Article Text

Abstract
Much work has been done to identify susceptibility genes in schizophrenia and bipolar disorder. Several well established linkages have emerged in schizophrenia. Strongly supported regions are 6p24–22, 1q21–22, and 13q32–34, while other promising regions include 8p21–22, 6q16–25, 22q11–12, 5q21–q33, 10p15–p11, and 1q42. Genomic regions of interest in bipolar disorder include 6q16–q22, 12q23–q24, and regions of 9p22–p21, 10q21–q22, 14q24–q32, 13q32–q34, 22q11–q22, and chromosome 18. Recently, specific genes or loci have been implicated in both disorders and, crucially, replicated. Current evidence supports NRG1, DTNBP1, DISC1, DAOA(G72), DAO, and RGS4 as schizophrenia susceptibility loci. For bipolar disorder the strongest evidence supports DAOA(G72) and BDNF. Increasing evidence suggests an overlap in genetic susceptibility across the traditional classification systems that dichotomised psychotic disorders into schizophrenia or bipolar disorder, most notably with association findings at DAOA(G72), DISC1, and NRG1. Future identification of psychosis susceptibility genes will have a major impact on our understanding of disease pathophysiology and will lead to changes in classification and the clinical practice of psychiatry.
- bipolar disorder
- genetics
- nosology
- psychosis
- schizophrenia
Statistics from Altmetric.com
Mental disorders are now recognised as leading causes of morbidity and affect human populations around the world.1 Genetic factors are known to play an important role in influencing susceptibility to many of the most common and important disorders.2 The functional psychoses are the most severe psychiatric disorders of adult onset and for over 100 years have been divided into two main diagnostic categories: schizophrenia and bipolar disorder.
Schizophrenia is a common disorder with a lifetime morbid risk of 1% (roughly equal in males and females), and more if spectrum disorders are included.3 It is a major cause of morbidity and consumes a great deal of long term medical and social care. It is characterised by psychotic features (delusions and hallucinations), disorganisation, dysfunction in normal affective responses, and altered cognitive functioning. Bipolar disorder is a severe mood disorder with a lifetime prevalence of 1% in both males and females.4 It is characterised by disturbances in mood ranging from extreme elation (mania) to severe depression often accompanied by psychotic features and cognitive changes. It is associated with high morbidity, service utilisation, and suicide risk. There is substantial evidence from family, twin, and adoption studies for the importance of genes in influencing susceptibility to schizophrenia and bipolar disorder and replicated findings for each disorder are now accruing from molecular genetic studies. Within this review we will briefly summarise the evidence that genes are involved in each disorder and then review the findings from molecular genetic linkage and association studies. We will end by considering the overlap in genetic findings in bipolar disorder and schizophrenia.
SCHIZOPHRENIA
Genetic epidemiology
Schizophrenia has been subjected to detailed genetic epidemiological investigation. The results of numerous family, twin, and adoption studies show conclusively that risk of illness is increased among the relatives of affected individuals and that this is the result largely of genes rather than shared environment.5,6 In the children and siblings of individuals with schizophrenia, the increase in risk is around 10-fold, and somewhat less than this in parents. The latter finding is probably explained by a reduction in the reproductive opportunities, drive, and possibly fertility of affected individuals. Five recent systematically ascertained studies using modern diagnostic criteria report monozygotic (MZ) concordances estimated at 41–65% compared with dizygotic (DZ) concordances of 0–28%, resulting in an estimated broad heritability of 85%.7
While the twin and adoption literature leave little doubt that genes are important, they also point to the importance of environmental factors, since the concordance for schizophrenia in MZ twins is typically around 50% and heritability estimates are less than 100%. Moreover, we should also note that liability resulting from gene-environment interaction tends to be attributed to heritability in most genetic epidemiological studies.
It is clear from genetic epidemiology that the mode of transmission is complex.8,9 The number of susceptibility loci, the disease risk conferred by each locus, the extent of genetic heterogeneity, and the degree of interaction among loci all remain unknown. Risch10 has calculated that the data are incompatible with the existence of any single locus conferring a relative risk in siblings (λS) of more than 3 and, unless extreme epistasis exists, models with at least two or three loci of λS⩽2 are more plausible. It should be emphasised that these calculations are based upon the assumption of homogeneity and refer to population-wide λS. It is quite possible that alleles of larger effect are operating in some groups of patients, for example families with a high density of illness. However, high density families are expected to occur by chance even under polygenic inheritance and their existence does not prove the existence of disease alleles of large effect.9
Linkage findings in schizophrenia
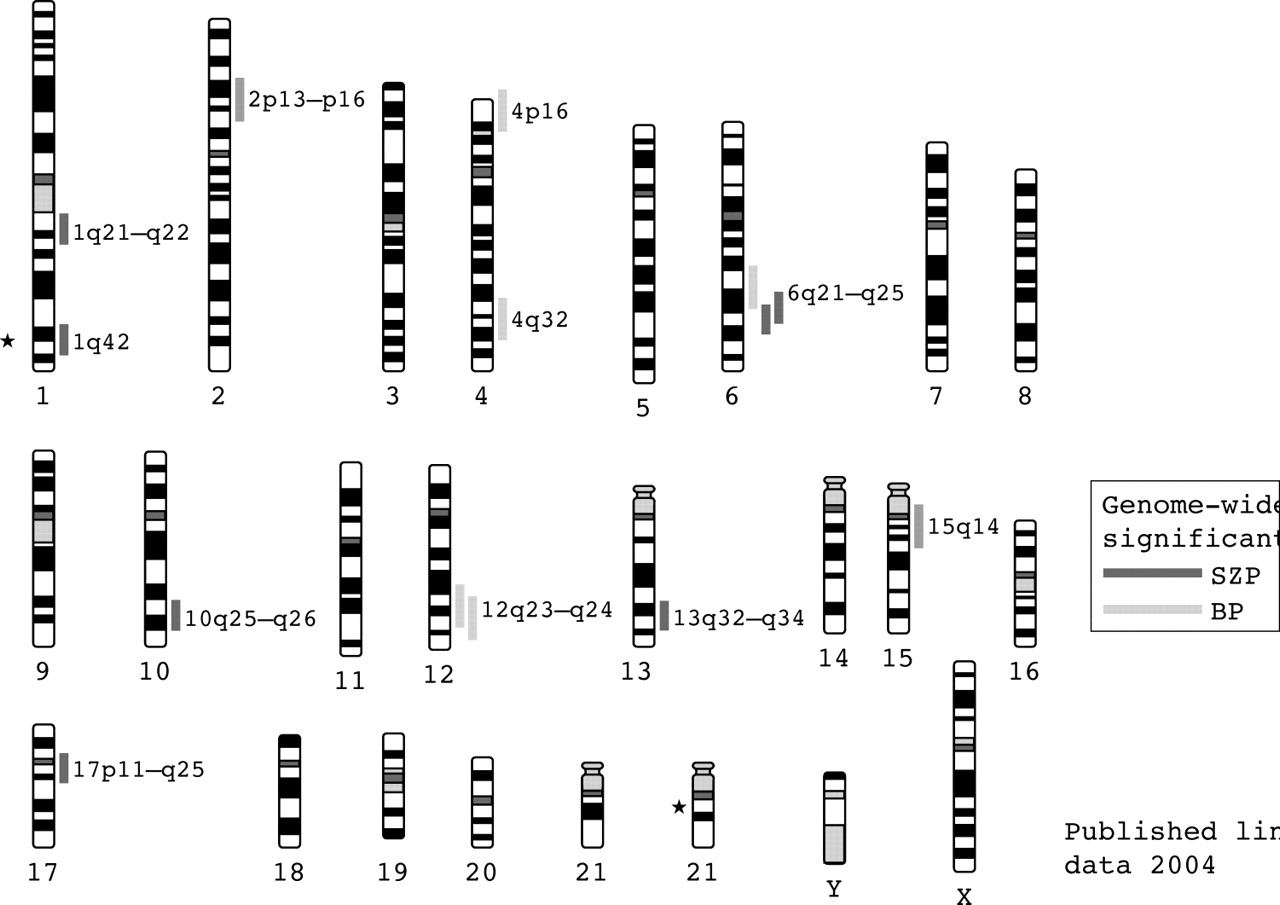
Several studies of schizophrenia pedigrees have found evidence for linkage at genome-wide significant level11 (see fig 1). Nevertheless, until recently, the results had, perhaps, seemed disappointing, with most positive studies falling short of genome-wide significance, and failures to replicate even the most interesting loci being the rule rather than the exception. This is probably attributable to a combination of small genetic effects and inadequate sample sizes.12 However, as the data from more than 20 genome-wide studies have accumulated and sample sizes increased, some consistent patterns have emerged.
{kind=link}
Chromosome ideograms showing locations of genome-wide significant linkages in schizophrenia and bipolar disorder. Asterisks mark the locations of chromosomal abnormalities associated with schizophrena.
Recently, two meta-analyses of schizophrenia linkage have been reported. Each used different methods and, unsurprisingly, obtained overlapping but somewhat different results. The study of Badner and Gershon13 supported the existence of susceptibility genes on chromosomes 8p, 13q, and 22q, while that of Lewis et al14 most strongly favoured 2q, but also found that the number of loci meeting the aggregate criteria for significance was much greater than the number of loci expected by chance (p<0.001). Support was also obtained for regions on chromosomes 5q, 3p, 11q, 6p, 1q, 22q, 8p, 20q, and 14p. Thus the 8p and 22q regions were supported by both meta-analyses but eight other regions were supported by only one. Furthermore, the region most strongly supported by the Lewis et al analysis, that on chromosome 2q, is not one that has received strong support previously, but it is now clearly worthy of further investigation.
Previously, the economics and practicalities of hunting a susceptibility gene within a linked chromosomal region dictated that one should have virtually definitive evidence that the linkage was a true positive. With recent improvements in our knowledge of genome anatomy and in genome analysis technology, the task of positional cloning has been transformed, and, in our view, favours bolder endeavours predicated on less stringent evidence. How much evidence will be dictated largely by research funding bodies, but it seems not unreasonable to the authors to pursue a locus based upon a true type I error rate of 0.05 even in the absence of replication, with the proviso that the error rate has been rigorously assessed by simulation and allows for examination of multiple models. For many of the above regions, such studies are underway. While some of the above loci will inevitably be false positives, many will not and it is likely that several genes cloned by positional approaches will be claimed in the next few years. In the next section, we discuss those genes that have been claimed so far.
Candidate gene findings in schizophrenia
Functional candidate genes
Most of the recent findings of interest in schizophrenia have emerged from positional cloning studies, but there is also a large schizophrenia functional candidate gene literature which we will only touch on in this review. In the main, the findings have either not been replicated or have seemed so unconvincing that no attempts to do so have been made. However, meta-analyses suggest that the dopamine receptors DRD3,15DRD2,16 and the serotonergic receptor HTR2A17 might confer risk, albeit with extremely small (<1.2) effect sizes. We should, however, remember that if the putative associations are the result of linkage disequilibrium between the assayed markers and the true susceptibility variant, it is possible that the latter might contribute somewhat greater effects, depending upon the degree of linkage disequilibrium (LD) between the two. Among the recent claims, reports of association between GRM3 (the gene encoding metabotropic glutamate receptor type 3) and schizophrenia are among the more interesting,18–20 although in our view, the genetic data remain weak. Further study of GRM3 in particular and the glutamatergic system in general is warranted.
Positional candidate genes
Several detailed mapping studies of linked regions have implicated specific genes. The quality of the data has been variable and a number of putative susceptibility genes have yet to be clearly replicated. Here we focus on the genes where, at the time of writing, there are published follow up studies, or where we are aware of unpublished data that allow us to make a judgement about whether the gene is likely to be a true positive.
Dysbindin (DTNBP1)
Evidence implicating dystrobrevin binding protein 1 (DTNBP1), also known as dysbindin, in schizophrenia was first reported by Straub et al,21 who undertook association mapping across the linkage region on chromosome 6p22.3. Support for association rapidly followed from a largely German sample22 and in two large samples, one case-control from the UK and Ireland23 and the other a parent-proband from Bulgaria24 studied by our own group. Although there have also been some studies in which no association was found,25 significant associations have now been published in 10 samples26,27 including an initially negative Irish sample,28 when one additional marker that defined the risk haplotype in our own UK sample was genotyped.29 Thus, the evidence in favour of DTNBP1 as a susceptibility gene for schizophrenia is strong. However, there are inconsistencies between the specific risk alleles and haplotypes between studies suggesting the presence of multiple susceptibility and protective alleles,23 or that a single susceptibility allele is carried on a remarkable diversity of haplotypes even in closely related populations. As yet, no causative variant has been identified, but the absence of associated non-synonymous alleles23 suggests that disease susceptibility may depend upon variation affecting mRNA expression. This is indirectly supported by evidence for as yet unknown cis-acting polymorphisms affecting DTNBP1 expression in human brain,29 and more directly by two recent studies showing reduced levels of expression of the mRNA30 and protein31 in postmortem brain samples from patients with schizophrenia.
Although its functions are largely unknown but probably many, there is evidence that variation in DTNBP1 might influence risk of schizophrenia through effects on pre-synaptic glutamate function.26,31
Neuregulin 1 (NRG1)
NRG1 was first implicated in schizophrenia in the Icelandic population after LD mapping across 8p21–22 revealed association between schizophrenia and a multi-marker haplotype at the 5′ end of NRG1.32 Strong evidence for association with the same haplotype was subsequently found in a large sample from Scotland,33 with considerably weaker support in our own UK sample.34 Further positive findings have emerged from Irish,35 Chinese,36–39 Bulgarian (Kirov et al, unpublished data), and South African40 samples. However, some negative findings have also been reported.40–43 Only the two deCODE studies32,33 and our own UK study have implicated the specific Icelandic haplotype, perhaps reflecting differences in the LD structure across NRG1 in European and Asian samples.38
NRG1 encodes many mRNA species and proteins. Despite detailed resequencing,32 specific susceptibility variants have not been identified, but the Icelandic haplotype points to the 5′ end of the gene, once again suggesting that altered expression or perhaps mRNA splicing might be involved. It is even formally possible at this stage that NRG1 is not the susceptibility gene, as intron 1 contains another expressed sequence35 whose function is unknown. However, in so far as it is possible to model schizophrenia in animals, behavioural analyses of NRG1 hypomorphic mice support the view that the association is related to altered NRG1 function or expression,44 a hypothesis supported by the observation of alteration in the ratios of NRG1 mRNA species in schizophrenic brain.45 Just as for DTNBP1, the mechanisms by which altered NRG1 function might lead to schizophrenia are unclear. Mediation of glutamatergic mechanisms may be involved32; however, NRG1 is thought to encode around 15 proteins with a diverse range of functions in the brain, including cell-cell signalling, Erbb receptor interactions, axon guidance, synaptogenesis, glial differentiation, myelination, and neurotransmission.46 Any of these could potentially influence susceptibility to schizophrenia.
d-Amino-acid oxidase (DAO) and d-amino-acid oxidase activator (DAOA)
Chumakov and colleagues47 undertook association mapping in the linkage region on chromosome 13q22–34. They found associations in French Canadian and Russian populations in markers around two novel genes, G72 and G30, which are overlapping but transcribed in opposite directions. G72 is a primate-specific gene expressed in the caudate and amygdala. Using yeast two-hybrid analysis, evidence for physical interaction was found between G72 and d-amino-acid oxidase (DAO). DAO is expressed in human brain where it oxidises d-serine, a potent activator of NMDA glutamate receptor. Co-incubation of G72 and DAO in vitro revealed a functional interaction with G72 enhancing the activity of DAO. Consequently, G72 has now been named d-amino-acid oxidase activator (DAOA). In the same study, DAO polymorphisms were shown to be associated with schizophrenia in one of the samples, and analysis of DAOA and DAO variants revealed modest evidence for a statistical interaction between the loci and disease risk. Given the three levels of interaction, the authors concluded that both genes influence risk of schizophrenia through a similar pathway, and that this effect is likely to be mediated through altered NMDA receptor function.
Associations between DAOA and schizophrenia have subsequently been reported in samples from Germany,48 China,49 Ashkenazi Jews,50 and both US and South Africa,40 as well as a small sample of very early onset psychosis subjects from the US.51 As before, and conceivably for similar reasons, there is no consensus concerning the specific risk alleles or haplotypes across studies. The German group also reported association between DAO and schizophrenia, although with no evidence for a statistical interaction between the loci and risk of schizophrenia. At present, the published genetic evidence in support of this gene is weaker than for DTNBP1 and NRG1. In our own large UK case-control sample, detailed direct and indirect association analysis (unpublished) provides support for association between schizophrenia and DAO but not DAOA. It is possible that this reflects a weak effect size at DAOA as we do find evidence for an interactive effect on risk of disease between the same markers at each locus as did Chumakov and colleagues.47
Regulator of G-protein signalling 4 (RGS4)
RGS4 maps to the putative linkage region on chromosome 1q22, but it was targeted for genetic analysis52 following a microarray based gene expression study in which decreased RGS4 expression was found in schizophrenic postmortem brain. Independent evidence for association between schizophrenia and a haplotype at the 5′ end of the gene was found in two samples from the USA, and while not providing significant evidence alone, inclusion of a sample from India added to the overall level of support. Positive findings have subsequently been reported by several other groups,53–55 but the level of support in each has been modest and the pattern of association differs between samples. RGS4 is a negative regulator of G protein coupled receptors. The relationship between RGS molecules and receptor function is a promiscuous one, but of possible interest to schizophrenia. There is evidence that RGS4 modulates activity at certain serotonergic56 and metabotropic glutamatergic receptors,57 while its own expression is modulated by dopaminergic transmission.58 Moreover, RGS4 interacts with ErbB3,59 which may be of relevance as ErbB3 is a neuregulin 1 receptor whose expression is down regulated in schizophrenic brains.46
Others
Finally, association analysis in single studies of CAPON (C-terminal PDZ domain ligand of neuronal nitric oxide synthase),60PPP3CC (protein phosphatase 3, catalytic subunit),61 and TRAR4 (trace amine receptor 4),62 which map, respectively, to the 1q22, 8p21.3, and 6q23.2 linkage regions, has suggested these as possible susceptibility genes. As discussed in the original articles, each of these genes can be plausibly related to the pathogenesis of schizophrenia, but at the time of writing we are not aware of any replication data to support these hypotheses.
Chromosomal abnormalities
There have been numerous reports of associations between schizophrenia and chromosomal abnormalities63 but with two exceptions none provides convincing evidence for the location of a susceptibility gene.
22q11
Several studies have shown that adults with 22q11 deletions have a high risk for schizophrenia,64–66 with the largest study of adult patients to date (n = 50) estimating this at 24%.66 The deletion cannot account for a high proportion of schizophrenic cases,67 but reports of linkage to 22q11 suggest that variants in genes mapping to this region might contribute to more typical cases. Current candidates include catechol-O-methyltransferase (COMT), proline dehydrogenase (PRODH), and zinc finger- and DHHC domain-containing protein 8 (ZDHHC8).
COMT has been intensively studied because of its key role in dopamine catabolism. Most studies have focussed upon a valine to methionine change at codon 158 of the brain predominant membrane-bound form of COMT (MB-COMT) and codon 108 of the soluble form (S-COMT). The valine allele confers higher activity and thermal stability to both forms of COMT68 and has been fairly consistently associated with reduced performance in tests of frontal lobe function.69,70 The results in schizophrenia have been mixed, with a recent meta-analysis71 reporting no overall evidence for association with the valine allele. Since the preparation of the COMT meta-analysis, as in an earlier study,72 four studies have found stronger evidence for association between haplotypes at COMT than for the Val/Met locus alone.68,73–75 However, we have been unable to find evidence for association between the single nucleotide polymorphisms (SNPs) or haplotypes that were positive in three of the studies,68,73,75 including the Val/Met polymorphism, in a study of more than 2800 individuals, including almost 1200 with schizophrenia. Moreover, it is difficult to reconcile the stronger evidence for association with haplotypes at COMT with the observation that COMT activity, at least in peripheral tissues,76 is largely dictated by the Val/Met locus, a finding that may also be true in brain.68 Thus, while the picture is confused, we consider that the evidence does not support a simple role for Val/Met 158 in susceptibility to schizophrenia, although a small effect on susceptibility cannot be excluded, nor can a role in phenotype modification. However, it remains a possibility that variation elsewhere in COMT, or in a neighbouring gene such as ARVCF,74,75 confers susceptibility.
PRODH is also located at 22q11 and is another functional candidate gene given that a mouse loss of function mutant exhibits behavioural abnormalities in sensorimotor gating that are analogous to those observed in schizophrenic subjects,77 and because proline dehydrogenase influences the availability of glutamate. In addition, a heterozygous deletion of the entire PRODH gene was found in a family including two schizophrenic subjects, and two heterozygous PRODH missense variants were detected in three of 63 schizophrenic patients studied by Jacquet et al.78 Evidence in favour of association between PRODH and schizophrenia has been reported by Liu and colleagues79 and by Li and colleagues.80 We have been unable to replicate either of these findings in large case-control and family based association samples81 and unpublished data. Moreover, we and others78,81 have also found that the missense mutations reported by Liu and colleagues79 are no more common in schizophrenic cases than controls, while in a very large sample of Japanese subjects, PRODH deletions were not associated with schizophrenia.82 Thus the case for PRODH as a susceptibility gene for schizophrenia does not seem strong to us. However, intriguingly, the study of Jacquet and colleagues83 while failing to find association with schizophrenia (or bipolar disorder), did report association between hyperprolinaemina and schizoaffective disorder. This may suggest that PRODH may have some role in phenotype modification although this conclusion is highly tentative as the results of this study are based on a small sample and are not significant after correction for multiple testing.
Finally, there is weak genetic evidence that an SNP in ZDHHC8, a gene which encodes a putative transmembrane palmitoyl transferase, directly confers susceptibility to schizophrenia but only in females.84 The findings were particularly interesting in that study because the weak genetic evidence was bolstered by evidence that the associated SNP was functional and that sexually dimorphic effects could be seen in ZDHHC8 knockout mice, with only female mice exhibiting the phenotypes used to model schizophrenia. However, while providing supportive evidence for association, Chen and colleagues85 found that the associated allele was the opposite to that of Mukai and colleagues, thus undermining the functional data, while also showing the effect apparently in a gender non-selective fashion, thus undermining the mouse model analogy. The only other study we are aware of is unpublished data from ourselves and others based upon almost 2000 schizophrenic subjects that do not support the finding. The findings are at present inconclusive, but given the large sample sizes studied by ourselves and the German group, together with the failure of the Chinese group to replicate the original findings in two key respects, we consider the balance of evidence for this gene still favours the null hypothesis. With somewhat more certainty, the two replication studies effectively exclude a direct role for the SNP notwithstanding the circumstantial support that it may affect the splicing of an mRNA expressed in brain regions relevant to schizophrenia.84
1q42
The other major finding based upon a chromosomal abnormality comes from an extended pedigree in which a balanced chromosomal translocation (1;11)(q42;q14.3) showed strong evidence for linkage to a fairly broad phenotype comprising schizophrenia, bipolar disorder, and recurrent depression.86 The translocation was found to disrupt two genes on chromosome 1: DISC1 and DISC2.86,87DISC2 contains no open reading frame and may regulate DISC1 expression via anti-sense RNA.87 Interestingly DISC1 and 2 are located close to the chromosome 1 markers implicated in two Finnish linkage studies.88,89 There is evidence from basic science that the function of DISC1 might be relevant to schizophrenia. For example, the truncated product in the translocation family might contribute to schizophrenia by affecting neuronal functions dependent upon intact cytoskeletal regulation such as neuronal migration, neurite architecture, and intracellular transport.90,91 While interesting hypotheses, it is important to remember that translocations exert effects on genes other than those directly disrupted. For example, there are several mechanisms by which a translocation can influence the expression of neighbouring genes. Thus, in order to unequivocally implicate DISC1 and/or 2 in the pathogenesis of schizophrenia, it is necessary to identify mutations or polymorphisms that are associated with schizophrenia in another population and are not in linkage disequilibrium with neighbouring genes. Four published studies have attempted to do this. Negative studies were reported by the Edinburgh group who originally identified DISC1 and 2,92 and by a group who focussed on the 5′ end of the gene in a large Japanese sample.93 However, positive findings have been reported in a large Finnish sample94 and in US samples with schizophrenia, schizoaffective disorder, and bipolar disorder.95
BIPOLAR DISORDER
Genetic epidemiology
Family, twin, and adoption studies conducted over the last 20–30 years have provided an impressive and consistent body of evidence supporting the existence of genes determining predisposition to bipolar disorder and show a gradation of risk of mood disorder as the genetic relatedness to a proband diminishes (reviewed in detail in Craddock and Jones96). There are methodological impediments to precise quantification, but the approximate lifetime risk of narrowly defined bipolar disorder in relatives of a bipolar proband are: unrelated member of the general population: 0.5–1.5%; first degree relative 5–10% (corresponding to λS∼8); monozygotic co-twin 40–70% (corresponding to λMZ∼60).97 Further, relatives of bipolar probands are also at a more modest increased risk of unipolar depression (that is, depressive episodes without mania) of the order of a 2–3-fold increase of risk for a first degree relative compared with the general population.98 Estimates of heritability are high: 89% in a recent hospital register study of 67 twin pairs in the UK99 and 93% in a population register study of 19 124 same-sex twin pairs in Finland.100
Occasional families may exist in which a single gene plays the major role in determining susceptibility, but most bipolar disorder involves the interaction of multiple genes (epistasis) or more complex genetic mechanisms (such as dynamic mutation or imprinting).96 Analyses using mathematical modelling suggest bipolar susceptibility genes are likely to have modest effect sizes (gene-specific sibling recurrence risk: λS⩽2)101 and this is consistent with findings emerging from molecular genetic studies. Bipolar disorder has not yet received the same degree of intensive study as schizophrenia, but a number of chromosomal regions have been repeatedly implicated in linkage studies and replicable findings have now been reported at some genetic loci.
Linkage findings in bipolar disorder
Systematic genome screens have been, and are being, conducted on a variety of sample sets, ranging from large densely affected pedigrees in genetic isolates to large numbers of affected sib pairs. The pattern of findings emerging is consistent with there being no gene of major effect to explain the majority of cases of bipolar disorder but several regions have been implicated repeatedly.
Two meta-analyses have been conducted with bipolar genome scan data. Badner and Gershon13 found the strongest evidence for susceptibility loci on 13q and 22q when examining seven published genome scans for bipolar disorder. However, the more extensive and detailed meta-analysis of Segurado et al102 conducted using the same bin ranking methodology as used in the meta-analysis of schizophrenia scans14 did not find genome-wide significant evidence for linkage but provided a more modest level of support for regions on chromosomes 9p22.3–21.1, 10q11.21–22.1, 14q24.1–32.12, and regions of chromosome 18. This meta-analysis demonstrated lesser consistency in the findings from bipolar scans than from schizophrenia scans.14 One likely contributor to this difference is the substantially smaller number of pedigrees that were available for inclusion in the bipolar meta-analysis compared with that for schizophrenia: 370 compared with 1208 for narrowly defined illness, respectively. Another consistent difference is that it has been usual to test a broader range of (non-independent) diagnostic models in bipolar scans which require correction for multiple testing, thus reducing power for a given sample size. A further consistent difference is that a greater proportion of larger, extended pedigrees has been investigated for bipolar disorder than for schizophrenia. This may reflect a difference in the genetic architecture of the disorders. If this is the case and if there was consequently a greater heterogeneity between studies in bipolar disorder this would make detection of consistent signals in meta-analyses more difficult, a particular problem using the bin ranking method.
Four bipolar genome scans were not included in the meta-analysis of Segurado et al102 because they included less than 20 genotyped affected individuals (and, hence, failed to meet the predefined inclusion criteria used for study selection). These studies used extended pedigrees and two reported genome-wide significant linkage signals. They are shown in table 1.
Genome scans excluded from meta-analysis because of the small number of genotyped affected individuals (<20)
Since publication of the meta-analyses several further genome-wide scans in independent samples have been published, with several regions identified that meet genome-wide significant or suggestive evidence for linkage criteria (table 2). Of particular note is the 6q16–q25 region which was not implicated in either meta-analysis but which is supported by one genome-wide significant and three genome-wide suggestive signals, making it one of the best supported regions for bipolar disorder.
Bipolar genome scans reported subsequent to the meta-analyses
In fig 1 we show the regions that have received genome-wide significant support in at least one scan. Of particular note are the 6q16–q25 region mentioned above and the 12q23–q24 region which has two genome scans reporting genome-wide significance103,104 and is also supported by linkage analysis in two pedigrees that segregate both bipolar spectrum mood disorder and Darier’s disease (an autosomal dominant skin disease caused by mutations at ATP2A2 which maps at 12q23–q24.1) showing maximum LOD>4 at markers in this region.105–107
Gene studies of bipolar disorder
In contrast to schizophrenia, to date there has not been unambiguous demonstration of a susceptibility gene identified for bipolar disorder by positional cloning. Potentially interesting findings have come from study of functional candidates and most recently investigation of genes first implicated in schizophrenia (some of which map in linkage regions of interest in bipolar disorder). However, none of the findings yet achieve the level of support that dysbindin and NRG1 have received in schizophrenia.
Functional candidates
Functional candidate gene studies depend crucially on the choice of candidates. Inevitably this depends on the current level of understanding of disease pathophysiology. For bipolar disorder, most such studies have focussed on neurotransmitter systems influenced by medications employed in the management of the disorder, namely the dopamine, serotonin, and noradrenaline systems (reviewed in Craddock et al108). Amongst the “traditional” candidates, the genes encoding monoamine oxidase A (MAOA), tyrosine hydroxylase, catechol-O-methyltransferase (COMT), and the serotonin transporter (5HTT) have received particularly extensive attention. While the usual pattern for each of these genes has been for some positive studies along with perhaps an even greater number of negative replications, meta-analyses of polymorphisms of known functional relevance in three of the genes are significant at the p<0.05 level: MAOA,109COMT,110 and 5HTT,111,112 all with modest effect sizes (odds ratios (OR)«2). It is of interest that COMT has been also been implicated in schizophrenia73 and has received support from the same group in a modestly sized study of bipolar disorder.113 These findings remain to be tested in the independent, large samples that will be required to determine unambiguously whether and to what extent variation within these genes contributes to susceptibility to bipolar disorder.
Many of the candidate gene reports in the literature describe studies in modestly sized samples (a few hundred individuals). As for all complex disorders, the trend in candidate gene studies of bipolar disorder is for the use of larger samples with increased power to detect modest or small effect sizes and examination of candidate genes predicated on more sophisticated models of pathogenesis or directed by positional information from linkage studies. Replicable positive findings have started to emerge from these approaches over the last 2–3 years.
d-amino acid oxidase activator DAOA(G72)/G30 locus
At least five independent datasets contribute evidence that variation at the DAOA/G30 locus on chromosome 13q influences susceptibility to bipolar disorder. As described in the schizophrenia sections above, this locus was implicated initially as being involved in susceptibility to schizophrenia. Subsequently linkage disequilibrium at this locus was reported also with bipolar disorder in two US family samples114 and this was replicated in a further US family sample,115 a German case-control sample,48 and our own large UK case-control sample.116 In all studies evidence for LD came from both individual SNPs as well as multilocus haplotypes, although there is variation between studies in the SNPs and haplotypes showing LD. No pathologically relevant variant has yet been identified and the biological mechanism remains to be elucidated (see schizophrenia section above). DAO has been examined in only one study of bipolar disorder48 which found no evidence of LD. However, DAO lies in the 12q23 region of linkage interest (see fig 1) and warrants more thorough study in bipolar disorder.
Brain derived neurotrophic factor (BDNF)
A functional candidate gene that has attracted a great deal of recent interest is brain derived neurotrophic factor (BDNF).117 BDNF is a member of the neurotrophin superfamily. Neurotrophins are synthesised in neurons as proforms that can be cleaved intra- or extracellularly and both their synthesis and secretion depends upon neuronal activity. BDNF plays an important role in promoting and modifying growth, development, and survival of neuronal populations and, in the mature nervous system, is involved in activity dependent neuronal plasticity,118 processes that are prominent in the synaptic plasticity hypothesis of mood disorder which focuses on the functional and structural changes induced by stress and antidepressants at the synaptic level. The BDNF gene lies on the reverse strand of chromosome 11p13 and encodes a precursor peptide (proBDNF), which is cleaved proteolytically to form the mature protein.119 The 11p13 chromosomal location of BDNF has been implicated in some linkage studies of bipolar disorder but not in meta-analyses of linkage studies.
Consistent with the strong evolutionary conservation of the BDNF coding sequence across species, only one frequent, non-conservative polymorphism in the human BDNF gene has been identified, a single nucleotide polymorphism (SNP) at nucleotide 196 within the 5′proBDNF sequence that causes an amino acid substitution of valine to methionine at codon 66 (Val66Met). There is cross-species conservation of the precursor portion of proBDNF which is consistent with potential functional importance and it is possible that the common Val66Met polymorphism could itself have a functionally relevant effect by modifying the processing and trafficking of BDNF.120
There have been three positive reports using family based association studies of Caucasian bipolar disorder samples of European-American origin and the Val66Met SNP: two are in adult bipolar samples121,122 and one is with a small childhood onset sample.123 All have shown over-transmission of the common Val allele. Evidence with multilocus haplotypes was stronger in one study.122 There have been four case-control association studies (of European,124,125 Chinese,126 and Japanese origin127) to date, in which there is no evidence for an allelic or genotypic association. In our own UK case-control study of close to 1000 bipolar cases we found no significant evidence for association of the Val allele (or multilocus haplotypes) with bipolar disorder but some evidence for association within phenotypic subsets of the data (unpublished data).
Substantial additional genetic and biological work will be required to confirm (or refute) the role of BDNF in influencing susceptibility to bipolar disorder. Systematic study of variation across the whole gene is required with study in further independent samples.
Other genes
We will briefly mention three other genes that have recently been reported as “bipolar genes”. Two of these are in the 22q chromosome region of interest: G-protein receptor kinase 3 (GRK3) was implicated through positional follow up of a linkage signal in a set of US pedigrees and was supported also by expression data in a rodent model of mania.128 However, this has not yet received independent support. XBP1, a pivotal gene in the endoplasmic reticulum (ER) stress response, was reported to show association at a promoter polymorphism with bipolar disorder susceptibility in two small association samples.129 Some degree of circumstantial biological support for a functional role for this polymorphism came from a cellular model of the action of mood stabiliser medications. However, this report is highly likely to be a type I error because the putative functionally relevant variant was found to have no influence on susceptibility in independent family based and case-control association samples six times larger than those in the initial report.130 More promisingly, but as yet not widely tested, is the report that P2X7, in the 12q24 region of linkage interest, influences susceptibility to both bipolar disorder and unipolar depression.131
Closer attention to the phenotype: clinical covariates and sub-types
As discussed elsewhere96 bipolar disorder researchers have been taking an interest in a variety of clinical sub-types and covariates over recent years as a way of testing subsets of cases with increased clinical (and hopefully genetic) homogeneity. Examples include rapid cycling (illness characterised by a high recurrence rate—at least four distinct episodes per year),132 lithium responsiveness,133 bipolar affective puerperal psychosis (triggering of bipolar episodes in females by parturition),134,135 early age at onset,136 and occurrence of psychotic features during illness.137–139
Consideration of the occurrence of psychotic features in bipolar disorder brings us to consider the interesting and biologically important issue of the overlap in genetic findings in bipolar and schizophrenia.
THE OVERLAP IN FINDINGS BETWEEN BIPOLAR DISORDER AND SCHIZOPHRENIA
It will be clear to the reader that some of the same regions and genes have figured prominently in the sections on both schizophrenia and bipolar disorder above. Traditionally, psychiatric research in general, and the search for predisposing genes in particular, has proceeded under the assumption that schizophrenia and bipolar disorder are separate disease entities with separate underlying etiologies (and treatments), the so-called “Kraepelinian dichotomy”. This distinction has pervaded Western psychiatry since Emil Kraepelin’s influential nosological writings140 and survives in current operational classification systems such as DSM-IV141 and ICD10,142 although some workers, such as Crow, have argued for a continuum approach to psychosis.143 The clinical reality is that many individuals with severe psychiatric illness have features that fall between these two “extremes” and have both prominent mood and psychotic features (often classified as “schizoaffective disorder” or some similar atypical diagnosis), raising the possibility, perhaps likelihood, that there is not a neat biological distinction between schizophrenia and bipolar disorder. This possibility finds support in several observations from genetic research, including those discussed below.
Family studies
Although family studies have demonstrated that schizophrenia and bipolar disorder tend to “breed true”,144–146 families are known in which multiple cases of schizophrenia, bipolar disorder, and cases with both psychosis and mood disorder occur.147 Further, some studies have also shown statistically significant evidence that bipolar disorder occurs at increased rate in the relatives of probands with schizophrenia148 and that bipolar disorder occurs at increased frequency in the relatives of bipolar probands.149 Moreover, schizoaffective disorder has been shown to occur at increased rate in the families of probands with schizophrenia150 and bipolar disorder151 and both schizophrenia and bipolar disorder have been shown to occur at increased rate in the families of probands with schizoaffective disorder.151 Together, these family data argue for a more complex relationship between the psychoses than is reflected in the conventional dichotomous view.
Twin studies
Only one twin study has used an analysis that was unconstrained by the diagnostic hierarchy inherent in current classification systems (that is, the principle that schizophrenia “trumps” mood disorder in diagnosis). This study demonstrated a clear overlap in the genetic susceptibility to syndromally defined mania and schizophrenia.152 The findings suggested the existence of some susceptibility genes that are specific to schizophrenia, others that are specific to bipolar disorder, and yet others that influence susceptibility to schizoaffective disorder, schizophrenia, and bipolar disorder. A particularly graphic illustration of the varied expression of the same set of susceptibility genes is provided by the Maudsley triplets—a set of genetically identical triplets, two of whom had a lifetime diagnosis of schizophrenia and the third a lifetime diagnosis of bipolar disorder.153
Linkage studies
As indicated above, genetic linkage studies have identified some chromosome regions that show convergent or overlapping regions of interest in both disorders, including regions of 13q, 22q, 18,13,154 and 6q (fig 1). There are, however, difficulties in interpreting overlaps in linkage findings from different phenotypes because of the poor localisation of linkage signals for complex disorders and the consequent difficulty of measuring co-localisation of signals and assessing statistical evidence for significant co-occurrence. However, the hypothesis that loci exist that influence susceptibility across the schizophrenia-bipolar divide receives further support from the observation that a genome scan using families ascertained on the basis of a proband with schizoaffective disorder (a form of illness with prominent features of both schizophrenia and bipolar disorder) demonstrated genome-wide significance at 1q42 and suggestive linkage at 22q11, with linkage evidence being contributed equally from “schizophrenia” families (that is, where other members had predominantly schizophrenia) and “bipolar families” (that is, where other members had predominantly bipolar disorder).155
Gene studies
Most persuasively, several recent reports implicate variation at the same loci as influencing susceptibility to both schizophrenia and bipolar disorder.
G72(DAOA)/G30
Currently the best supported locus for bipolar disorder is G72(DAOA)/G30 on chromosome 13q48,114–116 which also has positive association reported in schizophrenia.47–50
DISC1
The DISC1 locus at 1q42 receives linkage support in both schizophrenia87–89 and bipolar disorder156 and, although it has been named “Disrupted in Schizophrenia”, the family in which the translocation was observed contained cases of both psychosis and mood disorder; indeed the formal linkage evidence was actually somewhat greater for mood diagnoses than for psychosis diagnoses.87 Evidence for allelic association at polymorphisms at this locus has been reported for schizophrenia, bipolar disorder, and schizoaffective disorder.95
NRG1
Neuregulin 1 is one of the best supported schizophrenia susceptibility genes with several studies showing evidence that a so-called Icelandic “core haplotype” is associated with increased risk in Icelandic, Scottish, and UK populations.32–34 We have found that this same haplotype is significantly associated with risk for bipolar disorder and that it may exert a specific effect in the subset of functional psychosis that has both manic and mood incongruent psychotic features.157 Further support for the role of NRG1 across the mood-psychosis spectrum comes from a Dutch sample in which variation at NRG1 was found to influence susceptibility only to the subset of schizophrenia cases with predominantly good outcome.158 This is the subset that typically has more features in common with mood disorders.
COMT
We have discussed the COMT locus in relation to both schizophrenia and bipolar disorder. It lies at 22q11, a region implicated in both disorders.13 It is extremely likely that genetic variation in this region influences susceptibility across the psychosis spectrum, although it is not yet clear that COMT itself is the (or the major) susceptibility gene at this locus (see discussion in schizophrenia section).
These gene findings provide strong evidence that, as suggested by the family and twin data, there are genetic loci that contribute susceptibility across the Kraepelinian divide to schizophrenia, bipolar disorder, and schizoaffective disorders. These findings have important implications for classification of the major psychiatric disorders because they demonstrate an overlap in the biological basis of disorders that have, over the last 100 years, been classified as distinct entities.159 Thus, we can expect that over the coming years molecular genetics will catalyse a re-appraisal of psychiatric nosology as well as providing a path to understanding the pathophysiology that will facilitate development of improved treatments. For example, current genetic findings suggest that rather than classifying psychosis as a dichotomy, a more useful formulation may be to conceptualise a spectrum of clinical phenotype with susceptibility conferred by overlapping sets of genes.159 Already it is possible to recognise some common biological features amongst the genes implicated by current studies and tentative models have been advanced that postulate the key role of synaptic function.160
In the past psychiatric genetics has often attracted the pessimistic view that it is an area of endeavour that is so complex that advances were unlikely. However, the heritabilities of the major psychiatric disorders—schizophrenia and bipolar disorder—are amongst the highest of human illnesses. To place it in perspective, they are similar to that of height161 and type 1 diabetes,162,163 and greater than breast cancer,164 coronary heart disease in males,165 and type II diabetes.166 Moreover, in the last 2–3 years findings have been accruing (table 3) that will allow psychiatric genetics to take its place amongst the more successful areas of complex human disease genetics.
Summary of current weight of evidence supporting several of the more promising genes implicated in the pathogenesis of schizophrenia and/or bipolar disorder
These developments can be expected to continue. This will change the practice of clinical psychiatry and have enormous potential to benefit our patients.
Acknowledgments
The authors are indebted to all the individuals who have participated our studies.
REFERENCES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
Footnotes
-
The authors are grateful for support for their research in the genetics of schizophrenia and bipolar disorder to the UK Medical Research Council and the Wellcome Trust
-
Competing interests: none declared