpain binding to the protein. The involvement of protein phosphorylation in the “activation” or unmasking of a PEST sequence has been suggested for other proteins, e.g., PKC, as well (for review, see Rechsteiner and Rogers, 1996). It should be noted that although some proteins are actually cleaved in the PEST region, there is also evidence that binding of calpain to (activated) PEST sequences results in cleavage of the protein at a different position. For example, calpain cleavage of human brain α-fodrin involves recognition of a PEST sequence at a site that is 80 to 90 amino acids from the actual cleavage site (Wang et al., 1989). In addition, there is evidence that tyrosine phosphorylation is involved in the calpain-dependent degradation of some proteins (Huang et al., 1997). In T lymphocytes a rapid increase in tyrosine phosphorylation occurs after exposure to H2O2 (Fig. 4C, top;Carballo et al., 1999). Our data suggest that tyrosine phosphorylation is involved in the H2O2-induced decrease in GRK2 protein levels, because the tyrosine kinase inhibitor genistein partially prevents the decrease in cellular GRK2 after exposure to H2O2 (Fig. 4B). However, we do not detect any change in tyrosine phosphorylation of GRK2 after exposure of T cells to H2O2(Fig. 4C, bottom). Although we cannot exclude that tyrosine phosphorylation of GRK2 leads to its rapid degradation so that the phosphorylated GRK2 protein is no longer detectable, we suggest that tyrosine phosphorylation of one or more other proteins modulates the reduction of the GRK2 after exposure of cells to H2O2. Interestingly, in other systems GRK2 protein levels are controlled via the proteasome pathway. In human embryonic kidney-293 cells transfected with the β2-AR and GRK2, β2-AR agonists induce degradation of GRK2 that can be prevented by the proteasome inhibitor lactacystin and is associated with polyubiquitination of the GRK2 protein (Penela et al., 1998). More recently, we have shown that β-arrestin-mediated Src recruitment and subsequent GRK2 tyrosine phosphorylation is involved in GPCR-modulated GRK2 degradation by the proteasome pathway in both transfected and endogenous experimental systems (Penela et al., 2001). We do not know at present whether the use of the proteasome or the calpain pathway for regulation of cellular GRK2 protein levels is cell type or stimulus-specific. The molecular mechanisms involved and the functional interactions between these proteolytic pathways are interesting issues for future research.
Oxidative stress is known to activate a number of other kinase signaling pathways. Exposure of T lymphocytes to H2O2 is known to activate PKC, MAP kinase, and PI3-kinase. Activated PKC can phosphorylate GRK2, resulting in increased kinase activity (Chuang et al., 1995). Moreover, culture of lymphocytes with the PKC activator phorbol 12-myristate 13-acetate increases GRK2 levels after 24 to 48 h (De Blasi et al., 1995). Interestingly, inhibition of PKC does not affect basal GRK2 levels nor does it interfere with the H2O2-induced decrease in cellular GRK2 (Fig. 4A). In addition, specific inhibitors of MAP kinase or PI3-kinase do not have any effect on H2O2-induced decreases in GRK2 protein. Therefore, we conclude that above-mentioned kinases are not involved in the regulation of GRK2 protein levels after exposure to H2O2.
Previous reports showed that GRK2 is the predominant GPCR kinase involved in agonist-induced receptor sequestration of the β2-AR. Moreover, studies in transfected cell systems suggest that changes in the intracellular level of GRK2 will alter the rate and extent of sequestration of the β2-AR (Aramori et al., 1997; Menard et al., 1997; Ferguson et al., 1998; Penela et al., 1998). In addition, we have data showing that the 50% reduction in GRK2 protein expression in spleen cells from GRK2 heterozygous knockout mice results in a 70 to 80% decrease in agonist-induced β2-AR internalization (M. S. Lombardi, A. Vroon, A. Kavelaars, and C. J. Heijnen, unpublished observations). Our present data demonstrate that exposure of T cells to H2O2 results in an ∼50% decrease in GRK2 expression and an ∼70% reduction in agonist-induced internalization of the β2-adrenergic receptor as well (Fig. 8). These data indicate that the H2O2-induced decrease in T-lymphocyte GRK2 level has physiological consequences. There is indirect evidence that calpain may also be involved in the in vivo modulation of GRK2 levels in other situations. First, calpain expression and activity are increased in the spleen of rats with the inflammatory disease experimental autoimmune encephalomyelitis (Shields et al., 1999). In line with these data, we have shown that GRK2 levels are decreased in splenocytes from rats with experimental autoimmune encephalomyelitis (A. Vroon, A. Kavelaars, P. M. Cobelens, M. S. Lombardi, and C. J. Heijnen, unpublished data). Second, calpain activity is decreased in cardiac tissue of spontaneously hypertensive rats (Cicilini et al., 1995). Interestingly, spontaneously hypertensive rats show a significant increase in immunodetectable GRK2 both in lymphocytes and vascular myocytes compared with normotensive rats (Gros et al., 2000). Our present data suggest that there may be a causal relationship between alterations in intracellular calpain activity and GRK2 protein levels in inflammatory autoimmune diseases and hypertension. In conclusion, our results show another important function of ROS in autoimmunity: ROS may change the functioning of GPCRs during disease processes via the calpain-dependent regulation of cellular GRK2 levels.
Footnotes
- Received January 22, 2002.
- Accepted May 1, 2002.
-
This work was supported by Grant I/72032 from the Volkswagen Foundation, Germany (to C.J.H.), and Ministerio de Ciencia y Tecnologı́a PM98-0020 and F.R.Areces grants (to F.M.).
-
M.S.L. and A.K. contributed equally to this work.
Abbreviations
- GRK
- G protein-coupled receptor kinase
- GPCR
- G protein-coupled receptor
- β2-AR
- β2-adrenergic receptor
- PBMC
- peripheral blood mononuclear cell
- IL
- interleukin
- IFN-γ
- interferon-γ
- TNF
- tumor necrosis factor
- ROS
- reactive oxygen species
- NF-κB
- nuclear factor-κB
- IκB
- inhibitory κB
- PD98059
- 2′-amino-3′-methoxyflavone
- H7
- 1-5(isoquinoline sulfonyl)-2-methylpiperazine
- PP2
- [4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrrazolo[3,4-d]-pyrimidine
- DRB
- 5,6-dichloro-1-β-ribofuranosyl-benzimidazole
- CGP12177
- (4-[3-[1,1-dimethylethyl) amino]2-hydroxypropoxy]1,3-dihydro-2H-benzimidazole-2-one
- CYP
- cyanopindolol
- PBS
- phosphate-buffered saline
- Con A
- concavalin A
- SOD
- superoxide dismutase
- PAGE
- polyacrylamide gel electrophoresis
- PEST
- proline-glutamic acid-serine-theonine
- rGRK2
- recombinant G protein-coupled receptor kinase 2
- Ab
- antibody
- PI-3
- phosphatidylinositol 3-kinase
- MAP
- mitogen-activated protein
- aa
- amino acid
- PH
- pleckstrin-homology
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}