


Abstract
The regulation of presynaptic, voltage-gated calcium channels by activation of heptahelical G protein-coupled receptors exerts a crucial influence on presynaptic calcium entry and hence on neurotransmitter release. Receptor activation subjects presynaptic N- and P/Q-type calcium channels to a rapid, membrane-delimited inhibition—mediated by direct, voltage-dependent interactions between G protein βγ subunits and the channels—and to a slower, voltage-independent modulation involving soluble second messenger molecules. In turn, the direct inhibition of the channels is regulated as a function of many factors, including channel subtype, ancillary calcium channel subunits, and the types of G proteins and G protein regulatory factors involved. Twenty-five years after this mode of physiological regulation was first described, we review the investigations that have led to our current understanding of its molecular mechanisms.
I. Introduction
Depolarization-mediated calcium influx via voltage-gated calcium channels elicits a range of cytoplasmic responses, including the contraction of cardiac muscle, the initiation of calcium-dependent gene transcription, cellular proliferation, the activation of calcium-dependent enzymes, and the release of hormones and neurotransmitter molecules (Tsien et al., 1988; Wheeler et al., 1994; Dunlap et al., 1995; Martin-Moutot et al., 1996; Sutton et al., 1999; Dolmetsch et al., 2001; Reid et al., 2004). However, excessive calcium entry produces deleterious effects and may result in cell death. So it is essential for cells to carefully buffer intracellular calcium and to precisely regulate calcium entry via calcium-permeant membrane proteins such as voltage-gated calcium channels.
Multiple subtypes of voltage-gated calcium channels have been identified in mammalian tissues and classified, on the basis of their pharmacological and electrophysiological properties, into T-, L-, N-, P-, Q-, and R-types (Tsien et al., 1988, 1991; Snutch et al., 2005). Based on their thresholds of activation, these channel subtypes can be more grossly divided into low-voltageand high-voltage-activated (LVA1 and HVA, respectively) channels (Catterall et al., 2005). However, it is important to note that this criterion is not absolute, as the activation ranges of most calcium channels are modulated by alternate splicing, subunit composition, and interactions with regulatory elements. LVA channels comprise the family of T-type channels, which typically require only small membrane depolarizations to open. They activate and inactivate rapidly and are partially inactivated at normal neuronal resting potentials (Perez-Reyes, 2003). HVA calcium channels comprise all other channel subtypes named above. Relative to LVA channels they require stronger membrane depolarizations for activation and inactivation, hence showing a greater availability for opening at normal resting potentials.
Members of the HVA class are well distinguished by their pharmacological profiles. L-type channels are sensitive to dihydropyridine agonists and antagonists, although some L-type channel isoforms are less effectively inhibited by dihydropyridines than others (Fox et al., 1987; Xu and Lipscombe, 2001). N-type calcium channels are potently and selectively blocked by ω-conotoxins GVIA, MVIIA, and CVID, peptides isolated from various fish hunting cone snails (Olivera et al., 1984; Reynolds et al., 1986; Mintz et al., 1991; Feng et al., 2003). Both P- and Q-type channels are inhibited by ω-agatoxin IVA, a toxin isolated from the North American funnel-web spider, Agelenopsis aperta (Mintz et al., 1992; Adams et al., 1993). R-type channels were defined as such because they represent an HVA current that is resistant to the above blockers (Randall and Tsien, 1995). However, SNX-482, a peptide toxin isolated from a species of giant tarantula is now considered a potent and semiselective inhibitor of these channels (Newcomb et al., 1998; Bourinet et al., 2001).
Why are there so many subtypes of calcium channels, if they only function to allow passage of calcium ions into excitable cells? As suggested by their differences in biophysical properties, cellular expression pattern, and subcellular distribution, the channel subtypes also differ in the cellular functions they support (see below). Furthermore, different subtypes of voltage-gated calcium channels are subject to differential regulation by cytoplasmic messenger molecules, including protein kinases and G proteins.
The modulation of voltage-gated calcium channels is a vast field with many fascinating details, too extensive to review comprehensively in one article. Herein, we focus on the modulation of presynaptic calcium channels by G proteins. As notably illustrated by the action of morphine—a μ-opioid receptor agonist, which mediates potent analgesia by virtue of inhibition of N-type calcium channels and activation of potassium channels (Altier and Zamponi, 2004)—this type of regulation has important physiological implications.
II. Molecular Structure and Distributions of Voltage-Gated Calcium Channels
A. α1 Subunit
The core of every functional voltage-gated calcium channel and main determinant of channel subtype is the α1 subunit (Fig. 1A). Each calcium channel contains a single α1 subunit, which in turn consists of homologous domains (I, II, III, and IV, linked by cytoplasmic loops referred to as the I-II, II-III, and III-IV loops) and cytoplasmic N- and C-terminal regions (Fig. 1A) (Catterall, 1993, 2000). As outlined below, these cytoplasmic regions are key sites for second messenger modulation and for association with regulatory and adaptor proteins. Each homologous domain contains six transmembrane spanning helices (termed S1 through S6), plus a reentrant p-loop structure between S5 and S6 that is believed to form the ion-selective pore region of the channels. The S4 helices have characteristic positively charged amino acid residues at every third sequence position, which allows the channel to sense membrane depolarization and respond with channel opening (Catterall, 2000).
Ten different genes encoding calcium channel α1 subunits in higher mammals have been identified and functionally characterized (Fig. 1B). These genes fall into three homologous families, termed Cav1, Cav2, and Cav3 (Snutch et al., 2005). The Cav1 family has four genes that encode L-type calcium channels with various physiological functions: Cav1.1, expressed exclusively in skeletal muscle, forms the voltage-sensor for excitation-contraction coupling (Tanabe et al., 1987); Cav1.2 and Cav1.3 are widely expressed, including in the pancreas, the heart, and the brain (Mikami et al., 1989; Williams et al., 1992b; Tomlinson et al., 1993); and Cav1.4 is expressed in retina and is functionally linked to photo-transduction (Bech-Hansen et al., 1998; Koschak et al., 2003; McRory et al., 2004). The Cav2 family is widely expressed in neurons (further discussed below) and includes Cav2.2 [encoding the N-type channel (Dubel et al., 1992; Williams et al., 1992a)], Cav2.3 [encoding the R-type channel (Soong et al., 1993; Williams et al., 1994; Randall and Tsien, 1995)], and Cav2.1, with alternate splice isoforms of the latter giving rise to P- and Q-type channels (Bourinet et al., 1999). The three members of the Cav3 family encode different isoforms of T-type channels, with Cav3.1 and Cav3.2 expressed across numerous tissues and Cav3.3 confined to neuronal tissue (Cribbs et al., 1998; Perez-Reyes et al., 1998; Lee et al., 1999; Monteil et al., 2000a,b; McRory et al., 2001; Chemin et al., 2002).

A, schematic representation of structural features of the α1 subunit of the voltage-gated calcium channel. Predicted transmembrane helices are shown as shaded cylinders. Internally homologous domains are indicated by roman numerals above the domain; each domain comprises six predicted transmembrane helices (S1 through S6). Intracellular amino- and carboxy-terminal loops and loops connecting the homologous domains are labeled as N′, C′, I-II, II-III, and III-IV, respectively. + symbols in the S4 helices indicate positively charged amino acid residues that contribute to the voltage sensing mechanism of the channel. Reentrant loops between S5 and S6 helices line the pore of the channel and compose the ion selectivity filter of the channel. B, summary of different types of α1 subunits, including names of genes, plus corresponding electrophysiological subtype, pharmacology, and tissue distribution. C, schematic representation of the subunit composition and membrane topology of a typical high-voltage-activated calcium channel (note that LVA channels probably only contain the α1 subunit).
The specialized roles of individual calcium channel α1 subunits are apparent from the phenotypes of knockout mice deficient in these genes (Miller, 2001). Cav1.1-/- mice die at birth of asphyxiation caused by lack of skeletal muscle contraction and, thus, the inability to move their diaphragms (Strube et al., 1996). Cav1.2-/- mice die before birth because of an inability to contract cardiac muscle (Seisenberger et al., 2000). Cav1.3-/- and Cav1.4-/- mice are viable, but lack key aspects of sensory signal transduction, such that Cav1.3-/- mice are deaf (Platzer et al., 2000) and Cav1.4-/- mice display blindness due to compromised rod photoreceptor function (Mansergh et al., 2005). Cav1.3-/- mice also display cardiac arrhythmias. Mice lacking the Cav2.1 gene are severely ataxic and show absence seizures (Jun et al., 1999), whereas Cav2.2-/- mice are viable and show hyposensitivity to pain, as well as reduced anxiety and alcohol withdrawal symptoms (Hatakeyama et al., 2001; Kim et al., 2001; Saegusa et al., 2001; Newton et al., 2004). Cav2.3-/- mice are also viable, and show reduced response to certain pain stimuli, as well as reduced seizure activity in certain types of seizure models (Saegusa et al., 2000; Weiergräber et al., 2006). Cav3.1-/- mice are resistant to baclofen-induced seizures (Kim et al., 2001), and finally Cav3.2-/- mice show compromised vascular function (Chen et al., 2003).
The physiological consequences of gene knockout are consistent with the cellular and subcellular distributions of these channels in the central nervous system. For example, Cav3.1 channels are expressed on cell bodies and dendrites where they contribute to regulate cellular excitability (Molineux et al., 2006), which is consistent with their involvement in spike wave discharges. By contrast, Cav2.3 channels are localized to proximal dendrites and presynaptic nerve termini (Wu and Saggau, 1995; Yokoyama et al., 1995; Wu et al., 1998). Cav2.1 and Cav2.2 channels are also located at presynaptic nerve terminals, where they contribute to the release of neurotransmitters (Westenbroek et al., 1992, 1995, 1998). Yet, as noted above, individual knockouts of the Cav2.1 and Cav2.2 channels yield very different phenotypes, suggesting that the channels are not created equally in terms of coupling to the neurotransmitter release machinery. Indeed, Cav2.1 channels seem to preferentially contribute to the release of excitatory neurotransmitters, whereas Cav2.2 channels are more frequently linked to inhibitory synaptic transmission (although this linkage is by no means absolute) (Burke et al., 1993; Potier et al., 1993; Doroshenko et al., 1997; Caddick et al., 1999; Leenders et al., 2002). It should also be noted that these channels may serve functions other than simply triggering neurotransmitter release, e.g., regulation of gene transcription, as has been suggested for Cav2.1 (Sutton et al., 1999). Given the above arguments and evidence for a clear contrast in the physiological roles of Cav2.1 and Cav2.2 channels, the existence of second messenger/signaling mechanisms that would allow differential modulation of these channels seems essential.
B. Ancillary Calcium Channel Subunits
Since the first purification of the Cav1.1 calcium channel from skeletal muscle (Curtis and Catterall, 1984), it has been evident that these channels are complexes of multiple subunits (Fig. 1C). In skeletal muscle, the Cav1.1 α1 subunit copurified with ancillary β, α2-δ, and γ subunits (Catterall, 2000). We now know that all subtypes of HVA calcium channels contain at least one β and one α2-δ subunit, but it remains unclear whether γ subunits associate with nonskeletal muscle HVA channels (Dolphin, 2003). There is recent evidence that certain types of γ subunits can bind to Cav3 channels directly, but these investigations are ongoing (Best et al., 2006). In contrast, members of the LVA calcium channel family do not seem to associate physically with α2-δ and β subunits; however, coexpression of these subunits with members of the Cav3 family does seem to regulate channel density (Lambert et al., 1997; Leuranguer et al., 1998; Dolphin et al., 1999; Hobom et al., 2000; Dubel et al., 2004). How this occurs mechanistically remains unclear.
Vertebrates express four genes that encode different types of calcium channel β subunits (β1, β2, β3, and β4), with further heterogeneity arising from alternate splicing (Dolphin, 2003; Richards et al., 2004). With one exception—the β2a subunit, which is palmitoylated and thereby plasma membrane-anchored (Qin et al., 1998)— these subunits are cytoplasmic proteins. Their overall architecture encompasses two highly conserved regions, flanked and separated by more variable domains (Stotz et al., 2004). Recent crystal structure data indicate that these subunits contain guanylate kinase and SH3 domains, which interact with each other to form functional β subunits (Chen et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004). Biochemical studies have revealed that the β subunits bind to a highly conserved region within the I-II loop of the HVA calcium channel α subunit (Pragnell et al., 1994). Termed the α-interaction domain (AID) and not found in the LVA channels, this region fits into a hydrophobic groove on the surface of the β subunit. There is also evidence of a second calcium channel β subunit-binding domain, localized to the C-terminal region of certain subtypes of HVA calcium channels, but its functional role is unclear (Qin et al., 1997). Whereas the α1 subunit contains the minimal machinery to form a functional channel, the coexpression of a β subunit modulates a number of functional properties of the α1 subunit, resulting in a massive up-regulation in current densities, changes in the midpoint of the current voltage relations and steady-state inactivation curves, and altered activation and inactivation kinetics (Pragnell et al., 1994; Chien et al., 1995; Bichet et al., 2000a,b; Yasuda et al., 2004).
Four vertebrate genes encoding α2-δ subunits (termed α2-δ1 through α2-δ4) have been identified and characterized (Klugbauer et al., 1999; Arikkath and Campbell, 2003). Each α2-δ isoform is encoded by a single gene, translated as a single peptide, and post-translationally cleaved into α2 (extracellular) and δ (single transmembrane helix) portions, which are then reconnected via a disulfide bond (De Jongh et al., 1990). The consequences of α2-δ coexpression include an enhancement of peak current amplitude, altered channel pharmacology, and slightly altered channel gating (Klugbauer et al., 2003; Yasuda et al., 2004). The physiological role of α2-δ subunits is exemplified by mouse models of absence epilepsy in which the α2-δ2 subunit is truncated; in these mice, in addition to the epileptic phenotype, cerebellar Purkinje cell morphology is altered, and P/Q-type channel activity is diminished (Barclay et al., 2001).
To date, eight different γ subunits (termed γ1 through γ8) have been isolated, all of which comprise four transmembrane helices with intracellular N and C termini (Arikkath and Campbell, 2003; Black, 2003). The γ1 subunit is specific to skeletal muscle, but the other subunits can all be detected in the brain. With the exception of the γ7 subunit, which drastically reduces the activity of Cav2.2 channels in heterologous expression systems (Moss et al., 2002), coexpression of the remaining γ subunits with any of the other neuronal voltage-gated calcium channel α1 subunits mediates only small effects on channel kinetics (Rousset et al., 2001). That said, premature truncation of the γ2 subunit (also known as stargazin) results in absence seizures in mice, consistent with an important role in modulation of calcium channel function (Letts et al., 1998). However, this subunit has also been shown to associate with and regulate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (Chen et al., 2000; Tomita et al., 2005), and, therefore, the in vivo role of the γ2 subunit in the calcium channel complex, if any, remains unclear.
The multiple subtypes of calcium channel α1 subunits, the splice isoforms thereof, and the many potential combinations of ancillary subunits collectively imply a vast diversity in terms of the calcium channels that can be generated. This diversity is an important consideration for issues of channel function and for second messenger regulation of channel function, as described below.
III. G Protein-Coupled Receptor Signaling—A Brief Overview
To fully appreciate regulation of calcium channels by G proteins, we will briefly review some key aspects of G protein-coupled receptor (GPCR) signaling. GPCR signaling is a very extensive topic, too much so for comprehensive review herein. Thus, we give a brief synopsis and refer the reader elsewhere for further detail (Ferguson, 2001; McCudden et al., 2005; Perez and Karnik, 2005).
A. Activation of G Proteins via G Protein-Coupled Receptors
GPCRs are a family of seven transmembrane helix receptors that are activated by a variety of physiological stimuli, in most cases extracellular neurotransmitters and hormones. Over 350 different types of GPCRs have been identified; for many of these, cellular roles have not yet been defined (Landry et al., 2006). GPCRs have a common transmembrane topology: an extracellular N terminus, three cytoplasmic loops (connecting transmembrane helices I and II, III and IV, and V and VI), three extracellular loops (connecting helices II and III, IV and V, and VI and VII), and a cytoplasmically localized C-terminal region (Fig. 2A). GPCRs also have a common mechanism of signal transduction: when activated by agonists, GPCRs interact with heterotrimeric complexes of G protein αβγ subunits and stimulate exchange of Gα-bound GDP for cytoplasmic GTP. Nucleotide exchange in turn favors dissociation of the heterotrimeric complexes into Gα-GTP and heterodimers of Gβγ, each of the latter being active signaling entities that modulate various downstream effector systems, often with crucial consequences for cellular function (Fig. 2A). The intrinsic GTPase activity of the Gα subunit hydrolyzes bound GTP back into GDP + Pi, thus terminating Gα activity and promoting the reassembly of the inactive Gα-GDPGβγ complex (Fig. 2B). In the continued presence of agonist, each receptor is able to activate multiple Gαβγ subunits, which in turn may modulate multiple effector molecules, resulting in signal amplification.
Details of the molecular events of agonist-mediated GPCR activation are the subjects of extensive study, in many cases using the GPCR rhodopsin as a model (Gether et al., 2002; Perez and Karnik, 2005) and will not be reviewed here. By contrast, although mutagenesis- and chimera-based studies have defined many molecular determinants of the interactions between activated receptors and inactive G protein heterotrimers, the molecular and thermodynamic details of these interactions remain largely unclear, an obvious issue to address in future investigations (Gether, 2000).
B. Subtypes of G Protein Subunits
In the human genome, 16 genes encode different types of Gα subunits (McCudden et al., 2005); the products of these genes are classified into 5 groups based on their abilities to activate various cell signaling systems (Table 1). Gαs proteins activate adenylyl cyclase; the Gαi proteins, which include Gαo and Gαz, generally inhibit adenylyl cyclase; Gαt proteins (a group which includes Gαgust) are found in sensory transduction pathways and activate cyclic GMP phosphodiesterase activity; Gαq proteins are activators of phospholipase Cβ; and, finally, Gα12 proteins are regulators of sodium-proton exchange. Most known Gα subunits have molecular masses of 39 to 52 kDa and are post-translationally modified with palmitoyl lipid functionalities; Gαi proteins often carry myristoyl functionalities as well, and both of these lipid functionalities are thought to contribute to proper subcellular localization of Gα subunits (Chen and Manning, 2001).
Pharmacology and signaling properties of the major G protein subunit types
There are also five known subtypes of G protein β subunits [plus potential alternate splice isoforms, with molecular masses of 35-39 kDa (Clapham and Neer, 1997; Fletcher et al., 1998)], and 12 different subtypes of Gγ subunits (Ray et al., 1995; Clapham and Neer, 1997; Huang et al., 1999). Gβ subunits contain tryptophanaspartate repeats (WD repeats) and an N-terminal amphipathic helix, which is known to interact with the Gγ subunit (Wall et al., 1995; Lambright et al., 1996). The Gγ subunits (Gγ1-5,7-13) are smaller molecules (molecular mass of ∼6-8 kDa) that are isoprenylated at the C terminus, which results in the plasma membrane anchoring of this subunit. Because Gβ and Gγ subunits typically exist as a complex, this results in membrane localization of the Gβ subunit.

A, schematic representation of the key events that occur during heterotrimeric G protein activation and signaling. 1, extracellular ligand binds to a seven-transmembrane helix G protein-coupled receptor, inducing receptor activation and allosteric changes in conformation. Next, allosteric changes in the conformation of the G protein α subunit, resulting from (2a) receptor activation or (2b) interaction with an AGS protein, lead to (2c) guanine nucleotide exchange, replacing Gα-bound GDP with GTP, and result in dissociation of the heterotrimeric G protein complex into two active signaling complexes, Gα-GTP (3a) and Gβγ (3b). B, schematic representation of the basic events of G protein signaling inactivation via reformation of the inactive Gαβγ heterotrimer. Gα-bound GTP is hydrolyzed to GDP, either due to intrinsic GTPase activity of the Gα subunit (4a) or enhanced GTPase activity of the Gα subunit resulting from direct interaction with an RGS protein (4b). Allosteric changes in the conformation of Gα, resulting in GTP hydrolysis, lead to reassociation of Gα with Gβγ (5), ultimately resulting in an inactive heterotrimeric complex (6). C, schematic representation of events leading to inactivation of G protein signaling due to internalization of GPCRs. 1, Gβγ heterodimers bind to G protein-coupled receptor kinases (GRKs) and recruit them to nearby GPCRs, thus enabling phosphorylation of the intracellular C-terminal loop of the GPCR (2). In turn, the newly phosphorylated motif of the receptor recruits and interacts with β-arrestin (β-arr). This leads to a cascade of interactions with the clathrin-based endocytic machinery, resulting in the endocytic internalization of the receptor and the desensitization of the cell to further stimulation by extracellular receptor ligand. GIRK, G protein-activated inwardly rectifying K+ channel.
One could propose >1000 unique, potential combinations of Gα, Gβ, and Gγ subunits. However, because of specific cellular and subcellular expression patterns and unfavorable thermodynamics of binding, one may expect many of these to be rare or not to exist at all as functional, heterotromeric complexes (further discussed below in section VII.). Furthermore, GPCR-G protein coupling is preferential according to Gα subunit type (Schoneberg et al., 1999; Cabrera-Vera et al., 2003) and also according to Gβγ subtypes (Taylor et al., 1994; Kisselev et al., 1995a,b; Yasuda et al., 1996; McIntire et al., 2001; Cabrera-Vera et al., 2003; Jones et al., 2004). And, finally, many types of GPCRs are known to homo- and heterodimerize, which can alter the specificity of GPCR-Gα coupling (George et al., 2000) or affect receptor internalization (AbdAlla et al., 2000). Collectively, this process provides a tremendous potential for linking GPCR activity to specific signaling pathways, as illustrated by the requirement for specific Gβγ subunit compositions for direct regulation of N- and P/Q-type calcium channels (described below).
C. Regulation of G Protein Activity
Various endogenous modulators can regulate the activities of Gα subunits independently of GPCR activation. Activator of G protein signaling (AGS) proteins can directly stimulate guanine nucleotide exchange of the Gα subunit (Fig. 2A), resulting in receptor-independent activation of Gα (Blumer et al., 2005). Regulator of G protein signaling (RGS) proteins comprise a large family of proteins with >20 different members. A primary function of RGS proteins is to stimulate the intrinsic GTPase activity of Gα, thus accelerating inactivation of these subunits (Berman et al., 1996) (Fig. 2B). In turn, this promotes the reformation of the inactive G protein heterotrimer and the consequent termination of Gβγ action on effector molecules (Doupnik et al., 1997). There is also evidence that some types of RGS proteins can directly interfere with the interactions between activated Gα subunits and their effectors (Berman and Gilman, 1998). We also note that certain RGS proteins contain a G protein γ-like domain and associate with Gβ subunits by replacing the Gγ subunit (Snow et al., 1998).
G protein activity can also be regulated by pharmacological means: intracellular application of the nonhydrolyzable GTP analog GTPγS results in permanent activation of all known types of Gα subunits and, at the same time, a massive increase in free Gβγ subunits. In contrast, guanosine 5′-O-(2-thio)diphosphate, once bound to Gα subunits, renders the subunits permanently inactive: they cannot exchange this GDP analog for GTP and thus remain permanently associated with Gβγ. Aluminum fluoride produces a permanently active Gα-GDP subunit by mimicking the effects of the γ phosphate group of GTP. Cholera toxin permanently activates Gαs subunits via ADP ribosylation, which drastically reduces their ability to hydrolyze GTP. In contrast, pertussis toxin (PTX) permanently inhibits Gαi subunits (with the exception of Gαz) by blocking their abilities to interact with GPCRs, again via ADP ribosylation (Fields and Casey, 1997). Gαt is sensitive to both toxins; Gαgust is presumed to be sensitive to both on the basis of amino acid sequence, but has only been experimentally demonstrated to be sensitive to PTX (Spielman et al., 1994; Fields and Casey, 1997; Ming et al., 1998).
D. Receptor Desensitization and Internalization
Many GPCRs display intrinsic desensitization mechanisms that allow them to terminate their activities during the sustained presence of receptor agonist. There are multiple mechanisms by which GPCRs desensitize. Heterologous desensitization involves protein kinase A- and/or protein kinase C-dependent phosphorylation of the third intracellular loop of the receptors, irrespective of whether agonist is bound. These phosphorylation events block the interaction of the receptor with the Gα subunit, effectively terminating GPCR-mediated signaling, and serving as a general mechanism by which receptors are desensitized. Homologous desensitization involves the phosphorylation of specific residues in the C terminus of the receptor (in its agonist bound state) by specific G protein-coupled receptor kinases (GRK) (Diverse-Pierluissi et al., 1996). These GRKs are activated by Gβγ subunits and translocate from the cytoplasm to the plasma membrane. Once phosphorylated, the C terminus becomes available for binding to arrestins, which then block receptor-G protein coupling. Another means of terminating GPCR activity in the presence of agonist is internalization of GPCRs into cytoplasmic vesicular compartments (Fig. 2C). This process is enhanced by GRK-dependent phosphorylation and the binding of arrestins (Ferguson, 2001); internalization can either be reversible, allowing the reinsertion of the receptors into the plasma membrane, or irreversible, leading to receptor degradation in lysosomal compartments. The net result in the above cases is termination of GPCR-mediated signaling and, hence, the return of effectors such as voltage-gated calcium channels to their basal, nonmodulated activity. In the context of voltage-gated calcium channels, these desensitization and internalization mechanisms are important means for restoring normal calcium channel function in the continued presence of receptor agonists.
IV. Discovery and Characterization of Direct G Protein Inhibition of Cav2 Calcium Channels
G protein inhibition of voltage-gated calcium currents was first described 25 years ago in two seminal articles by Dunlap and Fischbach (1978, 1981). These authors showed that the contribution of calcium channels to somatic action potentials in chick dorsal root ganglion (DRG) neurons was reduced in response to activation of GABAB, serotonin, or adrenergic receptors (see Fig. 3A, top), thus shortening action potential duration (Dunlap and Fischbach, 1978). These authors subsequently showed that this effect was due to a robust, receptor-mediated inhibition of HVA calcium currents in chick DRG neurons, currents now known to be carried almost entirely by N-type channels (Dunlap and Fischbach, 1981) (Fig. 3A, bottom). Numerous studies followed, revealing that many types of GPCRs—muscarinic, opioid, somatostatin, and dopamine receptors among them— have the propensity to inhibit native calcium currents (Forscher et al., 1986; Holz et al., 1986b; Bean, 1989; Ikeda and Schofield, 1989; Kasai and Aosaki, 1989; Lipscombe et al., 1989; Beech et al., 1992; Bernheim et al., 1992; Ikeda, 1992; Golard and Siegelbaum, 1993; Mintz and Bean, 1993; Shapiro and Hille, 1993; Zhu and Ikeda, 1993; Caulfield et al., 1994). A unifying property of this GPCR-mediated inhibition of calcium currents was its sensitivity to pertussis toxin, thus implicating Gαi and/or Gαo proteins (Holz et al., 1986a). The receptor-mediated inhibition was found to be blocked by the application of guanosine 5′-O-(2-thio)diphosphate (Holz et al., 1986a), hence further implicating Gα subunits. A role for Gα subunits in this mode of inhibition was thus established, yet not well understood.

A, top, original data illustrating traces of action potentials obtained from a dorsal root ganglion neuron held in a current clamp before and during exposure to norepinephrine (NE). The trace with the largest shoulder (shown to correspond to calcium influx) was collected before application of 0.1 mM extracellular NE (and is marked “-NA”); the trace with the smallest shoulder was collected after application of NE (marked “+NA”); traces with shoulders of intermediate sizes were collected at 10-s intervals after the initial application of NE, illustrating progressive desensitization of the preparation to NE. Image provided by Dr. Kathleen Dunlap. Bottom, paired traces of pharmacologically isolated HVA calcium currents, recorded from a voltage-clamped chick dorsal root ganglion neuron, before and after extracellular application of 0.1 mM noradrenaline (no label and “+NA”, respectively) (Dunlap and Fischbach, 1981). B, electrophysiological recording paradigms and whole-cell current recordings from a typical set of paired-pulse facilitation experiments using heterologously expressed N-type channels and Gβ1γ2 subunits. “-” indicates the transmembrane voltage protocol used for baseline sweeps wherein a depolarizing prepulse was not administered. “+” indicates the voltage protocol used for sweeps wherein test pulses were preceded by a 50-ms prepulse to +150 mV. Current recordings on the left show typical results obtained using alternating prepulse protocols on a cell coexpressing heterologous Cav2.2, β1b, and α2-δ subunits in the absence of GPCR activation or coexpressed Gβ1γ2. Paired recordings on the right show typical results in the same scenario but with coexpression of heterologous Gβ1γ2. Note that the prepulse relieves tonic G protein inhibition, thus resulting in a current with larger peak current amplitude and accelerated kinetics. C, results reproduced from a publication by Williams et al. (1997), demonstrating the effects of applying trains of action potential (AP)-like depolarizations before a 0-mV test pulse (see voltage protocol trace at bottom and lower left) during recordings of N-type currents from dissociated guinea pig cholinergic basal forebrain neurons. As indicated by the labels for the current traces on the right, the current amplitude increases with increasing numbers of AP-like depolarizations before the test pulse. Copyright 1997 by the Society for Neuroscience. D, reproduction of the compound state model used by Agler et al. (2005) for analysis of single-channel recordings from N-type channels. The model assumes two parallel sets of states for the channel, one set for channels inhibited by direct physical interaction with Gβγ heterodimers (labeled “Gβγ-bound”) and another set for channels not inhibited by such interactions (“Gβγ-unbound”). Each set of states is presumed to have differing kinetics (indicated by differently sized arrows) of G protein association and dissociation, with association favored in the deeper closed states. The G protein bound and unbound states, respectively, reflect the reluctant and willing gating modes of the channel.
The inhibition was then found to be membrane-delimited, i.e., to involve a second messenger molecule that remained associated with the plasma membrane, rather than diffusing to the channel via a cytoplasmic pathway (Forscher et al., 1986; Hille, 1994). Because the G protein βγ subunit is anchored to the plasma membrane (described above), these findings were consistent with direct, inhibitory physical interaction between calcium channels and membrane-tethered Gβγ subunits. This paradigm was left untested at first, because the dominant view of Gβγ subunit function was that it served only to capture GDP-bound Gα subunits. But it has since become clear that the physiological role of Gβγ subunits is much more complex and includes direct modulatory interaction with many target effectors, e.g., the G protein-coupled inwardly rectifying potassium channel, the first such effector identified (Logothetis et al., 1987; Ford et al., 1998; Albsoul-Younes et al., 2001; Mirshahi et al., 2002b). A direct role for Gβγ in the inhibition of voltage-gated calcium channels was first proposed by Bourinet et al. (1996) and later demonstrated in experiments testing the effects of overexpressed Gβγ subunits (Herlitze et al., 1996; Ikeda, 1996). The results of these experiments were fully consistent with the notion of a “membrane-delimited” pathway and were reported in back-to-back publications by the Ikeda and Hille groups (Herlitze et al., 1996; Ikeda, 1996).
The direct G protein inhibition of voltage-gated calcium channels occurs in a calcium channel subtype-dependent manner. N-type channels have long been considered a prime target for direct G protein inhibition (see above), and it is now established that P/Q-type calcium channels are also inhibited upon activation of Gαi- or Gαo-coupled receptors. However, P/Q-type channels typically undergo a smaller degree of inhibition relative to N-type channels (Currie and Fox, 1997). In contrast, other calcium channel subtypes expressed in native cells do not seem to be subject to direct G protein inhibition, suggesting that this type of modulation is confined to the two main presynaptic calcium channel isoforms. Similar findings have been obtained in expression systems (Bourinet et al., 1996; Toth et al., 1996; Page et al., 1997; Stephens et al., 1998; Beedle et al., 2004), although there have been reports of Gβγ-mediated inhibition of Cav2.3 (i.e., R-type) channels, particularly in the absence of the calcium channel β subunit (Mehrke et al., 1997; Qin et al., 1997; Shekter et al., 1997). More recently, a putative direct modulation of Cav3.2 calcium channels by Gβ2γ2 has been reported (Wolfe et al., 2003). However, the hallmarks of this modulation differ from those of the classic G protein inhibition described for HVA calcium channels (see below). Taken together, these considerations indicate that all members of the Cav2 channel family have at least some ability to undergo direct G protein inhibition, whereas other calcium channel subtypes typically do not.
A. Electrophysiological Hallmarks of Direct G Protein Inhibition
The membrane-delimited inhibition of voltage-gated calcium channels bears a number of distinct hallmarks. At the whole-cell level, peak current amplitudes are reduced in a voltage-dependent manner, with inhibition being stronger at more hyperpolarized potentials (Bean, 1989; Kasai and Aosaki, 1989; Lipscombe et al., 1989). This finding is reflected in a depolarizing shift in the midpoint of the activation curve of the channel. In addition, the time courses of current activation and inactivation can be slowed after receptor activation. All of the above effects are reversed after strong membrane depolarization (∼+100 mV) (Fig. 3B), hence the term “prepulse facilitation” or “prepulse relief” to describe the current increase that occurs when a depolarizing voltage pulse is applied before a test depolarization (Bean, 1989; Hille, 1994; Zamponi and Snutch, 1998a,b; Arnot et al., 2000). Such strong membrane depolarizations do not occur in normal mammalian physiology. However, rapid trains of action potentials (as well as increases in action potential duration) can lead to a similar recovery from G protein inhibition (Fig. 3C), leading to the suggestion that voltage-dependent G protein disinhibition may be important for synaptic function (Brody et al., 1997; Williams et al., 1997; Park and Dunlap, 1998). It has even been suggested that this phenomenon contributes to a novel form of paired-pulse facilitation observed in autaptic hippocampal cultures in the presence of GABAB or adenosine A1 receptor agonists (Brody and Yue, 2000). Moreover, in hippocampal slices, carbachol-induced inhibition of postsynaptic responses is relieved by application of paired presynaptic depolarizations (de Sevilla et al., 2002). A potential role of voltage-dependent disinhibition of G protein regulation in synaptic function is also supported by modeling studies (Bertram and Behan, 1999; Bertram et al., 2002, 2003).
Voltage-dependent G protein inhibition of N-type calcium channels has also been examined at the single channel level (Carabelli et al., 1996; Patil et al., 1996). In these studies, inhibited N-type channels show an increased first latency to opening, giving rise to the slowed activation kinetics observed in whole-cell N-type currents. Inactivation kinetics of the whole-cell currents also seem to be slowed, again due to the occurrence of delayed channel openings. Hence, the altered first latency to opening can account for the hallmark features of voltage-dependent G protein inhibition observed in whole-cell recordings. Mechanistically, the delay in opening can be explained by a stabilization of the closed state of Gβγ-bound channels, and this Gβγ-induced stabilization of a closed channel conformation is consistent with previous suggestions of “willing” (i.e., G protein free) and “reluctant” (i.e., G protein-bound) gating modes of the channel. (Bean, 1989; Kasai and Aosaki, 1989; Elmslie, 1992; Boland and Bean, 1993; Golard and Siegelbaum, 1993). Kinetic modeling (Patil et al., 1996) suggests that the transition from the reluctant to the willing gating mode involves the dissociation of the Gβγ complex from the channel (Fig. 3D), although reluctant N-type channel openings can also occur, albeit with very low probability (Colecraft et al., 2000; Lee and Elmslie, 2000). The significance of dissociation of Gβγ subunits from the N-type calcium channel for transition to a willing gating mode is also supported by experiments in which the free Gβγ concentration was varied (Zamponi and Snutch, 1998a). The kinetics of G protein reinhibition that follow a strong depolarizing prepulse become faster at increasing concentrations of free Gβγ, implying that Gβγ subunits must physically dissociate from the G protein complex during the prepulse and consistent with modeling work of Bertram and Behan (1999). These kinetic data are also consistent with a bimolecular interaction between Gβγ and the channel, thus resolving the extensively discussed issue of G protein-calcium channel stoichiometry (Kasai and Aosaki, 1989; Boland and Bean, 1993; Golard and Siegelbaum, 1993).
B. Does the Nature of the Gα Subunit Affect Voltage-Dependent Modulation?
As mentioned above, in the majority of early studies, the voltage-dependent modulation of N-type calcium channels seemed to be sensitive to PTX, thus implicating Gαi and/or Gαo subunits. However, there is evidence that receptors coupling to other types of Gα subunits can also mediate voltage-dependent modulation. For example, vasoactive intestinal peptide (VIP) mediates voltage-dependent inhibition of N-type calcium channels in sympathetic neurons via activation of Gαs and independently of protein kinase (PK) A (Zhu and Ikeda, 1994). Similarly, work from our own laboratory indicates that dopamine D1 receptors, despite coupling to Gαs, can mediate voltage-dependent modulation of heterologously expressed Cav2.2 calcium channels (Kisilevsky et al., 2006). Likewise, overexpression of Gαz in rat sympathetic neurons effectively rescues the loss of voltage-dependent modulation of N-type channels by adrenergic, adenosine, prostaglandin E2, and somatostatin receptors that occurs after incubation with PTX (Jeong and Ikeda, 1998). The ability of many types of G protein α subunits to couple N-type channels to Gβγ modulation is also supported by overexpression studies in which a wide range of Gα subunit subtypes (including Gα11 and Gαt, but interestingly not Gαz) were found to interfere with norepinephrine- or VIP-mediated inhibition of N-type channels in rat sympathetic neurons (Jeong and Ikeda, 1999). Together, the results described above show that many subtypes of Gα subunits can form heterotrimers with the same types of Gβγ subunits that are involved N-type channel modulation.
Because all GPCR types activate Gβγ irrespective of the types of Gα subunits to which they (the GPCRs) couple, then in principle any type of GPCR should be able to promote Gβγ-mediated inhibition of N-type channels. However, there are several additional considerations: First, the receptors and channels need to be localized in close proximity to allow for effective diffusion and binding of Gβγ subunits to the channel. Second, although Gα subunits may be able to biochemically interact with a wide range of Gβγ dimers, structural features of the GPCR itself can prevent coupling to certain combinations of Gβγ subunits—a crucial point, since the isoform of the G protein β subunit is a key determinant of voltage-dependent calcium channel modulation (as we discuss below). Finally, the activation of other intracellular signaling pathways by particular types of Gα subunits may interfere with the ability of Gβγ to inhibit N-type channel activity (also described below). Hence, control of the specificity of GPCR signaling to N-type calcium channels goes beyond the coupling of the receptor to a particular Gα subunit.
C. Voltage-Independent G Protein Inhibition
The free Gβγ heterodimers resulting from GPCR activation do not signal exclusively to voltage-gated calcium channels: they also modulate components of other cytoplasmic messenger systems such as phospholipase Cβ and adenylyl cyclase (Gao and Gilman, 1991; Tang and Gilman, 1991; Camps et al., 1992). Moreover, activated Gα subunits trigger various intracellular responses, which may converge on voltage-gated calcium channels to either up-regulate or inhibit their activities. Thus, in addition to mediating voltage-dependent inhibition of Cav2 calcium channels via Gβγ, a number of GPCRs trigger a concomitant inhibition of the channels via soluble second messenger pathways (Beech et al., 1991, 1992; Bernheim et al., 1991, 1992; Luebke and Dunlap, 1994; Surmeier et al., 1995; Delmas et al., 1998a,b; Shapiro et al., 1999; Kammermeier et al., 2000; Schiff et al., 2000; Beedle et al., 2004). Because this type of inhibition cannot be reversed by strong membrane depolarizations, it is referred to as voltage-independent.
The precise molecular mechanisms by which voltage-independent modulation occurs are incompletely understood, but evidence suggests it can be elicited by a number of distinct signaling pathways that may be tailored to particular types of GPCRs. For example, dopamine D1 receptors couple to Gαs and thereby activate PKA, which in turn phosphorylates protein phosphatase 1. This phosphatase has been shown to dephosphorylate residues on N- and P/Q-type calcium channels, resulting in voltage-independent current inhibition (Surmeier et al., 1995). In contrast, robust voltage-independent modulation of N-type channels in chick DRG neurons has been attributed to tyrosine kinase-dependent phosphorylation of the N-type calcium channel α1 subunit (Schiff et al., 2000) and involves classes of G proteins different from those involved in voltage-dependent modulation (Diverse-Pierluissi et al., 1995). Finally, muscarinic M1 and neurokinin 1 receptors both trigger voltage-independent inhibition of N-type channels via a Gαq, but also require the action of Gβγ subunits as part of the signaling cascade (Kammermeier et al., 2000). The regulation of voltage-gated calcium channels by kinases and phosphatases is a vast area of research that has been reviewed recently (Bannister et al., 2005) and will not be further described herein. However, the above examples serve to illustrate the fact that G protein-coupled receptors mediate more than just voltage-dependent, membrane-delimited inhibition of Cav2 calcium channels. And, as described below, there may be cross-talk between voltage-dependent and voltage-independent pathways that contributes to the overall complexity of GPCR signaling to voltage-gated calcium channels.
D. Regulator of G Protein Signaling and Activator of G Protein Signaling Proteins and Calcium Channel Inhibition
RGS proteins have recently emerged as important factors in the voltage-dependent modulation of N- and P/Q-type calcium channels (Jeong and Ikeda, 1998, 2000; Diverse-Pierluissi et al., 1999; Melliti et al., 1999, 2001; Mark et al., 2000). RGS2 proteins accelerate the recovery from inhibition of heterologously expressed P/Q-type calcium channels by M2 muscarinic receptors and alter the availability of Gβγ subunits for producing voltage-dependent inhibition of these channels (Mark et al., 2000). Likewise, by stimulating the GTPase activity of Gα (and thus reducing the amount of free Gβγ that is available to modulate the channels), overexpression of RGS3 and RGS8 proteins attenuates the muscarinic inhibition of heterologously expressed N-type calcium channels (Melliti et al., 1999). Interestingly, RGS2 and RGS12 also alter voltage-independent inhibition of N-type channels in a manner that seems to be independent of altered GTPase activity (Schiff et al., 2000; Melliti et al., 2001), suggesting that RGS proteins can act directly on effector molecules such as N-type channels (Richman et al., 2005). RGS proteins have also been shown to reduce adrenergic inhibition of native N-type currents (Jeong and Ikeda, 2000). When RGS4 proteins are depleted by intracellular dialysis of an RGS4 specific antibody, adrenergic modulation of these channels becomes enhanced (Diverse-Pierluissi et al., 1999). Both RGS4 and RGS10 accelerate the deactivation of Gαz-mediated noradrenergic modulation of N-type channels in rat sympathetic neurons (Jeong and Ikeda, 1998). Finally, RGS9 antagonizes dopamine D2 receptor-mediated modulation of N-type currents in rat striatal cholinergic interneurons (Cabrera-Vera et al., 2004). Hence, a host of different RGS proteins have been linked to regulation of voltage-dependent modulation of N- and P/Q-type calcium channels, presumably via their stimulation of GTPase activity. The fact that many endogenous types of RGS proteins can affect receptor signaling to voltage-gated calcium channels indicates that caution should be exercised when attempting to reconstitute receptor-channel signaling in transient expression systems.
A number of RGS proteins have been shown to regulate calcium channel activity independently of an action on the Gα subunit. For example, both RGS6 and RGS11 antagonize the modulation of human Cav2.2 calcium channels by coexpressed Gβ5 subunits (Zhou et al., 2000). Both of these RGS protein types have been shown to interact directly with Gβ5 by competing with the G protein γ subunit (Snow et al., 1999), presumably resulting in an RGS-Gβ5 complex that is incapable of inhibiting the channel. RGS6 does not seem to interact with any other G protein β subunit subtype, but both RGS7 and RGS9 also have G protein γ-like domains (Snow et al., 1998), raising the possibility that some of the RGS proteins could directly modulate the actions of other subtypes of Gβ subunits on voltage-gated calcium channels. Such a scenario needs to be investigated experimentally in expression systems.
AGS proteins serve as receptor-independent means of activating G protein signaling (Blumer et al., 2005). Hence, up-regulation of AGS protein expression may trigger receptor-independent inhibition of voltage-gated calcium channels, although such an action has yet to be demonstrated. The synaptic vesicle release protein cysteine string protein (CSP), although not an AGS protein, does contain a region capable of stimulating exchange of Gα-bound GDP for GTP in purified Gα subunits (Natochin et al., 2005), which may account for voltage-dependent modulation of N-type channels that results from overexpression of this region of CSP (Miller et al., 2003). Collectively, these findings indicate that G protein inhibition of Cav2 calcium channels, be it indirect or mediated by direct interaction between the channel and Gβγ, is subject to a number of regulatory mechanisms that occur upstream of the channel itself.
V. Calcium Channel Structural Determinants of G Protein Modulation
A. Calcium Channel α1 Subunit Structural Determinants
The calcium channel structural determinants that underlie Gβγ modulation of Cav2 calcium channels has been investigated in numerous studies (Fig. 4A). Zhang et al. (1996) created a series of chimeric calcium channels that combined the structural features of Cav2.1 and Cav2.2 calcium channels and expressed them transiently in Xenopus oocytes. Based on this chimeric approach, the authors identified two structural regions that accounted for the differential capacity of the parent channels for G protein modulation: domain I and the intracellular C-terminal region. However, the chimeric approach used in this structure-function study has an inherent limitation: it will not identify all regions involved in G protein modulation but rather only the regions responsible for the differences observed between the two channels, an issue of importance in this case, as both Cav2.1 and Cav2.2 are capable of undergoing voltage-dependent G protein inhibition. Moreover, these authors did not demonstrate Gβγ binding to these channel regions. In contrast, using in vitro translation of recombinant Gβγ in conjunction with binding assays, De Waard et al. (1997) demonstrated binding of Gβγ to two distinct regions within the domain I-II linker of Cav2.1. Similarly, Zamponi et al. (1997) showed that synthetic peptides corresponding to the same regions in the I-II linkers of Cav2.2 and Cav2.1 were able to block functional G protein modulation of transiently expressed Cav2.2 channel, and that fusion proteins of the Cav2.2 I-II linker, but not the C terminus, bound Gβγ in vitro. In addition, the differential voltage-dependent modulation observed for Cav2.1 and Cav2.2 is reversed by a chimeric swap of the I-II linker regions of the two channel subtypes (Zamponi et al., 1997).
Interestingly, one of the two Gβγ-interaction regions of the domain I-II linker partially overlaps with the AID region that is known to bind the calcium channel β subunit (Pragnell et al., 1994). This putative G protein binding domain also includes a QXXER motif that is found in the Gβγ binding domain of adenylyl cyclase, and mutagenesis studies revealed a potential role of this region in G protein modulation: substitution from QQIER to QQIEE in Cav2.1 increased G protein modulation, and a change to QQLEE affected reversibility of G protein modulation (Herlitze et al., 1997). Studies involving Cav2.2-Cav2.3 chimeras also point to an involvement of the I-II linker region in the kinetic slowing observed during G protein inhibition but suggest that other regions may also contribute to steady-state inhibition (Page et al., 1997; Simen and Miller, 1998, 2000). On the other hand, Qin et al. (1997) reported that the I-II linker was not an essential determinant of G protein inhibition of N-type channels and instead pointed to the C terminus of the channel. However, large parts of the C terminus of Cav2.2 can be deleted with only minor changes in G protein inhibition (Hamid et al., 1999), suggesting an auxiliary role for this region rather than a key requirement. Indeed, ensuing work by Li et al. (2004) revealed that the C terminus, although being able to bind to Gβγ subunits, serves mainly to enhance the affinity of the channel for Gβγ rather than being an absolute requirement. Several studies have attributed a key role to the N terminus of the channel in G protein inhibition (Page et al., 1998; Simen and Miller, 1998, 2000; Stephens et al., 1998; Canti et al., 1999). In particular, three individual amino acid residues in the Cav2.2 N terminus (Ser48, Arg52, and Arg54) were found to be critical for N-type calcium channel modulation (Canti et al., 1999). The mechanism by which the N terminus of the channel contributes to G protein inhibition of Cav2.2 was resolved recently by Agler et al. (2005). The authors showed that the N terminus of Cav2.2 and the I-II linker physically interact with each other, and this interaction is essential for G protein modulation to proceed. At this point, it is not entirely clear whether the interaction of the I-II linker and the N terminus is required to facilitate functional modulation of the channel by Gβγ, or, alternatively, whether Gβγ binding to the channel promotes an interaction between the two channel regions, which in turn inhibits channel activity allosterically. Nonetheless, these findings raise several issues concerning the interpretations of earlier results. For example, it is possible that the synthetic I-II linker peptides used by Zamponi et al. (1997) might have acted in part by blocking the interaction between the N terminus and the I-II linker, rather than only competing Gβγ away from the channel. Moreover, mutagenesis of residues or substitutions of the I-II loop in Cav2.2 could simply have resulted in a loss of the I-II linker-N terminus interaction. Finally, it may be worth noting that the Cav2.2 channels used in the earlier chimeric studies of Zhang et al. (1996) carried a portion of the Cav2.1 N terminus to promote membrane expression; which, given the role of this region in G protein modulation, complicates the interpretation of the results obtained with these chimeras. That said, the work of Agler et al. (2005) has served to reconcile the many previous structure-function studies that implicated the N terminus and I-II linker regions as key functional entities in the direct membrane-delimited inhibition of N-type channels. How the C terminus region affects G protein regulation at the molecular level remains open to investigation. It is possible that this region may interact with either the N terminus or the I-II loop to promote G protein inhibition. Moreover, given recent evidence of an interaction between the domain I-II and III-IV linker regions of Cav2.1 channels (Fathallah et al., 2002; Geib et al., 2002), it is possible that the overall Gβγ binding pocket on Cav2 calcium channels could be formed by the concerted action of multiple intracellular regions of the channel (Fig. 4A).

A, schematic representation of Cav2 channel α1 subunit regions involved in G protein modulation. Structural regions demonstrated to play a role in modulatory interactions with Gβγ in the N-terminal loop (Canti et al., 1999; Agler et al., 2005), the I-II loop (De Waard et al., 1997; Zamponi et al., 1997; Agler et al., 2005), and the C-terminal loop (Qin et al., 1997; Hamid et al., 1999; Li et al., 2004) are indicated by shaded and unshaded boxes and by amino acid residue numbers corresponding to the respective sequence regions (numbering based on rat Cav2.2 sequence). B, schematic representation of the Cav2 channel engaged in simultaneous modulatory interactions with the calcium channel β subunit and the Gβγ heterodimer, in contrast to the scenario proposed in C, wherein the calcium channel β subunit and the Gβγ heterodimer compete for interaction with the channel in a mutually exclusive manner. A shaded box on the I-II loop represents an area of overlap between a sequence region known to interact with the calcium channel β subunit and another region known to interact with Gβγ.
B. Role of the Calcium Channel β Subunit
As outlined above, the domain I-II linker contains the AID region, a sequence of residues that binds to the calcium channel β subunit and overlaps partially with one of two G protein βγ binding motifs found in the I-II linker. Crystal structure data showing the interface between the calcium channel β subunit and the AID region indicate that only a few residues of the AID motif would be sufficiently exposed to interact with Gβγ (Chen et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004), raising the question of whether Gβγ could in fact bind to the I-II linker in the presence of the calcium channel β subunit. Nonetheless, the putative Gβγ binding motifs identified by de Waard et al. (1997) and Zamponi et al. (1997) extend beyond the AID region. So it seems plausible to hypothesize either a stable quaternary complex of the Gβγ subunits with the calcium channel α1 and β subunits or a scenario of mutually exclusive binding of either the Gβγ heterodimer or the calcium channel β subunit to the α1 subunit.
Whatever the case, the proximity of the AID region and the putative target regions for Gβγ binding suggests that the calcium channel β subunit may regulate the modulation of Cav2 channels by Gβγ subunits. The first evidence supporting such a mechanism came from antisense experiments in which calcium channel β subunits were depleted in rat DRG neurons, leading to an augmentation of GABAB-induced N-type channel inhibition (Campbell et al., 1995). Bourinet et al. (1996) subsequently showed that omission of the calcium channel β subunit dramatically augmented the inhibition of transiently expressed Cav2.1 channels by μ-opioid receptors. Moreover, these authors showed that receptor activation produces a much more pronounced kinetic slowing when the channel complex contained the calcium channel β2a subunit (compared with channels containing other subtypes of calcium channel β subunits). Several additional studies indicate that G protein inhibition of N-type calcium channels is affected by the calcium channel β subunit isoform. Feng et al. (2001) reported that different types of calcium channel β subunits affected the extent of prepulse relief in the presence of overexpressed Gβ subunits. In particular, for Cav2.2 channels coexpressed with the calcium channel β2a subunit, the kinetic slowing induced by Gβγ subunits was dramatically enhanced relative to that observed in the presence of other calcium channel β subunits. Canti et al. (2000) reported that all four types of calcium channel β subunits antagonized the G protein-induced depolarizing shift in the current-voltage relation when examined in Xenopus oocytes. Only the calcium channel β2a subunit led to a reduction in the total extent of G protein inhibition, and all four calcium channel β subunits accelerated the rate of Gβγ dissociation from the calcium channel complex. Curiously, when the same group of investigators examined the effects of calcium channel β subunits on G protein modulation of Cav2.2 channels in Cos-7 cells, the authors found that the calcium channel β subunit was necessary for Gβγ to mediate voltage-dependent inhibition (Meir et al., 2000). Cos-7 cells are apparently completely devoid of endogenous calcium channel subunits, whereas Xenopus oocytes are known to express endogenous β subunits. It is thus possible that these conflicting results were due to the effects of an endogenous β subunit that renders the channels more sensitive to G protein inhibition compared with that observed upon coexpression with rat β subunits. However, the data of Meir et al. (2000) are more difficult to reconcile with the antisense experiments of Campbell et al. (1995), wherein depletion of β subunits resulted in an increase rather than a decrease in inhibition. It is possible that calcium channel β subunit regulation of G protein inhibition could be affected by other proteins (synaptic proteins, scaffolding proteins, etc.) that are known to be expressed in neurons, but perhaps are not present in expression systems.
Inconsistencies aside, the studies described above establish the following: as a function of the identity of the calcium channel β subunit in the Cav2 channel, both Gβγ-Cav2 dissociation kinetics and the Gβγ-induced slowing of Cav2 current activation kinetics can differ. This is consistent with the notion that calcium channel β subunits and Gβγ subunits can interact simultaneously with Cav2.2 and Cav2.1 channels, although not to the exclusion of other possibilities. For Cav2.1 channels, this was examined directly by using Foerster resonance energy transfer (FRET) between the calcium channel β subunit and the Cav2.1 α1 subunit. Hummer et al. (2003) showed that coexpression of G protein βγ subunits increased FRET between the calcium channel α1 and β subunits. This increase depended on the presence of an intact C terminus of the α1 subunit of the channel, indicating 1) that Gβγ causes a change in channel conformation, 2) that this change in conformation involves the C terminus of the α1 subunit, and 3) that calcium channel and G protein β subunits can bind to the Cav2.1 channel concomitantly (Fig. 4B). These findings are consistent with the functional data obtained with Cav2.1 channels by Bourinet et al. (1996). Interestingly, an analogous study by Sandoz et al. (2004) came to the opposite conclusion. These authors reported a loss of α1-β subunit FRET during G protein activation and suggested that the calcium channel β subunit must dissociate from the channel complex during G protein modulation (Fig. 4C). It is difficult to reconcile such opposing findings, but it is important to note that absence of FRET does not imply lack of proximity between two labeled proteins, as the orientation of the fluorophore used during the FRET experiments is critical for FRET coupling.
In summary, the calcium channel β subunit is an important determinant of G protein inhibition of Cav2 calcium channels. In consideration of the facts that many cell types express multiple isoforms of calcium channel β subunits and that calcium channel β subunit expression is regulated during development, this is yet another mechanism by which calcium entry can be precisely controlled to suit a specific cellular requirement.
VI. Modulation of G Protein Modulation
A. Cross-Talk between G Protein Inhibition and Protein Kinase C Modulation
Studies in neurons have revealed that the membrane-delimited inhibition of N-type calcium channels by a number of different types of G protein-coupled receptors can be reduced by the concomitant (or prior) activation of pathways that activate PKC (Swartz, 1993; Swartz et al., 1993; Barrett and Rittenhouse, 2000). Because the Gβ subunit cannot be phosphorylated, this suggested that the cross-talk between PKC and G protein inhibition might occur directly at the level of the calcium channel α1 subunit. The second putative Gβγ interaction site within the domain I-II linker (i.e., the one downstream of the AID region) contains two separate PKC consensus sites (Thr422 and Ser425) (Fig. 5A). Synthetic peptides corresponding to this region can be phosphorylated in vitro, and, when phosphorylated, they lose their ability to interfere with Gβγ modulation of N-type channels (Zamponi et al., 1997). Site-directed mutagenesis of Thr422, but not of Ser425 to alanine prevents the effects of PKC on G protein modulation of Cav2.2 channels, and mutagenesis of Thr422 to glutamic acid mimics the effects of PKC activation (Hamid et al., 1999). Hence, the PKC-G protein cross-talk can be localized to phosphorylation of a single residue in one of the putative G protein binding regions on the channel protein (Fig. 5, B and C). The ability of PKC to antagonize Gβγ-mediated inhibition depends on the G protein β subunit isoform, with only Gβ1 being capable of sensing PKC-dependent phosphorylation of the channel (Cooper et al., 2000) (Fig. 5B). Hence, PKC-mediated antagonism of GPCR-mediated, voltage-dependent inhibition of N-type calcium channels may be most pronounced for GPCR types that couple preferentially to Gβ1, such as the somatostatin receptor (Cooper et al., 2000). In passing, we also note evidence of another mode of interaction between Cav2 channels and PKC signaling in that certain isoforms of Cav2.2 channels respond directly to PKC activation via an up-regulation of current activity (Stea et al., 1995); in this case, phosphorylation of either of the two PKC consensus sites was found to mediate the full effect (Hamid et al., 1999).
The cross-talk between PKC and G protein pathways is also highlighted by an elegant study involving receptor-G protein tandems (Bertaso et al., 2003). These authors created cDNA constructs in which specific, PTX-insensitive Gαi or Gαo mutants were fused to the C terminus of the α-adrenergic receptor. In the presence of PTX, these tandem constructs were able to fully restore voltage-dependent inhibition of N-type channels. When a similar experiment was conducted by using a modified Gαq-receptor tandem construct, voltage-independent modulation ensued, and, furthermore, activation of the Gαq pathway antagonized the voltage-dependent modulation via the Gαo pathway, consistent with the action of PKC.
PKC-Gβγ cross-talk allows for the integration of multiple second messenger inputs directly at the level of the calcium channel α1 subunit. It is not known whether other kinases can mediate a similar effect on G protein-mediated inhibition of Cav2 calcium channels, but it seems likely that additional interactions among signaling pathways could occur upstream of calcium channels, with secondary affects on G protein inhibition (recall the role of RGS proteins).
B. Synaptic Proteins
Both N- and P/Q-type calcium channels are known to associate with several synaptic proteins—among them, syntaxin 1, SNAP-25, and synaptotagmin—at a synaptic protein interaction site found in the domain II-III linker region (Sheng et al., 1994, 1997; Jarvis and Zamponi, 2001b; Spafford and Zamponi, 2003). These interactions might serve multiple functions, e.g., to increase the coupling between synaptic calcium entry and the neurotransmitter release machinery (Mochida et al., 1996, 2003; Harkins et al., 2004).
Coexpression of syntaxin 1 with either N- or P/Q-type calcium channels in oocytes or tsA-201 cells results in a hyperpolarizing shift in the steady-state inactivation curve of the channels, reflecting a decreased availability of the channel to open (Bezprozvanny et al., 1995; Jarvis and Zamponi, 2001a). Treatment of neurons with botulinum toxin C1, which cleaves syntaxin 1A, removes this inhibitory effect (Bergsman and Tsien, 2000; Stanley, 2003). Intriguingly, though, treatment of chick giant calyces with botulinum toxin C1 also renders N-type calcium channels insensitive to G protein inhibition induced by the inclusion of GTPγS in the patch pipette, indicating a potential interplay between the vesicle release machinery and G proteins (Stanley and Mirotznik, 1997). Consistent with such interplay, we found that syntaxin 1A can physically bind to G protein β subunits in vitro (Jarvis et al., 2000). And, when coexpressed with Cav2.2 channels in tsA-201 cells, syntaxin 1A results in tonic, voltage-dependent G protein inhibition of N-type currents, independent of any exogenous receptor activation (Jarvis et al., 2000). A similar effect, although of smaller magnitude, has been observed for channels expressed in Xenopus oocytes (Hurley et al., 2004). Because distinct structural regions of syntaxin bind to the calcium channel and to Gβ, it is possible that syntaxin acts as a chaperone to colocalize the channel and Gβ subunits, thus promoting tonic G protein inhibition (Jarvis et al., 2002) (Fig. 6).

A, schematic representation of the Cav2 channel and the Gβγ heterodimer, illustrating the locus for the PKC consensus site formed by threonine 422 (T422) in the domain I-II linker. B, whole-cell current recordings from cells coexpressing N-type channels with heterologous Gβ1γ2 dimers, with the label “+PP” indicating traces collected after a depolarizing prepulse, and “-PP” indicating traces collected when no prepulse was applied before the test pulse. The recordings at the top show significant Gβγ-mediated, voltage-dependent inhibition of the N-type current, which is reduced after activation of PKC by application of 30 mM phorbol 12-myristate 13-acetate (Cooper et al., 2000). C, representation of the effects of PKC on G protein regulation of Cav2.2 channels. Activation of phospholipase C (PLC) by interaction either with Gαq-GTP and/or Gβγ results in the production of diacylglycerol (DAG). DAG in turn activates PKC, which phosphorylates the side chain of residue T422 of the N-type channel. This phosphorylation event blocks modulatory interaction between the channel and any Gβγ heterodimer containing the Gβ1 subunit, presumably due to steric interactions (between Gβ1 residues N35 and N36 and phosphorylated Cav2.2 residue T422), thus reducing Gβγ-mediated, voltage-dependent inhibition of the channel.
In binding studies between syntaxin and purified Gβγ subunits, the Gγ2 subunit could not be detected (Jarvis et al., 2002). This finding suggests that Gβγ complexes containing Gγ2 are unable to associate with syntaxin or perhaps that syntaxin may replace the Gγ subunit when associating with Gβ, which would be reminiscent of the role of certain RGS proteins. Experiments examining the 1B isoform of syntaxin provide more intriguing details: syntaxin 1B can also bind to Gβγ, but its coexpression with Cav2.2 channels alters channel availability without promoting a tonic G protein inhibition of Cav2.2 calcium currents (Lu et al., 2001), perhaps because syntaxin 1B adopts a conformation that is less amenable to inducing tonic G protein inhibition when bound to the channel, compared with the conformation adopted by complexes of the channel and syntaxin 1A (Fig. 6, A and B).

Representation of the modulatory interactions between presynaptic (N-type or P/Q-type) calcium channels, the Gβγ subunit, and the presynaptic proteins syntaxin 1A, syntaxin 1B, and CSP. A, presynaptic vesicle fusion protein syntaxin 1B interacts directly with the synprint region of the intracellular II-III loop of the channel (represented as thick gray doubling behind the dashed line). The interaction results in a hyperpolarizing shift in the voltage dependence of inactivation, thus reducing channel availability (not shown), but does not affect basal G protein modulation of the channel. B, in contrast, the formation of a complex of channel and syntaxin 1A results in reduced channel availability and leads to enhanced recruitment of Gβγ, such that Gβγ-mediated, voltage-dependent channel inhibition is observed even in the absence of GPCR activation. Interaction with the channel-syntaxin 1A complex may potentially result in the dissociation of Gβ and Gγ (Jarvis et al., 2002). C, phosphorylation (*) of the synprint region by activated PKC (PKC*) modifies the conformation of the syntaxin 1A-channel complex such that the hyperpolarization in steady-state inactivation is no longer observed (Jarvis and Zamponi, 2001a), whereas tonic G protein modulation can still persist. D, CSP enhances Gβγ-mediated channel inhibition in two ways: first, the C-terminal half of CSP simultaneously interacts with the synprint region of the channel and recruits Gβγ, with results reminiscent of those obtained with syntaxin 1A-channel complexes; second, the N-terminal half of CSP functions as an AGS protein, acting on Gαβγ-GDP complexes to facilitate guanine nucleotide exchange. This in turn promotes dissociation of Gαβγ complexes, resulting in Gβγ modulation of the channel.
Phosphorylation of the synaptic protein interaction site by PKC and calmodulin-dependent protein kinase prevents syntaxin binding in vitro (Yokoyama et al., 1997, 2005). Consistent with a regulatory role of PKC, the syntaxin 1-induced change in channel availability is removed after PKC activation, whereas the tonic G protein inhibition seems to persist (Jarvis and Zamponi, 2001a) (Fig. 6C). This result suggests that under physiological conditions, PKC is unable to fully dislodge syntaxin 1A from the channel, a conclusion also supported by recent binding studies (Yokoyama et al., 2005). The effects of PKC on syntaxin 1 action are reminiscent of the PKC cross-talk described above and consistent with an overall stimulatory role for this kinase with respect to Cav2 calcium currents. Illustrating a similar regulatory cross-talk, RGS12 has been shown to bind to the synaptic protein interaction site to regulate tyrosine kinase-mediated, voltage-independent modulation of N-type channels (Richman et al., 2005).
The coexpression of CSP also mediates a tonic G protein inhibition of transiently expressed Cav2.2 channels. Like syntaxin 1, CSP physically binds to both the synaptic protein interaction site and to Gβγ (Magga et al., 2000) (Fig. 6D). However, the mechanism of CSP modulation is more complex. Expression of the C-terminal half of CSP induces tonic G protein inhibition of the channel, but inhibition is eliminated upon overexpression of synaptic protein interaction site peptides (Miller et al., 2003). Hence, the C-terminal half of CSP is sufficient to produce a syntaxin 1-like effect in terms of G protein modulation of N-type channels. However, expression of the N-terminal half of CSP also triggers tonic G protein inhibition of Cav2.2 channels. By contrast, this inhibition does not rely on binding to the synaptic protein interaction site; rather, it results from stimulation of GDP-GTP exchange in Gα. It is unclear whether and to what extent the CSP-mediated modulation of N-type currents occurs under physiological conditions, as N-type calcium channel activity has not yet been assessed in CSP-deficient mice (these mice show normal P/Q-type channel activity) (Fernandez-Chacon et al., 2004).
Worth noting is the fact that the interaction between G protein βγ subunits and synaptic proteins may also play an important role downstream of N-type calcium channels. It has been reported that Gβγ, by interacting with syntaxin 1 and SNAP-25, can directly interfere with neurotransmitter release (Blackmer et al., 2001, 2005; Gerachshenko et al., 2005). Hence, the interactions between calcium channels, synaptic proteins, and G protein subunits provide multiple avenues for regulation of neurotransmitter release, which may occur independently of receptor activation and may involve other mechanisms besides a mere reduction in presynaptic calcium entry.
VII. G Protein Structural Determinants of N-Type Channel Modulation
The question as to which combinations of Gβ and Gγ subunit types associate to form functional heterodimers, ostensibly relevant for the issue of modulatory interaction of Gβγ with Cav2 channels, has been examined, although not exhaustively (Iniguez-Lluhi et al., 1992; Pronin and Gautam, 1992; Schmidt et al., 1992; Watson et al., 1994; Meister et al., 1995; Watson et al., 1996; Yan et al., 1996). Structural regions of the Gγ subunit that determine selectivity of Gβγ binding and complex formation have also been identified (Spring and Neer, 1994; Lee et al., 1995; Meister et al., 1995). However, for extensive review of these issues, the reader is referred to Clapham and Neer (1997), as the discussion below will be limited to consideration of interactions between Gβγ and the Cav2 channel.
A. Gβ Subtype Dependence
To date five subtypes of Gβ subunit have been identified in mammals; subtypes 1 to 4 show ∼80 to 90% amino acid identity, and subtype 5 shows ∼50% identity with respect to subtypes 1 to 4 (Fong et al., 1986, 1987; Levine et al., 1990; von Weizsacker et al., 1992; Watson et al., 1994; Downes and Gautam, 1999). Various studies have aimed to quantify the abilities of the five Gβ subtypes, when incorporated into Gβγ heterodimers, to modulate Cav2 channels directly. In the two earliest demonstrations that voltage-dependent modulation of Cav2 channels is mediated by Gβγ heterodimers (and not by GTP-Gα), inhibition was shown when Gβ1γ2, Gβ1γ3, Gβ1γ7, or Gβ2γ3 was coexpressed with the channels (Herlitze et al., 1996; Ikeda, 1996). Confirming and building on these results, in a later study Garcia et al. (1998) tested heterodimers of Gγ3 and the five Gβ subunits, heterologously expressed in rat superior cervical ganglion (SCG) neurons, and found that Gβ1 and Gβ2 mediated voltage-dependent inhibition of N-type channels most effectively, followed by weak inhibition mediated by Gβ5, whereas any inhibition mediated by Gβ3 and Gβ4 was undetectable. An ensuing study by Ruiz-Velasco and Ikeda (2000) showed that Gβγ heterodimers, composed of any combination of Gβ1, Gβ2, Gβ3, or Gβ4, with Gγ2, Gγ3, or Gγ4, could mediate significant voltage-dependent inhibition of calcium currents when heterologously expressed in rat SCG neurons. These authors also reported significant voltage-dependent inhibition mediated by Gβ5γ2 heterodimers but only when cDNAs encoding the heterodimer were injected at a 10-fold higher concentration than was used for experiments testing any of the other four Gβ subunits (Gβ5γ1 and Gβ5γ3 heterodimers were not tested in this manner). The major discrepancy between the results of Garcia et al. and Ruiz-Velasco and Ikeda is that the latter observed significant voltage-dependent inhibition mediated by Gβ subtypes 3 and 4, whereas Garcia et al. did not. Various differences in experimental conditions may account for this: 1) Garcia et al. used bovine Gβ2 and (presumably) mouse Gβ3, Gβ4, and Gβ5, whereas Ruiz-Velasco and Ikeda used human Gβ2 and Gβ3 and mouse Gβ4 and Gβ5; 2) Garcia et al. used 2 μM nifedipine to block L-type calcium currents, and Ruiz-Velasco and Ikeda did not; and 3) Garcia et al. used bovine Gγ3 cDNA, whereas Ruiz-Velasco and Ikeda used cDNAs encoding human Gγ2-4.
A further discrepancy involves voltage-dependent Cav2 channel inhibition by Gβγ heterodimers containing Gβ5. Like Garcia et al. (1998) and Ruiz-Velasco and Ikeda (2000), Zhou et al. (2000) observed that Gβ5 mediated comparatively slight inhibition of calcium currents, but in this case as part of human Gβ5γ2 heterodimers, acting on human N-type calcium channels, all heterologously expressed in HEK cells. In contrast with Garcia et al. (1998), Zhou et al. (2000) observed no inhibition of N-type currents by Gβ5γ3 heterodimers. Rather, they observed inhibition to be mediated by Gβ5γ2 heterodimers. As above, various differences in experimental conditions may account for the differing results: Garcia et al. measured the modulation of endogenous rat N-type channels by heterodimers of mouse Gβ5 and bovine Gγ3, whereas Zhou et al. reported results showing modulation of human N-type channels by heterologously expressed human Gβ5.
Arnot et al. (2000) reported yet another body of differing results. Using transient expression in tsA-201 (HEK) cells, these authors observed voltage-dependent inhibition of N-type channels by Gβγ dimers as follows: Gβ1γ2 = Gβ3γ2 > Gβ4γ2 > Gβ2γ2 with no significant inhibition by Gβ5γ2 (Arnot et al., 2000). Inhibition of the P/Q-type channels was observed to follow a different pattern (Gβ4γ2 > Gβ3γ2 > Gβ1γ2 > Gβ2γ2 > Gβ5γ2), with weak but significant voltage-dependent inhibition by Gβ5 (Arnot et al., 2000). As in other cases examined above, experimental conditions differed from those used by other authors: in this case rat N- and P/Q-type calcium channels, the rat Gβ5 subunit, and the human Gγ2 subunit were used (Arnot et al., 2000). Another potential source of discrepancy involves isoforms of Gβ5 resulting from alternate splicing: the sequences of the isoforms examined in the studies of Garcia et al. (1998) and Ruiz-Velasco and Ikeda (2000) are ∼40 amino acids longer than those examined in the studies of Zhou et al. (2000) and Arnot et al. (2000). Likewise, alternate splicing may account for differences between the results obtained by Arnot et al. (whose experiments tested channels of a single, defined channel isoform with a defined complement of ancillary calcium channel subunits, an inherent result of a method based on heterologous overexpression of channels in cells transfected with cDNA overexpression vectors) and those obtained by Garcia et al. and Ruiz-Velasco and Ikeda, whose experiments tested endogenous N-type channels of rat SCG neurons, which presumably are a considerably more heterogeneous population of calcium channels.
Collectively, these results emphasize the remarkable complexity of the inhibitory interaction between Cav2 channels and Gβγ dimers, a mode of interaction that is clearly sensitive to many minor structural features of the molecules involved. However, a unifying theme in these studies is the fact that N-type calcium channel modulation depends on the nature of the G protein βγ subunit complex and that N- and P/Q-type channels are differentially modulated by different types of G protein βγ subunits. This modulation, in turn, would allow for a mechanism by which different types of GPCRs, by virtue of preferential coupling to specific Gβγ combinations, could differentially couple to N- and/or P/Q-type channels.
B. Gβ Structural Determinants
Characterization of the structural determinants of interactions between Gβγ and its effector targets, including the N-type calcium channel, has been the goal of considerable efforts (Fig. 7). To date, such studies of Gβγ modulation of N-type channels have tested relatively narrow ranges of structural variation. No published study of the phenomenon has used comprehensive scanning mutagenesis, an approach often used in structure-activity relationship studies of small peptide toxins and hormones, for the purpose of exhaustive and detailed mapping of functional epitopes (Nadasdi et al., 1995; Kristensen et al., 1997; Froy et al., 1999; Tedford et al., 2001, 2004; Maggio and King, 2002). A major reason for this is arguably the size of the Gβ subunit, which is 10- to 50-fold larger than molecules normally studied with comprehensive scanning, making it a poor prospect in view of the resources that would be required.
In a structure-activity relationship study, Ford et al. (1998) tested a panel of mutations targeted to Gβ residues that directly contact the Gα subunit in the crystal structure of the heterotrimeric G protein complex. This was done on the assumption that Gαβγ complex formation prevents the action of at least some modulatory epitopes of Gβ by burying them beneath the solvent-accessible surface of the complex. Prepulse facilitation assays confirmed this assumption, showing that mutations of Gβ residues Lys78, Met101, Asn119, Thr143, Asp186, or Trp332 diminish modulation of the N-type channel, whereas mutations of Leu55 or Ile80 enhance modulation (Fig. 7E) (Ford et al., 1998). In other studies, chimeras of Gβ subtypes 1 and 5 were used, followed by limited scanning mutagenesis of nonconserved residues in regions shown to play an important role in effector modulation (Mirshahi et al., 2002a,b; Doering et al., 2004; Tedford et al., 2005). This limited scanning approach revealed that Gβ1 residues Tyr111, Asp153, and Ser189 contribute to inhibitory modulation. It also established that Gβ1 residues Asn35 and Asn36 contribute crucially to the above-described cross-talk between the G protein and PKC pathways of N-type channel modulation (Doering et al., 2004). Regarding the role of Asn35 and Asn36, we speculate that phosphorylation of residue Thr422 of the N-type channel by PKC disrupts allosteric or perhaps direct interactions between these Gβ1 residues and Thr422, such that the overall mode of interaction between Gβ1 and the channel becomes unfavorable to voltage-dependent inhibition (Hamid et al., 1999; Cooper et al., 2000; Doering et al., 2004).

Solvent contact surface renderings are used to present epitopes of the Gβ molecule that contribute to modulation of seven targets of Gβγ signaling. Modulatory epitopes for the respective targets are illustrated in matched pairs of images of the Gβ molecule. Images on the left include surfaces that interact with the Gα subunit during the formation of the heterotrimeric G protein complex (Wall et al., 1995; Lambright et al., 1996). Images on the right present the opposite side of the molecule, corresponding to 160 to 180° rotation of the molecule with respect to the orientation used on the left. As landmarks, solvent contact surfaces of the N′-terminal and C′-terminal residues are marked in royal blue and navy blue, respectively. Highlighting and numbering in the images indicate solvent-exposed surfaces of individual residues that contribute to modulation, as described below. Mutations at residues whose surfaces are shown in green enhance functional modulation of the respective Gβ target; mutations at residues shown in orange reduce target modulation. Unless otherwise indicated, surfaces of amino acid sequences that contribute significantly to effector modulation, but wherein individual residues make undetermined contributions, are shown in yellow. Surface renderings were generated using MOLMOL molecular imaging and analysis software (Koradi et al., 1996). A, adenylyl cyclase 1. Left image: 1, M101; 2, Y124. Right image: no significant residues identified. Modulation normally inhibits this effector (Chen et al., 1997). B, adenylyl cyclase 2. Left image: 1, D186; 2, N119; 3, L117; 4, M101; 5, W99; 6, Y124; 7, K78; 8, K89; 9, L55; 10, K57; 11, W332; 12, D246; 13, D228. Right image: no significant residues identified. Modulation normally activates this effector (Chen et al., 1997; Ford et al., 1998; Li et al., 1998). C, β-adrenergic receptor kinase. Left image: 1, D186; 2, T143; 3, L117; 4, Y59; 5, K57; 6, W332; 7, K89. Right image: no significant residues identified (Ford et al., 1998). Modulation normally activates this effector via membrane recruitment (Stoffel et al., 1997). D, Gβγ-regulated inwardly rectifying K+ channel (GIRK). Left image: 1, D228; 2, W99; 3, S98; 4, K78; 5, I80; 6, K89; 7, L55; 8, W332; 9, D246. A, residues 84 to 94; B, residues 126 to 132. Modulation stimulates this effector. Right image: 1, T128; 2, S67 (Ford et al., 1998; Albsoul-Younes et al., 2001; Mirshahi et al., 2002b). Yellow regions with red labels: A, residues 84 to 94; B, residues 126 to 132. Modulation normally activates this effector (Albsoul-Younes et al., 2001). E, N-type calcium channel. Left image: 1, D186; 2, T143; 3, N119; 4, M101; 5, K78; 6, I80; 7, L55; 8, W332. Right image: 1, Y111; 2, D153; 3, S189; 4, S67 (Ford et al., 1998; Mirshahi et al., 2002a; Doering et al., 2004; Tedford et al., 2005). Residues indicated in yellow: A, N35 to N36; these mediate disinhibitory cross-talk with the PKC pathway via interactions with residue T422 of the N-type channel. Gβγ modulation normally inhibits this effector (Doering et al., 2004). F, phospholipase C β2. Left image: 1, D186; 2, T143; 3, N119; 4, L117; 5, M101; 6, W99; 7, S98; 8, H91; 9, I80; 10, K89; 11, L55; 12, W332; 13, D246; 14, D228. A, 132 to 139; B, 46 to 50; C, 302 to 306. Right image: 1, H91. A, 132 to 139; B, 46 to 50. Modulation normally activates this effector (Ford et al., 1998; Li et al., 1998; Panchenko et al., 1998; Buck et al., 1999). G, phospholipase C β3. Left image: 1, D228; 2, L117; 3, W332. Right image: no significant residues identified. Modulation normally activates this effector (Li et al., 1998).
The picture emerges that residues that are important for N-type channel modulation are situated on both major surfaces of the G protein β subunit, i.e., the surface comprising sites of direct interaction with the Gα subunit and the opposite surface, observed when a structural model is rotated such that the Gα interaction sites are directed away from the viewer (Fig. 7). Given that multiple channel regions have been implicated in G protein inhibition/binding (i.e., the N terminus, I-II linker, and C terminus), it is possible that these regions form a Gβγ binding pocket with a number of microsites that interact with specific Gβγ residues. To confirm such a scenario, it will be necessary to systematically map the mutual interaction points between Gβ epitopes and residues of the N-type (or P/Q- or R-type) calcium channel. Such work is currently ongoing in our laboratory.
C. Gγ Subtype Dependence
There is relatively limited information concerning the Gγ subtype dependence of Gβγ-mediated N-type channel inhibition. Ruiz-Velasco and Ikeda (2000) reported that, of the 15 potential Gβγ heterodimers that may be composed using each Gβ subunit in combination with either Gγ2, Gγ3, or Gγ4, the following heterodimers, Gβ1γ2, Gβ2γ4, Gβ3γ2, Gβ4γ4, and Gβ5γ4, mediated more effective voltage-dependent inhibition of endogenous N-type calcium currents compared with any other combination of Gγ2, Gγ3, or Gγ4 with the respective Gβ subunits. However, Zhou et al. (2000) showed that, when expressed as part of heterodimers with Gγ2, all five Gβ subunits mediate more extensive inhibition of N-type calcium channels (relative to inhibition mediated by heterodimers including either Gγ1 or Gγ3). However, these results cannot easily be compared with those of Ruiz-Velasco and Ikeda because of various experimental differences, among them, the facts that different panels of Gγ subunits were tested and that calcium channels from different species were tested. More recently, Blake et al. (2001) reported that Gγ13, when coexpressed with either Gβ1, Gβ2, Gβ3, and Gβ4, results in Gβγ heterodimers that mediate significant voltage-dependent inhibition of N-type channels, but this modulation was less pronounced than that observed for heterodimers composed of Gγ2 and the respective Gβ subunit (Blake et al., 2001). Taken together, these data suggest that the interactions that occur during G protein inhibition of N-type channels do depend on Gγ subtype. However, the mechanism of this subtype dependence (i.e., the contribution of differential formation of functional Gβγ heterodimers composed of certain Gβ subunits with certain Gγ subunits versus the contribution of the participation of certain Gγ subunits in thermodynamically unfavorable interactions during Gβγ binding to the channel, versus other factors) is not fully clear.
VIII. Signaling Complexes Involving N-Type Channels and G Protein-Coupled Receptors
GPCRs often exist in complexes with other effectors, such as ionotropic receptors (Liu et al., 2000; Lee et al., 2002) and even L-type calcium channels (Davare et al., 2001). We have recently reported that Cav2.2 calcium channels form a physical signaling complex with nociceptin receptors through a direct biochemical interaction between the C-terminal regions (Beedle et al., 2004). Because the nociceptin receptor displays a low level of constitutive activity, there is constant production of free Gβγ subunits, which, because of the proximity resulting from receptor-channel complex formation, are effectively bound by the Cav2.2 channel. This results in a tonic, agonist-independent, voltage-dependent G protein inhibition of channels that are complexed with these receptors (Fig. 8A). As receptor density is increased, a greater fraction of N-type channels exist as part of receptor complexes, and thus tonic inhibition of whole-cell current is increased (Beedle et al., 2004). In the prolonged presence of agonist, nociceptin receptors are known to internalize (Spampinato et al., 2001). During the internalization process, any Cav2.2 channels that are complexed with these receptors are cointernalized, resulting in agonist-induced removal of N-type calcium channels from the plasma membrane into lysosomes and, hence, an irreversible reduction in N-type current (Altier et al., 2006) (Fig. 8B). This cointernalization constitutes a novel mechanism of GPCR-mediated down-regulation of N-type channels, a modulation that could be classified as voltage-independent as it cannot be relieved with membrane depolarizations.
A similar N-type channel internalization process seems to occur in response to GABAB receptor activation in chick DRG neurons (Tombler et al., 2006). In contrast with the nociceptin receptor-mediated effects, the effects of GABAB receptor activation occur much more rapidly (within tens of seconds) and spontaneously reverse as the receptors desensitize. Moreover, these effects do not seem to arise from a receptor-channel cointernalization, instead they seem to involve a disruption of the association of channels with cytoskeletal elements (i.e., the L1-CAM-ankyrin B complex), followed by internalization into clathrin-coated vesicles. Tyrosine kinase activity has been implicated in both the disruption and the internalization, consistent with the idea that these processes underlie voltage-independent modulation of N-type channels by GABAB receptors (Richman et al., 2005).
Receptor-mediated trafficking of N-type calcium channels is a phenomenon that has been discovered only recently, but the vast number of different types of GPCRs, together with the ability of many of these receptors to heterodimerize, suggests that this form of N-type calcium channel regulation may be more widespread. If confirmed, this would probably open many new avenues of research into the complex functional interactions between GPCRs and voltage-gated calcium channels.
IX. Concluding Remarks
In the 25 years since Dunlap and Fischbach's first description of membrane-delimited inhibition of N-type calcium channels, there have been many advances in our understanding of its underlying mechanisms and of the implications of the phenomenon for neurophysiology. The regulation of voltage-gated calcium channels by GPCRs is immensely complex and is modulated by many factors, including membrane potential, channel subtype and subunit composition, receptor type, G protein β subtype, cross-talk with other intracellular signaling pathways, interactions with synaptic proteins and cytoskeletal elements, and probably other factors as yet uncharacterized. As investigations into this remarkable field continue, it is likely that further details and additional regulatory mechanisms will be uncovered.
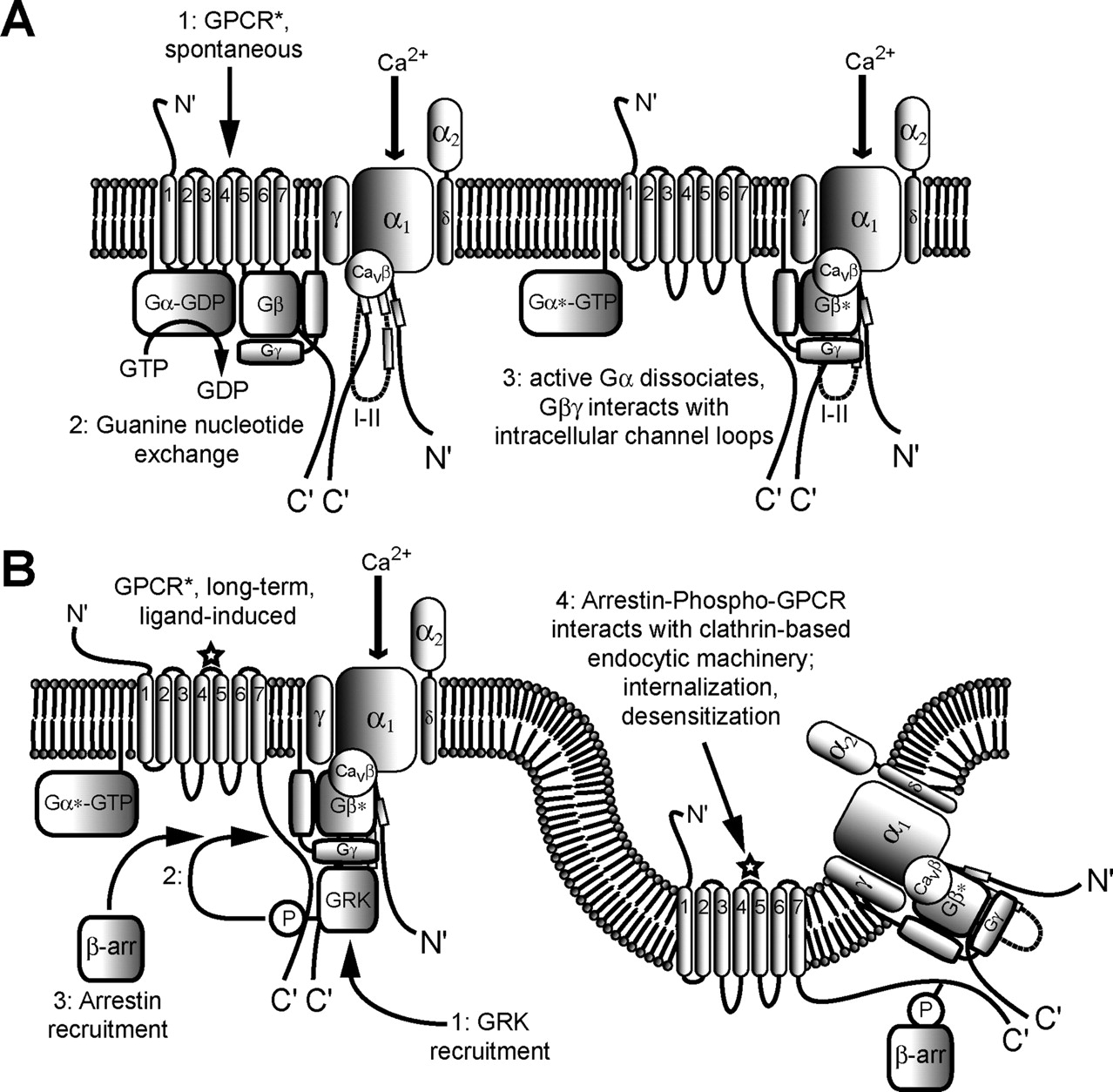
Diagrams illustrating the association of Cav2 channels and nociceptin receptors as components of signaling complexes, and their associated functional implications. A, (1) in a nociceptin receptor-Cav2 channel complex [formed by direct interactions between the C termini of the receptor and α1 subunit of the channel (Beedle et al., 2004)], constitutive receptor activity induces guanine nucleotide exchange in the associated Gαβγ-GDP complex (2), resulting in dissociation of the Gαβγ-GTP complex (3), which in turn allows for tonic Gβγ-mediated inhibition of the channel. B, long-term activation of nociceptin receptors by receptor agonists leads to high local levels of Gβγ heterodimers that recruit GRK (1), leading to phosphorylation of the C terminus of the receptor (2) and association of β-arrestin with the resulting phosphorylated site on the receptor (3). Finally, a cascade of interactions between arrestin and the clathrin-based endocytic machinery drives endocytic internalization of the channel-receptor complex (4), with two major physiological consequences: first, desensitization of the cell to further stimulation by extracellular ligand; second, voltage-independent inhibition of Cav2 currents, due to cointernalization and degradation of Cav2 channels as part of channel-receptor complexes and reduced current activity.
Although it is important to appreciate the complexity of G protein modulation, it is also important to recall that the basic outcome of G protein inhibition of presynaptic calcium channels is a reduction in the amount of calcium entering synaptic nerve terminals. The complexity of G protein modulation ultimately serves as part of an important cellular mechanism, by which calcium entry can be finely tailored to suit a particular physiological requirement, such as the correct amount of neurotransmitter to be released from a given presynaptic nerve terminal.
Acknowledgments
We sincerely thank Dr. Kathleen Dunlap for the generous contribution of the images of current and voltage traces shown in Fig. 3A, which represent two of the earliest experimental records of direct G protein modulation of Cav2 calcium channels.
Footnotes
-
↵1 Abbreviations: LVA, low-voltage-activated; HVA, high-voltage-activated; SH, Src homology; AID, α-interaction domain; GPRC, G protein-coupled receptor; AGS, activator of G protein signaling; RGS, regulator of G protein signaling; PTX, pertussis toxin; GRK, G protein-coupled receptor kinase; DRG, dorsal root ganglion; VIP, vasoactive intestinal peptide; PK, protein kinase; CSP, cysteine string protein; FRET, Foerster resonance energy transfer; SCG, superior cervical ganglion.
-
H.W.T. holds postdoctoral fellowship awards from the Heart and Stroke Foundation of Canada and the Alberta Heritage Foundation for Medical Research (AHFMR). G.W.Z. is an AHFMR Senior Scholar and a Canada Research Chair in Molecular Neurobiology.
-
Article, publication date, and citation information can be found at http://pharmrev.aspetjournals.org.
-
doi:10.1124/pr.58.4.11.
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
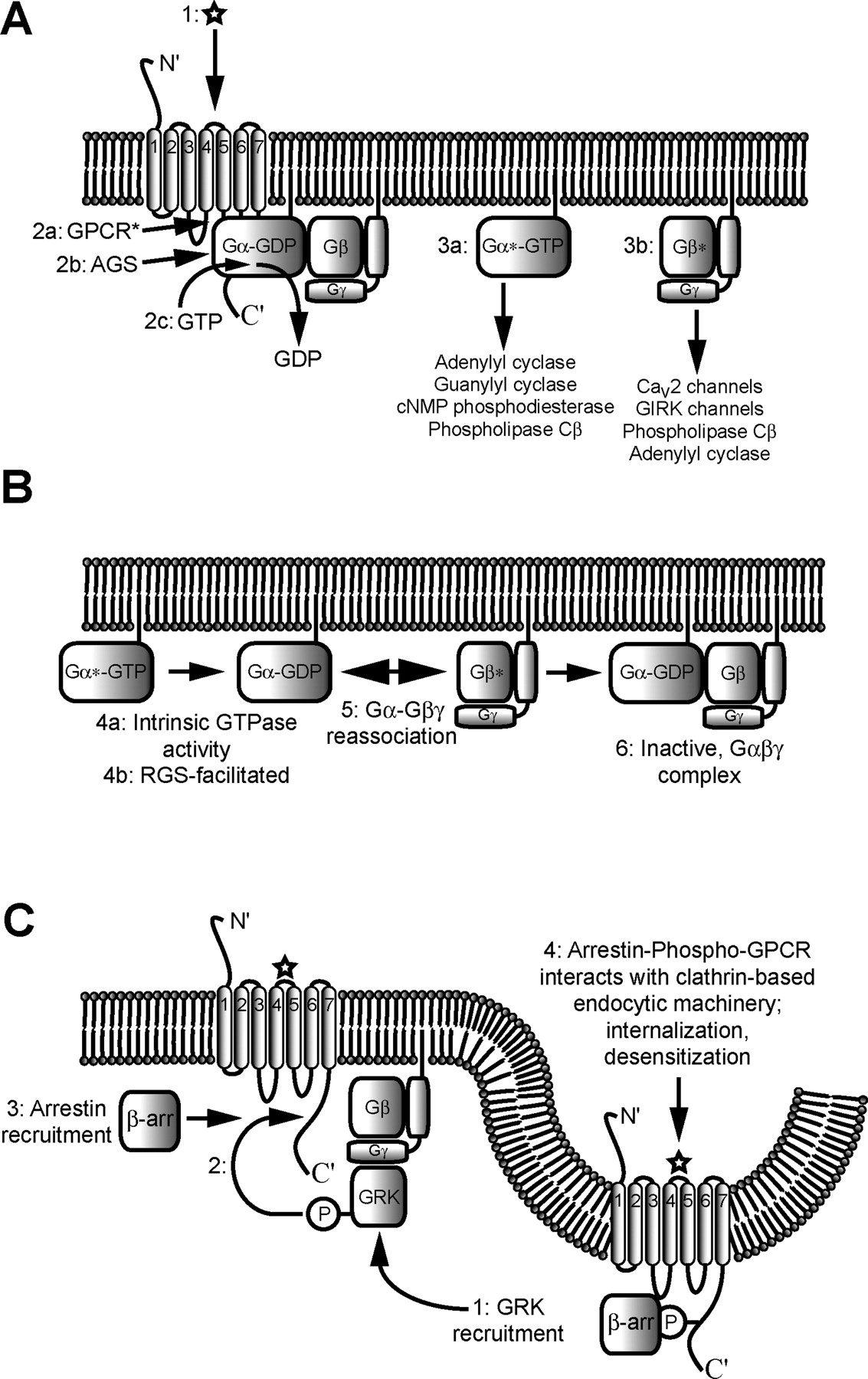{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Molecular Structure and Distributions of Voltage-Gated Calcium Channels
- III. G Protein-Coupled Receptor Signaling—A Brief Overview
- IV. Discovery and Characterization of Direct G Protein Inhibition of Cav2 Calcium Channels
- V. Calcium Channel Structural Determinants of G Protein Modulation
- VI. Modulation of G Protein Modulation
- VII. G Protein Structural Determinants of N-Type Channel Modulation
- VIII. Signaling Complexes Involving N-Type Channels and G Protein-Coupled Receptors
- IX. Concluding Remarks
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters