


Abstract
Phosphoinositide-specific phospholipase C (PLC) plays a pivotal role in the signal transduction of various cellular responses. However, although it is undeniably important that modulators of PLC activity be identified, no direct PLC activity modulator has been identified until now. In this study, by screening more than 10,000 different compounds in human neutrophils, we identified a compound that strongly enhances superoxide-generating activity, which is well known to be PLC-dependent. The active compound 2,4,6-trimethyl-N-(meta-3-trifluoromethyl-phenyl)-benzenesulfonamide (m-3M3FBS) stimulated a transient intracellular calcium concentration ([Ca2+]i) increase in neutrophils. Moreover, m-3M3FBS stimulated the formation of inositol phosphates in U937 cells, indicating that it stimulates PLC activity. The compound showed no cell-type specificity in terms of [Ca2+]i increase in the various cell lines including leukocytes, fibroblasts, and neuronal cells. We also ruled out the possible involvement of heterotrimeric G proteins inm-3M3FBS–stimulated signaling by confirming the following: 1) pertussis toxin does not inhibitm-3M3FBS–induced [Ca2+]iincrease; 2) m-3M3FBS does not stimulate cyclic AMP generation; and 3) the inhibition of Gq by the regulator of G protein-signaling 2 does not affect them-3M3FBS–induced [Ca2+]iincrease. We also observed that m-3M3FBS stimulated PLC activity in vitro. The purified isoforms of PLC that were tested (i.e., β2, β3, γ1, γ2, and δ1) were activated bym-3M3FBS and showed no isoform specificity. Taken together, these results demonstrate that m-3M3FBS modulates neutrophil functions by directly activating PLC. Becausem-3M3FBS is the first compound known to directly activate PLC, it should prove useful in the study of the basic molecular mechanisms of PLC activation and PLC-mediated cell signaling.
Phosphoinositide (PI) hydrolysis is one of the important early signals associated with the stimulation of leukocytes by diverse extracellular stimuli (Rhee, 2001). Phospholipase C (PLC) hydrolyzes phosphatidylinositol bisphosphate (PIP2) into inositol-1,4,5-triphosphates and diacylglcerol, which mediate intracellular calcium release or the activation of protein kinase C, respectively (Noh et al., 1995; Rhee, 2001). Intracellular calcium concentration ([Ca2+]i) increase and protein kinase C activation subsequently induce diverse intracellular signaling, such as the activation of phospholipase A2, phospholipase D, or mitogen-activated protein kinases. Finally, these intracellular signals result in the modulation of various cellular responses, including superoxide generation, secretion, and proliferation in leukocytic cells (Bae et al., 1999; Kim et al., 1999;McLaughlin and De Vries, 2001). Eleven isoforms of PLC are known (Rhee, 2001). Whereas the β isoforms are known to modulate GTP-binding proteins, the γ isoforms have been reported to activate the stimulation of growth factor receptors (Noh et al., 1995; Rhee, 2001). Although many extracellular ligands that stimulate cell surface receptors leading to the activation of PLC β or γ have been reported, no direct PLC activity modulator has been identified until now.
Recently many synthetic compounds have been reported to modulate diverse immune responses (Tian et al., 1998; Rosania et al., 1999; Zhang et al., 1999). Synthetic compounds are known to regulate cellular activity by modulating cellular target proteins (Tian et al., 1998; Rosania et al., 1999; Zhang et al., 1999). Whereas some of the compounds bind to cell surface receptors and induce receptor-mediated intracellular signals, others directly modulate intracellular target molecules after penetrating cells (Rosania et al., 1999; Strizki et al., 2001). The identification of compounds that modulate important physiological responses gives us two pieces of critical information: 1) they enable the development of synthetic compounds that modulate certain cellular functions and 2) their cellular target molecules can also be regarded as potential drug targets. In this respect, it is important not only to develop synthetic compounds that modulate cellular responses, but also to identify their target cellular proteins.
In this study, we screened a chemical library consisting of more than 10,000 different species in an effort to find a chemical that can stimulate superoxide generation in human neutrophils. We found that the compound 2,4,6-trimethyl-N-(meta-3-trifluoromethyl-phenyl)-benzenesulfonamide (m-3M3FBS) can stimulate human neutrophils and that this stimulation leads to superoxide generation. By studying the action mechanism of m-3M3FBS, we suggest that the compound stimulates neutrophil activity by directly activating PLC.
Materials and Methods
Materials.
Compounds were purchased from the Chembridge Corporation (San Diego, CA). Peripheral blood mononuclear cell separation medium (Histopaque-1077), dioleoyl-phosphatidylethanolamine, and tetracycline were purchased from Sigma Chemical (St. Louis, MO). RPMI 1640 and Dulbecco's modified Eagle's medium were from Invitrogen (Carlsbad, CA); dialyzed fetal bovine serum and supplemented bovine calf serum were from Hyclone Laboratories (Logan, UT). U-73122 and U-73343 were obtained from Sigma/RBI (Natick, MA). Hygromycin B and pertussis toxin (PTX) were from Calbiochem (San Diego, CA). [myo-2-3H]inositol (18.3Ci/mmol), [8-3H]adenosine 3′,5′-cyclic phosphate, and phosphatidylinositol-4,5-bisphosphate(inositol-2-[3H]) were from Amersham Biosciences (Piscataway, NJ). The AG 1-X8 resin was purchased from Bio-Rad (Hercules, CA).
Isolation of Human Neutrophils.
Peripheral blood leukocytes were donated by the Ulsan Red Cross Blood Center (Ulsan, Korea). Human neutrophils were isolated by standard dextran sedimentation, by the hypotonic lysis of erythrocytes, and by the use of a lymphocyte-separation medium gradient, as described previously (Bae et al., 2001). Isolated human neutrophils were used promptly.
Cell Culture and the Differentiation of HL60 Cells.
Human histiocytic lymphoma cells (U937), human promyelocytic leukemia cells (HL60), NIH Swiss mouse embryo fibroblasts (NIH 3T3), and rat adrenal pheochromocytoma cells (PC12) were obtained from the American Type Culture Collection (Manassas, VA), and human adenocarcinoma cells (HeLa) Tet-off cells were purchased from BD Biosciences Clontech (Palo Alto, CA) and maintained as recommended. The cells were maintained at approximately 1 × 106 cells/ml under standard incubator conditions (humidified atmosphere, 95% air/5% CO2, 37°C). HL60 cells were induced to differentiate into the granulocyte phenotype by adding dimethyl sulfoxide (final concentration, 1.25% v/v) for 5 days, as described before (Itoh et al., 1998).
Establishment of Cell Lines.
pRevTRE vector containing the cDNA of rat regulators of G protein signaling (RGS)-2-GFP was transfected into HeLa Tet-off cells using LipofectAMINE. Selection was performed in Dulbecco's modified Eagle's medium supplemented with 2 μg/ml of tetracycline and 500 μg/ml of hygromycin B. Two weeks later, several well-isolated colonies were picked out and analyzed by Western blotting to determine the expression level of RGS2 in the absence of tetracycline. For these experiments, HeLa cells were cultured for 48 h in the absence or presence of tetracycline.
Measurement of Superoxide Anion Generation.
Superoxide anion production was measured by monitoring chemiluminescence in the presence of the chemiluminogenic probe lucigenin (Bureau et al., 2001). Prepared neutrophils were plated in 96 wells and stimulated with chemicals at concentrations of 2.5, 5, 10, 15, 20, 25, and 50 μM, and then the lucigenin (40 μM) was added. Luminescence, measured with a Luminoskan (Labsystem, Helsinki, Finland), was integrated over 10-s intervals for a total of 3 min at room temperature.
Measurement of [Ca2+]i.
The chemically induced [Ca2+]i increase was measured using fura-2/acetoxymethyl ester (Grynkiewicz et al., 1985). Freshly prepared human neutrophils were incubated in serum-free RPMI 1640 medium with 3 μM fura-2/acetoxymethyl ester at 37°C for 30 min with continuous stirring. After washing with serum-free RPMI 1640 medium, the cells were suspended in serum-free RPMI containing 250 μM of sulfinpyrazone to prevent dye leakage. Approximately 2 × 106 cells were suspended in Ca2+-free Locke's solution (158.4 mM NaCl, 5.6 mM KCl, 1.2 mM MgCl2, 5 mM HEPES, pH 7.3, 10 mM glucose, and 0.2 mM EGTA) for each measurement. Changes in the fluorescence ratio were measured at an emission wavelength of 500 nm for dual excitation wavelengths at 340 nm and 380 nm. The calibration of the fluorescence ratio versus [Ca2+]i was performed as described by Grynkiewicz et al. (1985).
Measurement of the Formation of Inositol Phosphates in Cells.
The chemically induced formation of inositol phosphates was determined as described previously (Baek et al., 1996). Cells grown in culture were harvested by centrifugation, washed with inositol-free RPMI 1640 medium, and resuspended at a density of 2 × 106 cells/ml in the same medium. The cells were then labeled with [myo-3H]inositol (1 μCi/106 cells) for 24 h at 37°C and rinsed twice with inositol-free RPMI 1640 medium containing 0.5% fetal bovine serum, 20 mM of HEPES at pH 7.2, 20 mM of LiCl, and bovine serum albumin (1 mg/ml) and then resuspended at a density of 2 × 107 cells/ml. A portion (0.1 ml) of the cell suspension was transferred to a microcentrifuge tube and incubated at 37°C for 15 min. PIP2 hydrolysis was initiated by adding chemicals or solvents for the indicated times. Reactions were terminated by adding 200 μl of ice-cold 10% perchloric acid (HClO4). After 30 min in an ice bath, the tubes were centrifuged, and the supernatants were diluted 5-fold with distilled water and applied to Dowex AG 1-X8 anion exchange columns (Bio-Rad). Each column was then washed with 2 ml of distilled water, and this was followed by 10 ml of 60 mM ammonium formate containing 5 mM sodium tetraborate. Total inositol phosphates were eluted with a solution containing 1 M ammonium formate and 0.1 M formic acid. The radioactivity of the [3H]inositol phosphates was determined using a scintillation counter (Tri-Packard, Meriden, CT).
Measurement of Cyclic AMP Generation.
To measure the cAMP level, we used a radioreceptor assay (Pio et al., 2001), which was derived from the competition between unlabeled cAMP (in the sample) and a fixed quantity of 3H-labeled cAMP, for a protein with a high cAMP specificity and affinity. The amount of labeled protein-cAMP complex formed was inversely related to the amount of unlabeled cAMP present in the assay sample. Prepared cells were treated with m-3M3FBS (25 μM) and histamine (100 μM) and then lysed with Tris-EDTA containing 50 μM Ro20–1724, a phosphodiesterase inhibitor. After centrifugation, the supernatant was collected and [3H]cAMP and protein were added. The protein-bound cAMP was separated from the unbound nucleotide by adsorbing the free nucleotide onto coated charcoal and centrifuging. An aliquot of the supernatant was then collected for liquid scintillation counting. The amount of unlabeled cAMP (in the sample) was calculated by measuring the protein-bound radioactivity.
Measurement of Phosphoinositide Hydrolysis In Vitro.
PLC activity was assayed using [3H]PtdIns-4,5-P2 as a substrate (Min et al., 1993). PtdIns-4,5-P2–hydrolyzing activity was measured with the use of mixed phospholipid micelles containing 120 μM phosphatidylethanolamine, 30 μM PtdIns-4,5-P2, and 1 μCi/ml of [3H]PtdIns-4,5-P2. The lipids, in chloroform, were dried under a stream of nitrogen gas, suspended in assay buffer [20 mM HEPES, pH 7.0, 120 mM NaCl, 2 mM MgCl2, 40 or 100 μM CaCl2, and 1 mg/ml bovine albumin serum (Bayer, Leverkusen, Germany)], and sonicated. All proteins added to the reaction mixture were dialyzed overnight against the assay buffer. Incubation was performed for 10 min at 37°C in a 200-μl reaction mixture containing lipid micelles (5 μM [3H]PtdIns-4,5-P2, 20,000 cpm). The reaction was stopped by adding 2 ml of CHCl3/CH3OH/HCl (50:50:0.3, v/v). The inositol trisphosphates were extracted with 0.5 ml of 1 N HCl, and radioactivities in the upper aqueous phase were measured.
Results
Identification of a Synthetic Compound That Strongly Enhances Superoxide Generation in Human Neutrophils.
In this study, we screened approximately 10,000 chemicals in an effort to identify chemicals that stimulate superoxide generation in human neutrophils, and we found several chemicals that do so within the concentration range of 20 to 50 μM (data not shown). Among these, a chemical namedm-3M3FBS proved to be the most potent in terms of its ability to stimulate the generation of superoxide. Figure1A shows that m-3M3FBS greatly enhanced superoxide generation within the concentration range of 15 to 50 μM. Interestingly, however, 2,4,6-trimethyl-N-(ortho-3-trifluoromethyl-phenyl)-benzenesulfonamide (o-3M3FBS), which has a similar structure tom-3M3FBS except for the position of the trifloromethyl-phenyl group, did not affect superoxide generation up to 50 μM (Fig. 1A). Therefore, we used o-3M3FBS as an inactive analog of m-3M3FBS. Figure 1B shows the structures of m-3M3FBS and o-3M3FBS.

Effect of m-3M3FBS on superoxide anion generation in human neutrophils. Superoxide anion generation was measured by monitoring chemiluminescence in the presence of the chemiluminogenic probe lucigenin. Prepared neutrophils were stimulated with m-3M3FBS or o-3M3FBS at 2.5, 5, 10, 15, 20, 25, and 50 μM, and lucigenin (40 μM) was then added. Luminescence changes were measured using a Luminoskan (Labsystem). A, data are presented as means ± S.E.M. of four independent experiments. B, structures of m-3M3FBS ando-3M3FBS.
m-3M3FBS Stimulates [Ca2+]i Increase in Neutrophils.
Many extracellular agonists that stimulate superoxide anion generation in human phagocytic cells also increase [Ca2+]i (Liang and Huang, 1995; Bae et al., 1999). Therefore, we examined the effect ofm-3M3FBS on [Ca2+]i in human neutrophils. As shown in Fig. 2A,m-3M3FBS caused an increase in [Ca2+]i as a result of the transfer of calcium through the plasma membrane when Ca2+ levels were at physiological levels extracellularly, whereas the inactive analog, o-3M3FBS, failed to elicit this response. m-3M3FBS stimulated [Ca2+]i release in a concentration-dependent manner, showing the saturated maximal activity at a concentration of 50 μM (data not shown). Moreover,m-3M3FBS also evoked a [Ca2+]i increase in the extracellular calcium-depleted condition. These findings demonstrate that m-3M3FBS induces intracellular calcium increases as a result of both plasma membrane calcium entry and the release of intracellularly stored calcium.

Effect of m-3M3FBS on [Ca2+]i and the inhibition ofm-3M3FBS–induced calcium increase by U-73122. A, fura-2–loaded neutrophils were stimulated with m-3M3FBS or o-3M3FBS at 25 μM in extracellular calcium containing 2 mM or extracellular free calcium conditions. The results shown are representative of three independent experiments. B, prepared neutrophils were loaded with fura-2 and then stimulated with 25 μM ofm-3M3FBS in the absence or in the presence of 4 μM of U-73122 or U-73343. Changes in the 340/380 nm excitation ratio were monitored and converted into [Ca2+]i levels. Data are presented as means ± S.E.M. of three independent experiments.
Because intracellular calcium mobilization can be caused by the activation of PLC (Rhee, 2001), we undertook to prevent this compound response by blocking the activation of PLC with the membrane-permeable PLC inhibitor U-73122. A 3- to 5-min pretreatment of neutrophils with U-73122 has been documented previously to fully prevent PLC activation upon agonist stimulation (Hershfinkel et al., 2001). As shown in Fig.2B, pretreatment of neutrophils with 4 μM of U-73122 resulted in the complete inhibition of the calcium signal induced bym-3M3FBS. However, preincubating the same cells with U-73343, an inactive analog of U-73122, had no effect on them-3M3FBS–evoked response. This result indicates that an increase in [Ca2+]iinduced by m-3M3FBS is mediated by PLC activity. Preincubation of neutrophils with U-73122 in the presence of extracellular calcium also completely blockedm-3M3FBS–induced calcium signal (data not shown). The results suggest that intracellular calcium increases caused by plasma membrane calcium entry are mediated by intracellular calcium release.
m-3M3FBS Stimulates the Formation of Inositol Phosphates in U937 Cells.
From previous data, we expected thatm-3M3FBS would influence the activity of PLC. We next examined whether m-3M3FBS could stimulate PLC activation by measuring total inositol phosphate formation in U937 cells. After labeling with [myo-3H]inositol (1 μCi/106 cells), the cells were treated withm-3M3FBS or o-3M3FBS. As shown in Fig.3, the accumulation of inositol phosphates after treatment with m-3M3FBS increased gradually, giving a 2.5-fold increase at 50 μM ofm-3M3FBS. The concentration-dependence ofm-3M3FBS–induced inositol phosphate formation was closely correlated with that of m-3M3FBS–induced superoxide generation (Fig. 1A). In contrast, o-3M3FBS had no effect on PLC activity. This result indicates that m-3M3FBS stimulates PLC activation.

Effect of m-3M3FBS on in vivo PLC activity. U937 cells were labeled with [myo-3H]inositol (1 μCi/106cells) for 24 h at 37°C and then treated with various concentrations of m-3M3FBS or o-3M3FBS. Total inositol phosphates were eluted with a solution containing 1 M ammonium formate and 0.1 M formic acid. Radioactivity of the [3H]inositol phosphates was determined by counting in a scintillation counter. Data are presented as means ± S.E.M. of five independent experiments.
m-3M3FBS Has No Cell-Type Specificity.
Up to this point, we had observed that m-3M3FBS caused superoxide anion generation in a PLC-dependent manner, and we wondered whether this chemical could affect signaling molecules upstream of PLC. In the case of the PLC β series, ligand-specific receptor and heterotrimeric G proteins generally are located upstream of signaling molecules (Rhee and Bae, 1997; Rhee, 2001). First, we investigated whether m-3M3FBS has a specific receptor. As shown in Fig.4, m-3M3FBS induced a calcium increase in all cell lines (human neutrophils, HL60, differentiated HL60, U937, NIH 3T3, and PC12) examined, showing no cell-type specificity.

Effect of m-3M3FBS on [Ca2+]i in several cell lines. Neutrophils, differentiated HL60, HL60, U937, NIH 3T3, and PC12 cells were loaded with fura-2 and then stimulated with 25 μM ofm-3M3FBS. Changes in the 340/380 nm excitation ratio were monitored and converted into [Ca2+]ilevels. Data are presented as means ± S.E.M. of three independent experiments.
m-3M3FBS Desensitizes the Calcium Increase Induced by Other Agonists.
We next investigated the capacity ofm-3M3FBS to desensitize other extracellular agonists by examining its effect on ATP and a synthetic leukocyte chemoattractant peptide, Trp-Lys-Tyr-Met-Val-d-Met-CONH2(WKYMVm), which is known to stimulate PLC enzymes (Seo et al., 1997;Bae et al., 2000). In cross-desensitization experiments, stimulation of the cells with m-3M3FBS significantly reduced cellular responses to ATP and WKYMVm (Fig. 5). However, the administration of m-3M3FBS after stimulating cells with ATP or WKYMVm elicited still further [Ca2+]i increases (Fig.5). Therefore, the m-3M3FBS–induced [Ca2+]i increase was more potent than that induced by other agonists, demonstrating thatm-3M3FBS may more strongly affect the PLC enzyme(s) than other extracellular agonists. These results suggest that the target ofm-3M3FBS is a common mediator of cell surface receptors, such as PLC or heterotrimeric G protein.

Cross-desensitization experiments. Fura-2–loaded neutrophils were pretreated with m-3M3FBS (25 μM) and then rechallenged with ATP (500 μM) or WKYMVm (1 μM) in this or in the reverse order in the extracellular calcium-free condition. Changes in the 340/380 nm excitation ratio were monitored and converted to [Ca2+]i levels. The results shown are representative of three independent experiments.
m-3M3FBS–Induced Signaling Is Not G Protein-Dependent.
Several extracellular signals, including those caused by many chemoattractants, activate phagocytic cells via PTX-sensitive Gi proteins (Feniger-Barish et al., 2000; Mellado et al., 2001). Therefore, we investigated the involvement of PTX-sensitive Gi proteins uponm-3M3FBS–induced neutrophil activation. As shown in Fig.6A, formyl-methionyl-leucyl-phenylalanine, a chemoattractant that signals through PTX-sensitive Gi proteins (Jiang et al., 1996), induced a [Ca2+]iincrease, and this response was inhibited by preincubating neutrophils with PTX (1 μg/ml) for 90 min. However, the calcium increase caused by m-3M3FBS was insensitive to PTX, implying that Gi proteins are not involved inm-3M3FBS-induced [Ca2+]i increase.
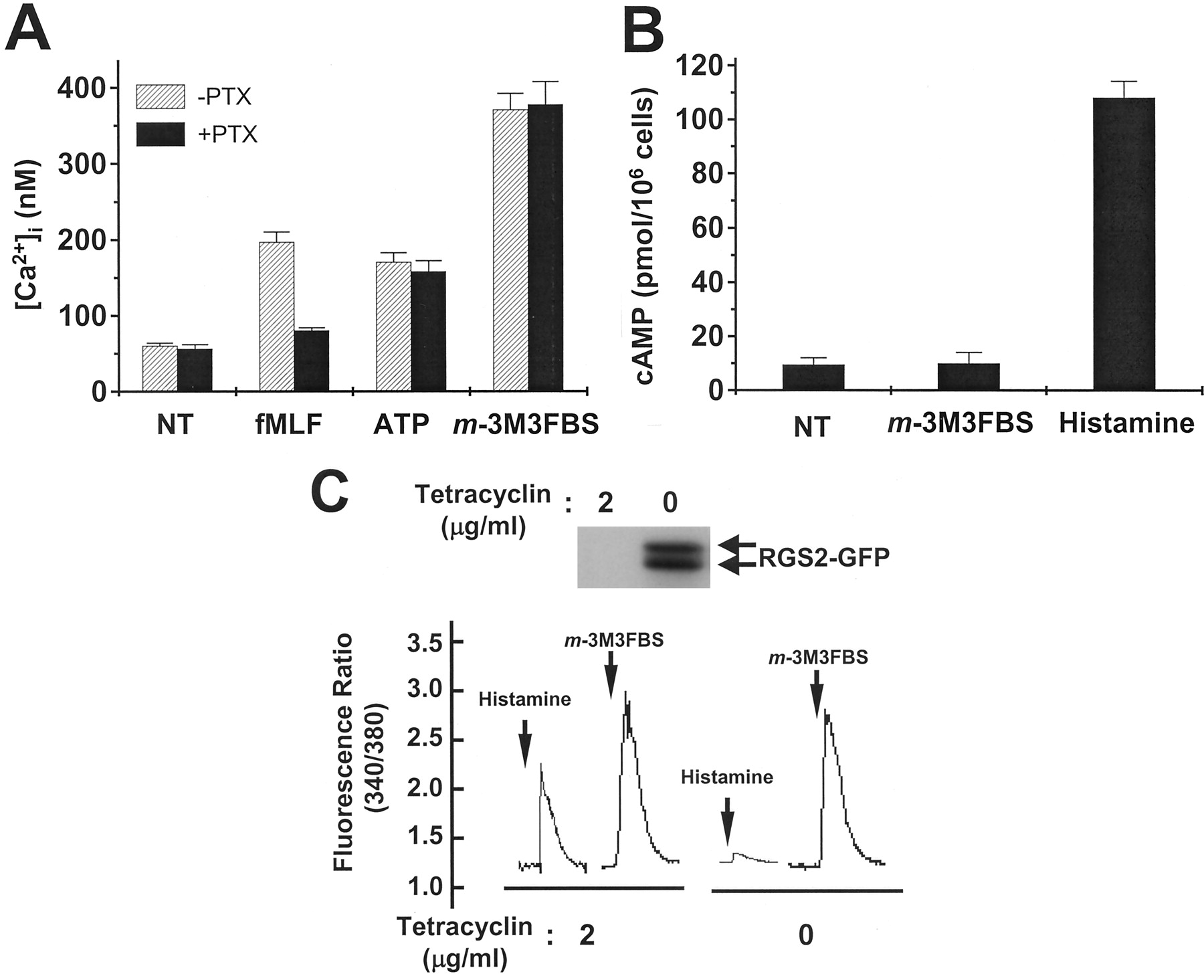
Effect of m-3M3FBS on the activity of heterotrimeric G proteins. A, isolated neutrophils were preincubated with PTX (1 μg/ml) or vehicle for 90 min at 37°C and stimulated with m-3M3FBS, formyl-methionyl-leucyl-phenylalanine, or ATP at concentrations of 25, 1, and 500 μM, respectively. Changes in the excitation ratios at 340/380 nm were monitored. Data are presented as means ± S.E.M. of two independent experiments. B, cAMP levels were determined by a radio receptor assay. Prepared cells were treated with m-3M3FBS (25 μM) or histamine (100 μM) and then lysed with Tris-EDTA. After removing the supernatant, [3H]cAMP and protein were added, and the amount of [3H]cAMP-protein complex was determined. The amount of unlabeled cAMP (in the sample) was then calculated by measuring the protein-bound radioactivity. Data are presented as means ± S.E.M. of two independent experiments. C, RGS2-GFP–inducible HeLa cells were cultured for 48 h in the absence or presence of tetracycline, and the induction of RGS2-GFP was confirmed by Western blot analysis using anti-GFP antibody. RGS2-GFP inducible HeLa cells were cultured for 48 h in the absence or presence of tetracycline and loaded with fura-2. Cells were treated with histamine (100 μM) orm-3M3FBS (25 μM), and changes in the 340/380 nm ratios were monitored and converted into [Ca2+]ivalues. The results shown are representative of three independent experiments.
Adenylate cyclase integrates positive and negative signals that act through Gs protein-coupled cell-surface receptors to finely regulate levels of cAMP within several types of cells (Simonds, 1999). We next examined whether m-3M3FBS could activate Gs proteins by measuring the change in the intracellular cAMP level. As shown in Fig. 6B, histamine is known to bind to Gs- and Gq-coupled receptors (Kuhn et al., 1996; Kilts et al., 2000), which potently induce cAMP production, but whenm-3M3FBS was added to human neutrophils, it did not change the cAMP level. These results suggest that m-3M3FBS cannot activate Gs proteins.
The RGS proteins are GTPase-activating proteins that inhibit signaling via heterotrimeric G proteins. Recently, it was reported that RGS2 is a selective inhibitor of Gq protein function (Heximer et al., 1997). Therefore, we checked the involvement of Gq proteins in m-3M3FBS–induced signaling using HeLa cells, which overexpress wild-type RGS2 under the control of an inducible tetracycline-regulated promoter. As shown in Fig. 6C, both histamine and m-3M3FBS caused [Ca2+]i increases in the presence of tetracycline. However, the calcium increases induced by histamine and m-3M3FBS were altered when RGS2 was overexpressed in cells, because whereas the histamine-induced calcium response was completely blocked by RGS2 induction, them-3M3FBS–induced Ca2+ response was unaffected. These results suggest that Gqproteins are not involved in the m-3M3FBS–mediated signaling pathway.
m-3M3FBS Directly Activates PLC In Vitro.
Because m-3M3FBS did not seem to act on cell surface receptor(s) or G protein(s), we investigated the effect ofm-3M3FBS on PLC activity directly. PLC activity was assayed using [3H]PtdIns-4,5-P2as substrate. Initially we examined the activity of PLC β2, because it is highly expressed in immune cells (Bertagnolo et al., 2002). As shown in Fig. 7A, althoughm-3M3FBS had no effect at low doses (approximately 2.5 and 5 μM), the PIP2-hydrolyzing activity of PLC β2 was enhanced at m-3M3FBS concentrations exceeding 10 μM. However, the inactive form of m-3M3FBS had no effect at this or higher concentrations (Fig. 7A). We also tested the effect ofm-3M3FBS on in vitro PLCβ2 activity using [3H]PtdIns as a substrate and found that PLCβ2 activity was significantly increased by the compound, showing a similar pattern with the experiments using [3H]PtdIns-4,5-P2 as substrate (data not shown).
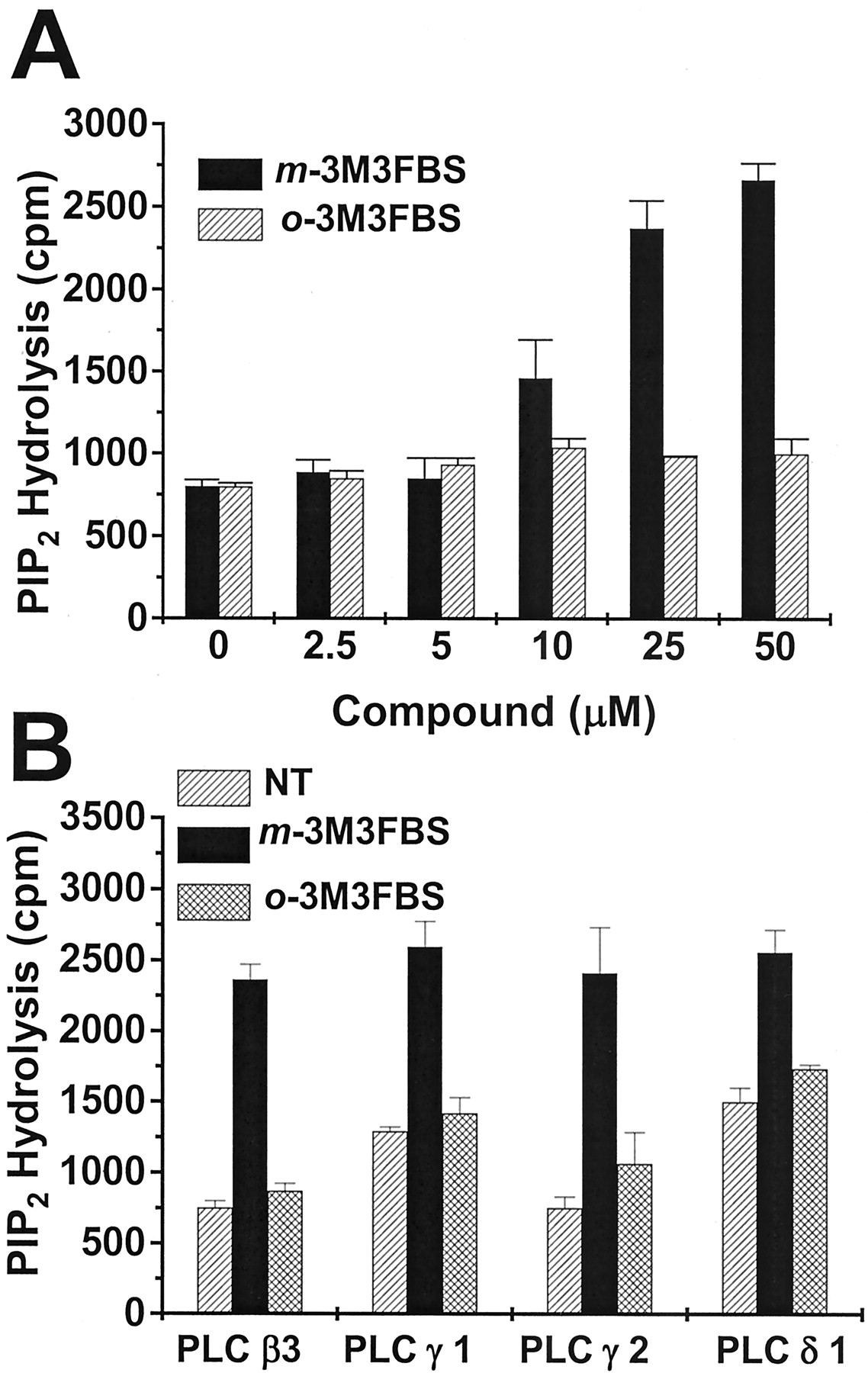
Effect of m-3M3FBS on the activity of PLC isotypes in vitro. The hydrolyzing activity of PtdIns-4,5-P2 was measured on mixed phospholipid vesicles containing 120 μM phosphatidylethanolamine, 30 μM PtdIns-4,5-P2, and 1 μCi/ml [3H]PtdIns-4,5-P2. Purified PLC β2, substrates, and the chemicals (2.5, 5, 10, 20, and 25 μM ofm-3M3FBS or o-3M3FBS) were coincubated (A), and 25 μM of m-3M3FBS or o-3M3FBS and phospholipid substrates were mixed with purified PLC β3, γ1, γ2, or δ1 (B). Reactions were performed for 10 min at 37°C in a 200-μl reaction mixture containing lipid micelles (5 μM [3H]PtdIns-4,5-P2, 20,000 cpm). Reactions were stopped by adding 1 ml of CHCl3/CH3OH/HCl (50:50:0.3, v/v). Inositol trisphosphates were extracted with 0.5 ml of 1 M HCl/5 mM EGTA, and the radioactivity in the upper aqueous phase was measured. A and B, data are presented as means ± S.E.M. of three independent experiments.
We next tested several other purified PLC isozymes (β3, γ1, γ2, and δ1) to check the isozyme specificity of m-3M3FBS. As shown in Fig. 7B, m-3M3FBS augmented the activity of all PLC isozymes tested, whereas o-3M3FBS did not affect PLC activity. These results indicate that m-3M3FBS elevates the PIP2-hydrolyzing activity of PLC in vitro without showing isotype specificity.
Discussion
In this study, we identified the synthetic compoundm-3M3FBS, which stimulates superoxide generation in human neutrophils. m-3M3FBS also evoked [Ca2+]i increases not only in neutrophils but also in several other cells, including neuronal cells and fibroblasts, and therefore, it showed no cell-type specificity. Through a study of its mode of action, we found that it directly activates many PLC isozymes, again without showing isozyme specificity.
Our experiment demonstrates that stimulation of human neutrophils withm-3M3FBS induces [Ca2+]i increases (Fig.2A), and that this effect of m-3M3FBS is inhibited byU73122, a specific PI-PLC inhibitor (Fig. 2B). The stimulation of U937 cells with m-3M3FBS also caused PI hydrolysis (Fig. 3). During experiments designed to investigate the effect ofm-3M3FBS on PLC activity, we observed that it activated several isoforms of PLC in vitro (Fig. 7, A and B). These results suggest that m-3M3FBS activates PLC directly, ruling out a possible non-PLC–dependent mechanism of the compound. Because all isoforms of PLC tested (β2, β3, γ1, γ2, and δ1) were activated by m-3M3FBS in vitro, it seems that the compound does not have any isoform specificity in terms of the activation of this enzyme (Fig. 7B). Although many different extracellular ligands are known to stimulate PLC activity by binding to their specific cell surface receptors, there has been no report on the direct activation of PLC until now. Generally, the activation of cell surface receptors induces diverse signaling pathways. PLC activation is one of the earliest responses downstream of receptor stimulation (Noh et al., 1995; Rhee, 2001). Several previous reports have suggested that PLC is involved in several important cellular functions, such as proliferation, differentiation, and apoptosis (Noh et al., 1995; Rhee and Bae, 1997; Rhee, 2001). However, the complications of cellular receptor-mediated signaling hinder our understanding of the natures of the signals and of the cellular responses regulated by PLC. Bearing this in mind, the identification of a molecule that can modulate PLC activity directly will undoubtedly be helpful for the elucidation of PLC-mediated cellular signaling and physiological responses. Furthermore, no ligand has been identified that stimulates the isoforms of PLC, including PLCδ1 and δ2. Because m-3M3FBS could stimulate PLCδ1 activity directly, it should be useful for the study of cellular signaling and functional events downstream of the enzyme.
In our in vitro experiments, we observed that m-3M3FBS stimulated the β, γ, and δ isoforms of PLC and that it showed no isoform-specificity (Fig. 7, A and B). Moreover, the primary structures of the several different isoforms of PLC are known (Noh et al., 1995; Rhee, 2001). PLC β and PLC δ have an NH2-terminal PH domain, an EF-hand, X and Y domains known to form the catalytic core, and a COOH-terminal C2 domain. In addition, PLC β has a long C-terminal tail beyond the C2 domain (Noh et al., 1995; Rhee and Bae, 1997; Rhee, 2001). PLC γ has three additional SH domains between the X and Y domains, but no long COOH-terminal tail (Noh et al., 1995; Rhee and Bae, 1997; Rhee, 2001). Our study shows that m-3M3FBS stimulates three subfamilies of the PLC isoforms (PLC β, γ, and δ) (Fig. 7, A and B). This suggests that the compound acts on a common conserved region of these three isoforms, thus ruling out the possible involvement of the SH domains and COOH-terminal tail of PLC β. For the proper activation of the PLC enzyme, calcium has been regarded as an essential requirement (Rhee and Bae, 1997). Calcium is required not only for the functioning of C2 domain that mediates the Ca2+-dependent binding to lipid vesicles, but also for the catalytic activity of the enzyme (Rhee and Bae, 1997). In our study, we found thatm-3M3FBS stimulated in vitro PLC activity in the presence or absence of Ca2+ (data not shown). The result suggests that m-3M3FBS may stimulate PLC activity with different mechanism from the calcium ion. Previously, Horstman et al. (1996) demonstrated that the addition of a purified X and Y domain in vitro showed lipase activity. An investigation of the effect ofm-3M3FBS on the lipase activity of an X and Y domain mixture will be required to confirm the possible action of m-3M3FBS on the two catalytic cores. Because m-3M3FBS is the first compound that directly stimulates PLC activity, the elucidation of the action mechanism of the compound will give useful information on the basic molecular mechanisms of the activation of PLC enzymes.
In Fig. 7, we demonstrated that all tested PLC isoforms were activated by m-3M3FBS. The result led us to check whether the compound acts specifically on PLC or on other enzymes that recognize phosphoinositols as substrates, such as phosphoinositide-3-kinase (PI3-kinase). For this, we tested the effect of m-3M3FBS on the Akt phosphorylation that is dependent on the PI3-kinase in U937 cells. A concentration of 50 μM m-3M3FBS could not significantly increase the phosphorylation level of Akt (data not shown). This indicates that m-3M3FBS has specificity for PLC but not for PI3-kinase. To check the effect of m-3M3FBS on other phospholipase, we also tested the effect of m-3M3FBS on the in vitro activity of phospholipase D. We observed that the compound did not affect on the activity of phospholipase D (data not shown). The results support our notion that m-3M3FBS acts specifically on PLC.
In conclusion, by screening a chemical library, we identified a small synthetic molecule that potently stimulates superoxide generation. This is the first report of a direct activator of PLC, and we believe that the compound will prove to be a useful agent for the study of PLC-mediated cell signaling.
Footnotes
-
This work was supported by grant 01-PJ4-PG4-01VN01-0319 from the Korea Health 21 R&D Project, Ministry of Health and Welfare, Republic of Korea.
- Abbreviations:
- PI
- phosphoinositide
- PLC
- phospholipase C
- PIP2
- phosphatidylinositol bisphosphate
- [Ca2+]i
- intracellular calcium concentration
- m-3M3FBS
- 2,4,6-trimethyl-N-(meta-3-trifluoromethyl-phenyl)-benzenesulfonamide
- PTX
- pertussis toxin
- o-3M3FBS
- 2,4,6-trimethyl-N-(ortho-3-trifluoromethyl-phenyl)-benzenesulfonamide
- RGS
- regulators of G protein signaling
- WKYMVm
- Trp-Lys-Tyr-Met-Val-d-Met-CONH2
- PI3-kinase
- phosphoinositide 3-kinase
- GFP
- green fluorescent protein
- Received September 13, 2002.
- Accepted January 22, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}