


I. Introduction
Intercellular communication in the central nervous system (CNS)2 requires the precise control of the duration and the intensity of neurotransmitter action at specific molecular targets. After they have been released at the synapse, neurotransmitters activate pre- and/or postsynaptic receptors (Fig.1). To terminate synaptic transmission, neurotransmitters are, in turn, inactivated by either enzymatic degradation or active transport in neuronal and/or glial cells by neurotransmitter transporters (Iversen, 1975). Once inside the neuronal cell, neurotransmitters can be further transported into synaptic vesicles by vesicular carriers (Fig. 1). These processes are responsible for the homeostasis of neurotransmitter pools within nerve endings (Fig. 1). Both at the plasma and the vesicular membranes, neurotransmitter influxes are directly coupled to transmembrane ion gradients which provide the energy for the retrotransport (Kanner and Schuldiner, 1987).

Schematic representation of the main neurotransmission steps at a synapse. The neurotransmitter is synthesized in the presynaptic neuron, stored in synaptic vesicles (by the VNT), and released by exocytosis. The neurotransmitter then acts at metabotropic and/or ionotropic receptors (RNT, E: effector), and is removed from the synaptic cleft by uptake in presynaptic or postsynaptic neurons and/or glial cells. The uptake process is carried out by plasma membrane-bound neurotransmitter transporters (PNT).
Neurotransmitter transporters can be classified in superfamilies, families, and subfamilies according to their primary structures and site of action. In particular, the latter criterion allows the distinction of two superfamilies: 1) the plasma membrane transporters and 2) the vesicular membrane transporters. The superfamily of plasma membrane transporters can be further divided into two families depending on their ionic dependence: 1) the Na+/Cl−-dependent transporters and 2) the Na+/K+-dependent transporters.
This review article describes the present status of the art about neurotransmitter transporters involved in the fine tuning of neuronal communication. Special attention is paid to their anatomical and cellular localization, pharmacological properties, and involvement in the physiology of the normal and pathological CNS.
II. Plasma Membrane Neurotransmitter Transporters
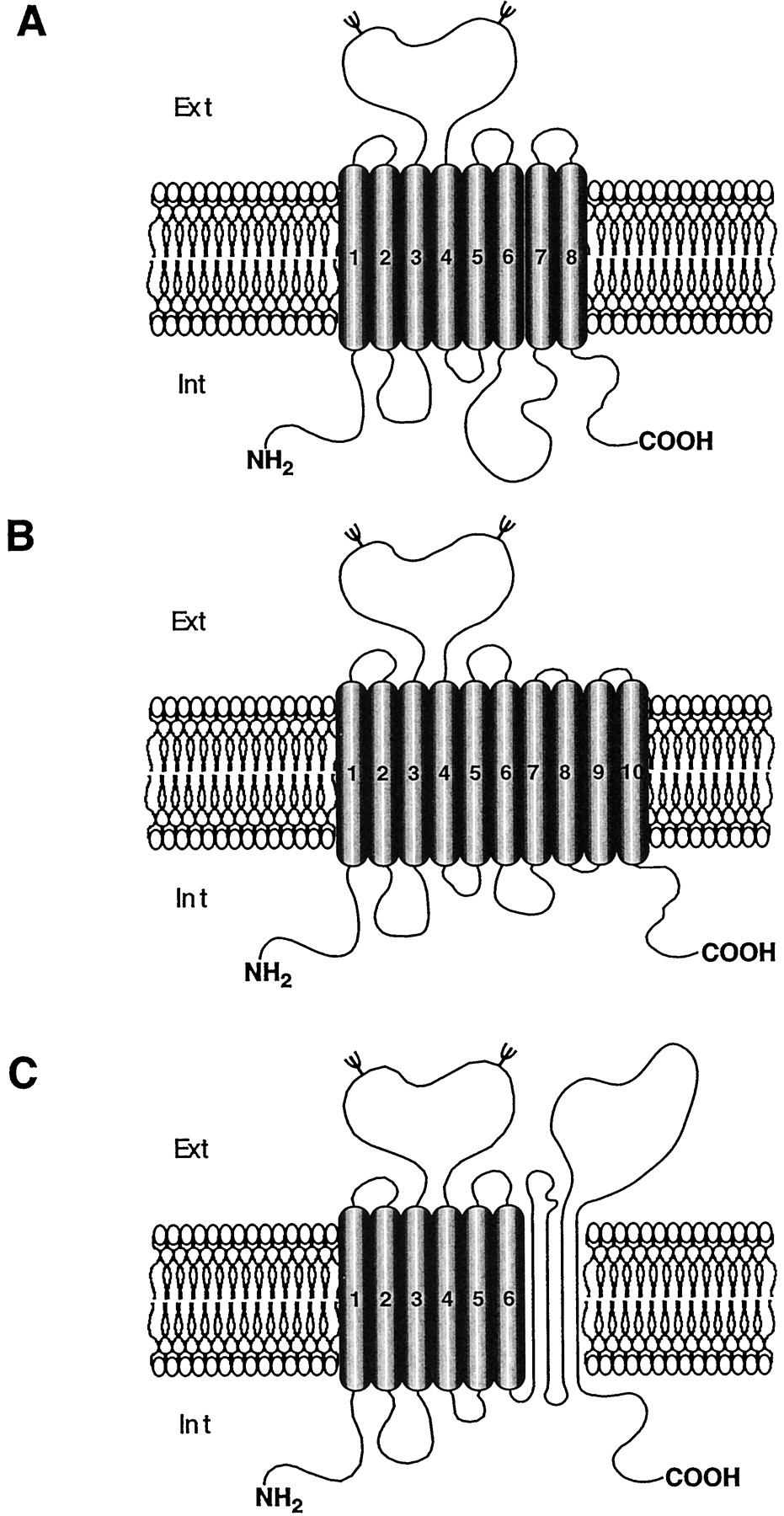
Plasma membrane neurotransmitter transporters are responsible for the high-affinity uptake of neurotransmitters by neurons and glial cells at the level of their plasma membrane. These membrane-bound proteins are all dependent on the Na+intracellular/extracellular gradient for their activity; in addition they also may require either Cl− or K+ (Kanner and Schuldiner, 1987; Kavanaugh et al., 1992; Zerangue and Kavanaugh, 1996). The advent of molecular cloning has allowed the pharmacological and structural characterization of a large family of related genes encoding Na+/Cl−-dependent neurotransmitter transporters (SCDNTs; Fig.2). The monoamine [dopamine (DA), norepinephrine and serotonin (5-HT)], amino acid [aa; γ-aminobutyric acid (GABA), glycine, proline, and taurine], and osmolite (betaine, creatine) transporters require Na+ and Cl− and possess 12 hydrophobic structural motifs (Fig. 2). In contrast, excitatory aa (glutamate and aspartate) transporters are Na+/K+-dependent. They belong to another transporter family whose members possess 6 to 10 hydrophobic (transmembrane) domains, and share no sequence homology with the Na+/Cl−-dependent carrier family (Amara, 1992).

Schematic structural organization of the different subfamilies of Na+ /Cl−-dependent transporters. A, classical Na+/Cl−-dependent transporters. On the left side, the topology is derived from hydropathy plot analyses; on the right side, the representation is derived from the work of Bennett and Kanner (1997) and Olivares et al. (1997). B, orphan transporters with an atypical structure. All of these proteins have an enlargement of their fourth extracellular loop. The gray horizontal band represents the plasma membrane. Ψ, potentiallyN-glycosylated asparagine residue.
A. Na+/Cl−-Dependent Neurotransmitter Transporters (SCDNTs)
The molecular characterization of neurotransmitter transporters began with the purification, aa sequencing, and cloning of the rat GABA transporter (Radian et al., 1986; Radian and Kanner, 1986; Guastella et al., 1990). The cDNA encoding the GABA transporter (GAT) was expressed in Xenopus oocytes to establish the Na+/Cl− dependence of the transport as well as its pharmacological characterization (Guastella et al., 1990). In parallel, the expression cloning of the human norepinephrine transporter (NET) was performed by Pacholczyk et al. (1991), and the high sequence homology between GAT and NET was unraveled. Consequently, these two clones were classified within the same gene family. Expression and homology cloning rapidly led to the enlargement of this family with the betaine, creatine, DA, glycine, proline, serotonin, and taurine transporters (Figs. 2 and3). Moreover, subtypes and isoforms of GABA and glycine transporters have been characterized. All of the data concerning the SCDNT family are summarized in Table1. Interestingly, these so called “high-affinity” neurotransmitter transporters have affinities for their respective substrates ranging from ∼320 nM (for rat SERT/serotonin) to 930 μM (for human BGT/betaine).

Alignment of the aa sequences of Na+ /Cl−-dependent transporters. Classical members are rat DA [rDAT (Giros et al., 1991; Kilty et al., 1991; Shimada et al., 1991)], human norepinephrine [hNET (Pacholczyk et al., 1991)], rat serotonin [rSERT (Blakely et al., 1991; Hoffman et al., 1991], rat GABA [rGAT1 (Guastella et al., 1990)], rat proline [rPROT (Fremeau et al., 1992)], rat glycine [rGLYT1 (Guastella et al., 1992)], rat taurine [rTaurT (Smith et al., 1992b)], and rat creatine [rCREAT (Mayser et al., 1992)] transporters. Orphan members are Rxt1/NTT4 (Liu et al., 1993; El Mestikawy et al., 1994), V-7-3-2 (Uhl et al., 1992), rB21a (Smith et al., 1995), and ROSIT (Wasserman et al., 1994). The putative α-helical membrane spanning domains (I-XII) are indicated by bars. Conserved residues are shaded.
Na+/Cl−-dependent transporters
Aside from these “classical” members, a new subfamily has emerged since 1993. The primary sequence and topology of the new members are clearly similar to those of prototypical Na+/Cl−-dependent transporters (Uhl et al., 1992) (Figs. 2 and 3). However, some structural differences as compared to classical members have been reported. Moreover, because their transported substrates are, up to now, unknown, they have been named “orphan” transporters (Table2).
Rat orphan transporter subfamily
1. Classical Members.
The SCDNTs DA transporter (DAT), 5-HT transporter (SERT), GABA transporter [GAT(1–3)], norepinephrine transporter (NET), proline transporter (PROT), taurine transporter (Taurt or rB16a), glycine transporter GLYT(1a, -b, -c, and -2) share the same topology and are 40 to 60% homologous. The hydropathicity analysis of these clones revealed 12 stretches of 15 to 25 hydrophobic aas which have been interpreted as forming α-helical transmembrane domains (TMs; Kyte and Doolittle, 1982). The N- and C-terminal regions are intracellular and the second large extracellular loop contains two to four potentialN-glycosylation sites (Fig. 2). This initial theoretical topology has been recently challenged. Using theN-glycosylation scanning method, a somewhat different structural organization for GAT1 and GLYT1 could be established (Fig.2) (Bennett and Kanner, 1997; Olivares et al., 1997). In this new model, the two thirds of the proteins on the C-terminal side (from TM4 to TM12) are organized as proposed in the initial theoretical topology. However, the introduction of N-glycosylation sites as reporters indicates that TM1 is not spanning the membrane. Bennett and Kanner (1997) suggest that this highly hydrophobic region might form a pore loop structure associated with the plasma membrane. Such a secondary structure has already been described for ion channels in which it can form a selectivity ion filter (MacKinnon, 1995). Consequently, 1) the former TM2 becomes the first transmembrane domain, 2) extracellular loop (EL) 1 is intracellular, and 3) an hydrophobic portion of EL2 becomes TM3 (see Fig. 2) (Bennett and Kanner, 1997;Olivares et al., 1997). Indeed this modified topology might well be relevant to all SCDNTs.
Interestingly, the former TM1/pore loop region corresponds to a relatively well conserved sequence within the SCDNT family (Worral and Williams, 1994). It has been speculated that highly conserved regions are pointing at structural elements important for the common functions of these transporters (Giros et al., 1994). In particular, the highly homologous N-terminal portion of these molecules (from TM1 to TM4) may be involved in Na+/Cl−transport. On the other hand, the divergent C-terminal region (from TM7 to TM12) may be responsible for substrate recognition and inhibitor binding (Zaleska and Erecinska, 1987; Kitayama et al., 1992; Buck and Amara, 1994; Giros et al., 1994).
2. The Orphan Transporter Subfamily.
Using the homology cloning strategy, four new members of the Na+/Cl−-dependent transporter family have been isolated: Rxt1 (also named NTT4), V-7-3-2, ROSIT, and rB21a (Uhl et al., 1992; Liu et al., 1993; El Mestikawy et al., 1994; Wasserman et al., 1994; Smith et al., 1995) (Figs. 2 and 3). Up to now, the four new members are still to be established as actual transporters since their respective substrates have not been identified. Nevertheless, they are usually referred to as “orphan” transporters. These four proteins have all of the classical features of Na+/Cl−-dependent transporters described above. However, they also exhibit original structural characteristics such as the enlargement of their fourth and sixth extracellular loops and the presence of an additional N-glycosylation site in the fourth extracellular loop (Fig. 2). The four transporter-like proteins have significant homology (∼20–30%) with the “classical” members such as the DA, serotonin, GABA, or glycine transporters. However, the overall percentage of homology among Rxt1, V-7-3-2, ROSIT, and rB21a is higher, averaging 50 to 60%. Because they are more closely related to each other than to any other Na+/Cl−-dependent transporter, they can be considered as forming a specific group within this family. In addition, their common structural features allow the hypothesis that these orphan transporters probably exhibit functional similarities. However, their transport activities have yet to be demonstrated.
3. Ionic Dependence and Electrogenic Properties.
All of the SCDNTs are utilizing, as primary driving force, the Na+ electrochemical gradient which is created and maintained by the (Na+/K+)-ATPase across the plasma membrane. They also required Cl− to transport their substrate against a concentration gradient from the extra- to the intracellular compartment. However, under physiological conditions, the energy derived from the Cl−electrochemical gradient is negligible when compared to that of Na+.
Before the advent of molecular cloning, determinations of the stoichiometry of native SCDNTs were performed in synaptosomes or in reconstituted vesicles. These studies were already suggesting that most SCDNT members are electrogenic (Kanner and Schuldiner, 1987). Accordingly, SCDNTs would carry one or several positive charges for each substrate molecule transported with a stoichiometry of 2 Na+/1 Cl−/1 zwitterion. However, because of the low turnovers of SCDNTs (0.5 to ∼3 s-1), the predicted microscopic current (10−17–10−19 A) is 5 to 7 orders of magnitude lower than the best resolution achieved with patch-clamp recording. To really provide experimental support to this inference, it was necessary to find cells with enough transporters expressed on their surface for the recording of macroscopic uptake-associated currents. These conditions were initially fulfilled in some amphibian glial cells for the recording of the activity of glutamate and GABA transporters (Brew and Attwell, 1987; Cammack and Schwartz, 1993) and in invertebrate neurons expressing a serotonin transporter (Bruns et al., 1993).
At the beginning of the 1990s, cloned SCDNTs could be successfully expressed in Xenopus oocytes at a density higher than 103/μm2 (as established from cryofracture experiments; Zampighi et al., 1995), allowing the measurement of currents associated with the transport of neurotransmitters. GAT1, the first cloned SCDNT, was also the first one to be characterized electrophysiologically. In GAT1 expressingXenopus oocytes, application of GABA evoked a steady-state inward current that could be recorded under voltage-clamp conditions (Kavanaugh et al., 1992; Mager et al., 1993). The ionic coupling was estimated at 1.29 charges per GABA molecule transported by determining the ratio of [3H]GABA uptake over the integral of the GABA-evoked current in the same transfected oocyte (Kavanaugh et al., 1992); these data were in reasonable agreement with the predicted stoichiometry of 2 Na+/1 Cl−/1 GABA (Kanner and Schuldiner, 1987). The uptake current was dependent on the presence of Na+ and Cl− ions and blocked by specific GABA uptake inhibitors such as SKF-89976A [N-(4,4-diphenyl-3-butenyl)-3-piperidine carboxylic acid]. Interestingly, classical inhibitors of GABA or glycine transport, such as nipecotic acid (for GAT1) or sarcosine (for GLYT1) were found to be substrates. Like GABA, these compounds evoked a current when applied alone (Kavanaugh et al., 1992; Supplisson and Bergman, 1997). Shortly after, the same type of measurements were extended to other SCDNTs such as NET, SERT, DAT, GLYT1, and GLYT2 (Mager et al., 1994; Galli et al., 1996a, 1997; Sonders et al., 1997; Lopez-Corcuera et al., 1998). However, in the case of both SERT and NET, the measured ionic coupling was in far excess when compared with the one predicted by the stoichiometry (Lin et al., 1994; Mager et al., 1994; Galli et al., 1996a,b, 1997). This discrepancy could not be explained by classical models of cotransport with an alterned access at both sides of the membrane. Rather, it suggested that some channel activity is associated with the transporter cycle (for review, see Lester et al., 1994;DeFelice and Blakely, 1996; Kavanaugh, 1998). This new feature of SCDNT transport activity received more direct support from the recording of single channel in cells expressing GAT1 or SERT (Cammack and Schwartz, 1993; Lin et al., 1996).
In addition to the electrogenic substrate translocation described above, neurotransmitter transporters can also generate uncoupled currents that can be blocked by uptake inhibitors or the susbtrate itself. These uncoupled currents were first described for the transport of glutamate in photoreceptor cells of the salamander (Sarantis et al., 1988). With the advent of molecular cloning, this initial observation has now been extended to other neurotransmitter transporters (for review, see Sonders and Amara, 1996). For example, relatively large current can be recorded by patch-clamping HEK293 cells that express GAT1 (Cammack et al., 1994). This leakage current was identified as resulting from the channel-like behavior of the transporter (Cammack et al., 1994). However, it should be noted that these observations were not reproduced with Xenopus oocytes (Mager et al., 1993).
In addition, capacitive currents that can be suppressed by substrates or inhibitors have been observed during voltage jumps for GAT1 (Cammack and Schwartz, 1983; Mager et al., 1993, 1994, 1996) and TAUT (Loo et al., 1996). Integration of the current and determination of the saturating charges movement (Qmax) allowed an estimate of the number of transporters expressed (Mager et al., 1993,1998; Loo et al., 1996). In addition, the slope of the relationship between the maximal uptake current (Imax) and the Qmax has permitted the determination of transporter turnover (for review, see Mager et al., 1998).
All of these data show that in addition to their known function as neurotransmitter transporters, SCDNTs have complex and not yet completely identified ion channel-like properties (Sonders and Amara, 1996).
4. Cellular and Subcellular Localization.
Thanks to the availability of their sequences, probes (cRNA and antibodies) have been produced for the determination of the detailed anatomical and cellular localization of SCDNTs. In particular, specific antibodies have been raised against DAT (Ciliax et al., 1995; Freed et al., 1995), GAT1–3 (Ikegaki et al., 1994; Minelli et al., 1995, 1996), GLYT1–2 (Jurski and Nelson, 1995; Zafra et al., 1995), NET (Bruss et al., 1995), PROT (Velaz-Faircloth et al., 1995), and SERT (Qian et al., 1995; Sur et al., 1996; Zhou et al., 1996). In summary, in situ hybridization and immunocytochemical data showed that DAT, NET, and PROT are present exclusively in neurons; GAT3 is found in glial cells; and GAT1, GLYT1–2, and SERT are synthesized both in neurons and astrocytes (Minelli et al., 1995; Zafra et al., 1995; Bel et al., 1997). On the other hand, GAT2 is expressed by arachnoid and ependymal cells (Ikegaki et al., 1994; Durkin et al., 1995).
Many SCDNTs [as well as excitatory aa transporter (EAAT) 3/EAAC1, and EAAT5 which are Na+/K+-dependent glutamate transporters (SKDGTs)] are found in the brain as well as in non-neuronal peripheral tissues (Uhl and Hartig, 1992; Amara and Kuhar, 1993; Kanai et al., 1993; Borden, 1996). Nonetheless, this article will concentrate on their distribution in the mammalian CNS.
DAT and NET are considered as specific markers of DAergic and noradrenergic neurons in the CNS, respectively. Similarly, SERT can be used as a marker of serotoninergic neurons because its synthesis in astrocytes (Bel et al., 1997) seems to be hardly detected in the CNS of adult intact (i.e., unlesioned) rats (F. C. Zhou, personal communication). Apparently, all GABAergic neurons express GAT1 mRNA; however, GAT1 is also synthesized in pyramidal glutamatergic cells in the hippocampus and the cerebral cortex (Minelli et al., 1995; Yasumi et al., 1997). GLYT1 is expressed in both glycinergic neurons in the brainstem and in the spinal cord and glutamatergic neurons within the forebrain (Zafra et al., 1995). Finally, the PROT is found in glutamatergic neurons (Fremeau et al., 1992).
Some SCDNTs seem to be addressed to specific subcellular compartments, whereas others are present all over the plasma membrane. For example, DAT, NET, and SERT are present on dendrites, perikarya, axons, and nerve endings of the corresponding monoaminergic neurons (Ciliax et al., 1995; Freed et al., 1995; Qian et al., 1995; Nirenberg et al., 1996, 1997b). Interestingly, previous results already demonstrated the existence of dendritic [3H]DA uptake in the substantia nigra (Gauchy et al., 1994) and somatodendritic [3H]5-hydroxytryptamine (5-HT) uptake in the dorsal raphe nucleus (Descarries et al., 1982). Accordingly, these ultrastructural data formally establish a fact that has long been suspected: SERT and DAT are not only present but are also functional over the entire surface of serotoninergic and dopaminergic neurons, respectively. In striatal dopaminergic terminals, which belong to neurons located in the substantia nigra pars compacta, DAT is found on the varicose and intervaricose plasma membrane but not in the active synaptic zones (Hersch et al., 1997; Sesack et al., 1998). Interestingly, in dopaminergic nerve endings of the rat prefrontal cortex (which belong to neurons located in the ventral tegmental area), very low levels of DAT immunoreactivity are observed, most of which being extrasynaptic (Sesack et al., 1998). These observations are in line with the fact that extracellular concentrations of DA are higher in the prefrontal cortex than in the striatum (Cass and Gerhardt, 1995).
In contrast, GAT1, GLYT2, and PROT seem to be restricted to axon terminals (Ikegaki et al., 1994; Jurski and Nelson, 1995;Velaz-Faircloth et al., 1995; Riback et al., 1996). These data allow the distinction of two groups of SCDNTs: those that are restricted to nerve terminals and those that are not specifically addressed to this cell compartment. Furthermore, it appears that a transporter such as DAT may have different subcellular localization in different neurons.
The cellular mechanisms and molecular signals that are responsible for the targeting of a given transporter are just beginning to be investigated. For this purpose, transfection in polarized cells such as the LLCPK-1 and MDCK-1 epithelial cell lines has proven to be a valuable method. The plasma membrane of these cells can be divided in two functionally and structurally different compartments. The basolateral membrane of an epithelial cell seems to correspond to the somatodendritic domain of a neuron, whereas the apical side is apparently equivalent to nerve terminals (Dotti and Simons, 1990). In line with their respective neuronal targeting (see above), DAT, NET, and SERT are addressed to the basolateral (in LLCPK cells) and apical (in MDCK cells) domains (Gu et al., 1996), whereas GAT1 is found only at the apical domain of the plasma membrane of transfected epithelial cells (Pietrini et al., 1994). Thus, it should be kept in mind, as mentioned above for neurons (Sesack et al., 1998), that the subcellular sorting of a transporter in heterologous systems is largely dependent on the cell type that is used (Gu et al., 1996). However, despite this important drawback, it should be feasible using such cellular models to determine the molecular mechanisms responsible for the specific targeting of a given transporter, using notably site-directed mutated and/or chimeric transporters.
In addition to the above-mentioned SCDNsT, the orphan transporter Rxt1/NTT4 has also been extensively studied with regard to its cellular and subcellular localization (Liu et al., 1993; El Mestikawy et al., 1994, 1997; Masson et al., 1995; Luque et al., 1996). In the rat CNS, Rxt1/NTT4 is expressed both in glutamatergic cells and in subsets of GABAergic neurons (such as reticular thalamic and Purkinje cells). At the subcellular level, this transporter-like protein is found almost exclusively in axon terminals. Surprisingly, Rxt1/NTT4 has recently been demonstrated to be located on synaptic vesicles (Masson et al., 1998), although it is clearly a member of the Na+/Cl−-dependent transporter family. Furthermore, the proline transporter was also found in small synaptic vesicles within subsets of presynaptic axons forming asymmetric excitatory synapses (Renick et al., 1999). Interestingly, in situ hybridization data support the idea that Rxt1/NTT4 and PROT are probably synthesized in the same subset of glutamatergic neurons (Fremeau et al., 1992; El Mestikawy et al., 1994;Velaz-Faircloth et al., 1995; Masson et al., 1997).
Although Rxt1/NTT4 and PROT are located in synaptic vesicular membranes, both proteins share no sequence homology with the vesicular proton-driven carrier. Moreover, as mentioned above, transporters of the SCDNT family use the plasma membrane Na+ionic gradient as energy source, whereas vesicular transport is coupled to the H+ electrochemical gradient. Indeed, to our knowledge, the existence of a Na+ gradient between the lumen of the vesicle and the neuronal cytoplasm has never been reported. Therefore, it can be hypothesized that in spite of their Na+/Cl−-dependent transporter-like primary structure, Rxt1/NTT4 and PROT are able to use the proton-generated energy to perform their presumed function in vesicles. However, PROT is unable to drive l-proline vesicular uptake in HEK293-transfected cells (Miller et al., 1997). Thus, alternatively, vesicular Rxt1/NTT4 and PROT might represent pools of “spare transporters” awaiting for a yet unidentified physiological signal to be addressed at the plasma membrane and to become functional.
5. Phosphorylation-Dependent Regulation of Transport.
Before the advent of molecular cloning, the reuptake process appeared to be less regulated than other important steps of the neurotransmission cascade (such as the biosynthesis and the release of neurotransmitters or their binding to specific receptors). However, in a few cases, protein kinase-dependent modulation of neurotransmitter reuptake was described. For example, in primary cultures of astrocytes and neurons, GABA uptake could be modulated by Ca2+/calmodulin-dependent protein kinase, protein kinase C (PKC), or cAMP-dependent protein kinase A (PKA) (Gomeza et al., 1991, 1994; Corey et al., 1994). In addition, DA uptake in mouse striatum was significantly affected by a wide-spectrum inhibitor of protein kinases (Simon et al., 1997). On the other hand, the transport of serotonin in choriocarcinoma cells, as well as that of DA in hypothalamic neurons, was stimulated by cAMP-dependent phosphorylation (Kadowaki et al., 1990; Cool et al., 1991).
The presence of conserved PKC and, in some cases, PKA consensus phosphorylation sites in cytosolic domains of all SCDNTs (see alignments in Fig. 3) supports the hypothesis of transport regulation by second messengers. Indeed, PKC-dependent negative modulation of DAT activity could be evidenced in transfected COS-7 and LLCPK-1 cells exposed to phorbol esters (Kitayama et al., 1994; Huff et al., 1997). Similarly, GLYT1b and SERT have been shown to be inhibited upon PKC activation in transfected HEK293 cells (Sato et al., 1995; Sakai et al., 1997; Ramamoorthy et al., 1998). In all cases, PKC-induced inhibition was associated with a reduction inV max with no modification inK m (Kitayama et al., 1994; Sato et al., 1995; Huff et al., 1997; Sakai et al., 1997), suggesting a decrease in the cell density of functional transporters. Indeed, rapid internalization of SERT could be observed in transfected cells with activated PKC (Blakely et al., 1998; Ramamoorthy et al., 1998). However, the down-regulation of glycine and serotonin uptake is still observed after site-directed mutagenesis of all five predicted PKC consensus sites in GLYT1b as well as in SERT. At first it was hypothesized that uptake inhibition was not due to a direct PKC-dependent phosphorylation of the transporters (Sato et al., 1995;Sakai et al., 1997). However, because a direct phosphorylation of SERT has been recently demonstrated, it is more likely that PKC-phosphorylated sites are noncanonical (Ramamoorthy et al., 1998).
Corey et al. (1994) have demonstrated that PKC activation markedly increased the activity of GAT1 expressed in Xenopus oocytes. Interestingly, this effect was associated with a shift in GAT1 subcellular localization from intracellular vesicles to the plasma membrane (Corey et al., 1994; Quick et al., 1997). Using site-directed mutagenesis and coinjection of various mRNAs, it was then found that the redistribution of this GABA transporter was dependent on both the presence of a leucine zipper motif in its second transmembrane domain and the level of syntaxin expression (Corey et al., 1994; Quick et al., 1997).
In summary, relevant studies clearly showed that the activity of SCDNTs can be modulated by protein kinases. The resulting changes generally concern the concentration of SCDNTs at the plasma membrane rather than their intrinsic transport activity.
6. Pharmacological and Functional Aspects.
Numerous psychiatric, neurological, and neurodegenerative disorders have been associated with alterations in the neurotransmission cascade. In this context, the complete elucidation of neurotransmitter transporter functions can be of strategic importance for the development of new therapies. Indeed, a wide range of pharmacological agents are known to interact with neurotransmitter transporters. Generally, these compounds act as transport inhibitors. This is particularly well illustrated with antidepressants and psychostimulants which act primarily as inhibitors of monoamine transporters (SERT, NET, and DAT).
The pharmacological properties of SCDNTs were first determined from uptake studies performed with brain synaptosomes. Subsequently, experiments were also performed using Xenopus oocytes injected with total mRNAs isolated from discrete rat brain regions (Sarthy, 1986; Blakely et al., 1988). However, interpretation of the data obtained with such approaches could be difficult because of the possible participation of more than one transporter type in the uptake process. Furthermore, these approaches are not especially appropriate for the study of human transporters.
Thanks to molecular cloning, the precise substrate specificity and pharmacological profile of each monoamine transporter could be determined in in vitro experiments performed on transfected mammalian cells or Xenopus oocytes (Giros and Caron, 1993). One interesting finding of such studies is that the human DAT can transport both DA and norepinephrine with K mvalues of 2.5 and 20 μM, respectively (Giros et al., 1992, 1994). Surprisingly, NET has a better affinity for DA (K m = 0.67 μM) than DAT itself, on one hand, and than for its proper substrate, norepinephrine (K m = 2.6 μM), on the other (Giros et al., 1994). The capacity of SCDNTs to transport more than one substrate is not unique to DAT and NET since the betaine transporter can take up not only betaine but also GABA with high affinity (Yamauchi et al., 1992; Borden et al., 1995a). Thus, the concept of “one transporter/one substrate” can no longer be considered as a general rule.
In the human brain, DAT, NET, and SERT are the primary binding sites of cocaine (Giros et al., 1992; Giros and Caron, 1993). Recent reports have established that inhibition of DA reuptake may be the key event leading to the rewarding action of cocaine and thus to addiction (Giros et al., 1994, 1996). However, knockout mice that do not express DAT (Giros et al., 1996) can still self-administer cocaine under certain conditions (Rocha et al., 1998). Thanks to this model, it could be unraveled that serotoninergic mechanisms, in addition to dopaminergic systems, play an important role in the development of addiction to cocaine (Rocha et al., 1998). However, using the place-preference paradigm, Sora et al. (1998) recently found that the appetitive properties of cocaine are lost neither in DAT nor in SERT knockout strains. Thus, neither DAT nor SERT seems to be absolutely required for the rewarding action of cocaine.
Among monoamine transporters, DAT seems to be implicated in the etiology of various neurological or psychiatric syndromes. Thus, as expected of the marked degeneration of dopaminergic neurons, a decrease of DAT is regularly observed in Parkinson’s disease (Boja et al., 1994; Miller et al., 1997). In addition, the amounts of DAT in striatal axon terminals are reduced in spinocerebellar ataxia of type 1 (Kish et al., 1997). Aberrant dopaminergic neurotransmission is also associated with disorders of the schizophrenic spectrum and Tourette’s syndrome (Pearce et al., 1989; Singer et al., 1991). However, no linkage was found to date between DAT alleles and hereditary pathogenesis of schizophrenia (Byerley et al., 1993a,b; Li et al., 1994; Persico et al., 1995).
Monoamine transporters are also the primary sites of action for tri- and heterocyclic antidepressant drugs (Blakely et al., 1994; Barker and Blakely, 1995). Indeed, both the reduced serotonin transport in platelets and, possibly, brain in depressed and suicidal patients (Meltzer and Lowy, 1987), and the efficacy of selective SERT inhibitors (fluoxetine, fluvoxamine, paroxetine, sertraline, citalopram, etc.) in the treatment of depression (Anderson and Tomenson, 1994) are compelling evidences in support of the involvement of this transporter in the etiology of mood disorders. Decreased serotonin brain levels in patients with disorders of the affective spectrum may reflect a structural defect and/or dysregulation of SERT (Perry et al., 1983). The gene coding for the human SERT has been cloned and localized on chromosome 17q11.2 (Ramamoorthy et al., 1993). This gene spans over 35 kilobases and is organized in 14 introns. No allelic variation has been observed in the coding region of the SERT gene in patients with affective disorders (Altemus et al., 1996; Di Bella et al., 1996). In contrast, multiple polymorphims are found in the 5′-flanking region and in the second intron (Lesch et al., 1994; Heils et al., 1996). Interestingly, the two variants in the 5′ region are associated with different rates of SERT expression and the one leading to the lowest transcription rate seems to be more frequent in subjects with anxiety-related personality traits (Lesch et al., 1996a) and in alcoholics with suicidal behavior (Gorwood et al., 1998). Moreover, allelic forms at the second intron locus seem to be associated with bipolar and unipolar disorders (Battersby et al., 1996;Collier et al., 1996; Ogilvie et al., 1996; Bellivier et al., 1997). Thus, during the last 2 years, numerous genetic studies have strengthened the “serotonin hypothesis” of mood disorders. However, molecular and genetic studies on neurotransmitter transporters in relation with psychiatric diseases are just on the starting line, and numerous investigations will have to be performed to get really definitive and clear-cut data regarding the actual association of a given polymorphism of the SERT gene and one or several of these diseases.
GABA is the major inhibitory neurotransmitter in the mammalian brain. Pharmacological compounds which modulate GABAergic neurotransmission, such as benzodiazepines and barbiturates, have proven to be efficient in the treatment of anxiety and epilepsy (During et al., 1995; Dalby and Nielsen, 1997b). Pharmacological data have long been pointing at the existence of two distinct GABA transporters in glial and neuronal cells (Borden, 1996). Indeed, with the advent of molecular cloning, five GABA transporter subtypes have been found: GAT1, GAT2, GAT3, betaine/GABA transporter 1, and rB16a (Table 1). Specific inhibitors at each subtype of GABA transporters are thus representing new potential therapeutic agents for the treatment of epilepsy and anxiety. Furthermore, reverse functioning of GABA transporters should allow the inhibition of excessive neuronal firing due to excitatory aas during stroke and seizures. Accordingly, pharmacological compounds able to release GABA by reversing GAT’s activity might represent a new class of neuroprotective agents.
The precise pharmacological profiles of the various cloned GABA transporters have been determined using stably transfected mammalian cell lines (Borden et al., 1994, 1995b, 1996). In these studies, cis-3-aminocyclohexanecarboxylate, CI-966 (1-[2-[bis 4-(trifluoromethyl) phenyl]methoxy] ethyl]-1,2,5,6-tetrahydro-3-pyridine carboxylic acid), nipecotic acid, NNC 05-711 (1-[2-[[[diphenylmethylene]amino]oxy]ethyl]-1,2,5,6-tetrahydro-3-pyridinecarboxylic acid), SK&F 89976-A (N-[4,4-diphenyl-3-butenyl]-3-piperidinecarboxylic acid), and tiagabine [Gabitril; (R)-1-[4,4-bis(3-methyl-2-thienyl)-3-butenyl]-3-piperidinecarboxylic acid] were shown to act selectively at GAT1 (Borden et al., 1994,1995b). The rank order potency of these compounds at the human GAT1 is as follows: NNC 05-711 > tiagabine > SK&F 89976-A > CI-966 (Borden, 1996). All of these compounds display anticonvulsive activity in relevant animal models (Dalby and Nielsen, 1997b). Tiagabine has even proven its efficiency in the treatment of complex and refractory myoclonic seizures (Dalby and Nielsen, 1997a). Other compounds have been found to act selectively at GAT2 and GAT3: β-alanine, hypotaurine, NNC 05-2045 (1[3-[9H-carbazol-9-yl]-1-propyl]-4-[4-methoxyphenyl]-4-piperidinol) and NNC 05-2090 (1-[3-[9H-carbazol-9-yl)-1-propyl)-4-(2-methoxyphenyl]-4-piperidinol) (Clark and Amara, 1994; Dhar et al., 1994; Borden et al., 1995b). Interestingly, the latter drugs also display anticonvulsive properties (Borden et al., 1994, 1995b; Dalby et al., 1997). Thus, in the last few years, the field of antiepileptic drugs has developed rapidly thanks to the availability, for relevant pharmacological studies, of mammalian cell lines expressing the various GABA transporters.
B. Na+/K+-Dependent Glutamate Transporters (SKDGTs)
Large amounts of l-glutamate are found in the mammalian CNS where this aa acts as the major excitatory neurotransmitter (Fagg and Foster, 1983; Fonnum, 1984). The neurotransmitter pool of glutamate is highly concentrated in nerve terminals, and low levels of the aa (below 1 μM) are normally found in the extracellular space (Ottersen and Storm-Mathisen, 1984). It is now widely accepted that elevated levels of extracellular glutamate can induce severe damages to target neurons. Because glutamate is such a potent excitotoxin, its removal from the synaptic cleft is of key importance to maintain the integrity of neuronal tissues. The Na+-dependent transport of glutamate into neurons and glial cells represents the prime mechanism by which this aa is removed from the synaptic cleft (McBean and Roberts, 1985; Nicolls and Attwell, 1990). This high-affinity transport is in fact dependent on both Na+ and K+, but does not require Cl− (Danbolt, 1994). Glutamate transporters were first studied and pharmacologically defined using brain synaptosomes. Then, in the early 1990s, molecular cloning of SKDGTs allowed the detailed characterization of the family of the Na+/K+-dependent glutamate transporters (Amara, 1992).
1. Molecular Cloning and Primary Structure.
Three glutamate transporters, named GLAST1, GLT-1, and EAAC1 (Table3), were cloned almost simultaneously but independently by different groups (Kanai and Hediger, 1992; Pines et al., 1992; Storck et al., 1992). GLAST1 was isolated using probes derived from the sequence of a protein copurified with UDP-galactose:ceramide galactosyl transferase (Storck et al., 1992). GLT-1 was first purified from the rat brain and then used to produce a specific antiserum (Danbolt et al., 1992) for immunoscreening of an expression library (Pines et al., 1992). On the other hand, an expression cloning strategy was successfully used inXenopus oocytes to clone EAAC1 from the rabbit small intestine (Kanai and Hediger, 1992). The human homologs EAAT1 (=GLAST1), EAAT2 (=GLT1), and EAAT3 (=EAAC1) have all been found in the motor cortex (Arriza et al., 1994). Two new members of the SKDGT family, named EAAT4 and EAAT5 (see Fig. 5), were subsequently identified in the human cerebellum and retina, respectively (Fairman et al., 1995; Arriza et al., 1997). Interestingly, molecular cloning has also permitted the isolation of two neutral aa transporters, named ASCT-1 and -2 (Arriza et al., 1993; Shafqat et al., 1993; Utsunomiya-Tate et al., 1996), which belong to the same X− A,G system (McGivan and Pastor-Anglada, 1994) as the acidic aa transporters of the SKDGT family.
Na+/Cl−-dependent glutamate transporter family

Hydropathy plots Kyte-Doolittle of the glutamate transporters. Putative α-helical transmembrane domains of 20 to 25 aa are numbered (1–6) above each sequence. LHCS, large hydrophobic conserved domain.
Comparison of the respective primary sequences revealed a relatively high degree of homology (40–60%, see Fig.4) among the five SKDGT (Amara, 1992;Kanner, 1993; Taylor, 1993). As can be seen in Fig. 4, similarity is particularly striking in the C-terminal moiety. Along with the neutral aa carriers (transporting alanine, cysteine, and serine; Arriza et al., 1993), the five subtypes of glutamate transporters define a new gene family (Danbolt et al., 1992; Kanai et al., 1993). From a structural point of view, this group of genes is completely distinct from the SCDNT family described above. For the sake of clarity, the five glutamate transporters are named EAAT-1 to -5 in the following sections of this review (see Table 3).
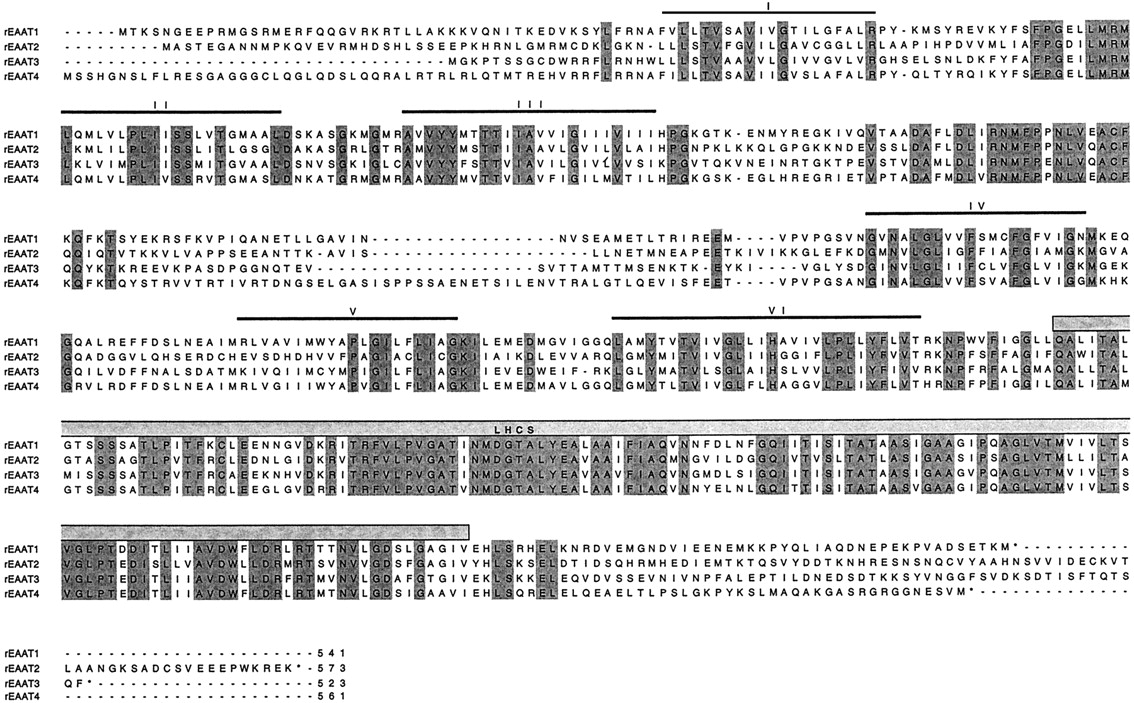
Alignment of the aa sequences of SKDGTs. Glutamate transporters are rat EAAT-1/GLAST1 (Storck et al., 1992), rat EAAT-2/GLT1 (Pines et al., 1992), rat EAAT-3/EAAC1 (Bjoras et al., 1996), and rat EAAT-4 (Fairman et al., 1995). The putative α-helical membrane spanning domains (I-VI) are indicated by bars. LHCS, large hydrophobic conserved domain. Conserved residues are shaded.
The sizes of EAAT-1 to -5 range between 523 and 573 aas. The precise transmembrane topology, predicted from hydrophobicity analysis, has been a matter of controversy (Gegelashvili and Schousboe, 1997). Hydropathy plots of the SKDGTs are rather homogeneous (Fig.5) and show six clear membrane-spanning α-helices (Kanai et al., 1993) in the amino-terminal portion (Figs. 4and 5). On the other hand, the carboxyl terminus (Fig. 5) is mostly formed of a long hydrophobic and highly conserved domain (spanning over ∼50 residues) (Kanai et al., 1993; Arriza et al., 1997). The motif R-F-V-L-P-V-G-A-T-I/V-A-A-I/V-F-I-A-Q-X-N-X-X-L-G-Q-I, which is found in the eighth transmembrane domain of the five EAAT cloned so far, can be considered as a SKDGT signature. The prediction of secondary structure as well as the consequences of deletions in this region are more in favor of its organization as four short [8–9 aas (aa)] β-sheets instead of two to four long (20–25 aa) α-helices (Arriza et al., 1994, 1997; Slotboom et al., 1996; Wahle and Stoffel, 1996). Nonetheless, it has to be pointed out that the controversy is not closed yet because a recent study is favoring the 10-α-helices model (Slotboom et al., 1996). In the β-sheet model, the C terminus has been proposed to interact with the plasma membrane as depicted in Fig. 6C. Accordingly, both the N and C termini, which are rather poorly conserved in this family, are supposed to be located in the cytoplasmic compartment. In addition, EAAT-1 to -5 have a long second extracellular loop, which is also poorly conserved, bearing one to three consensus N-linked glycosylation sites (N-X-S/T). As shown by their migration patterns in polyacrylamide gel electrophoresis and deglycosylation experiments, EAAT-1, -2, -3, and -4 are actually glycosylated proteins (Rothstein et al., 1994;Furuta et al., 1997; Kataoka et al., 1997).
Hypothetical topologies of glutamate transporters. The predictions of secondary structures of the identified glutamate transporters propose α-helices for the first six transmembrane domains N-terminal domain. The secondary structure of the C-terminal domain is a matter of controversy. Some models predict 8 A or 10 B transmembrane domains (Kanai et al., 1993; Lesch et al., 1996; Slotboom et al., 1996; Gegelashvili and Schousboe, 1997), but other data are more in favor of the existence of 6 transmembrane domains only and 4 β-sheets C (Wahle and Stoffel, 1996).
2. Ionic Dependence and Ligand-Gated Cl− Channel Properties.
Only a brief survey of this rapidly developing research area will be presented here. For more elaborate information, the reader can refer to the recent review of Palacin et al. (1998).
Extensive biochemical studies have shown that both low (K m > 500 μM)- and high (K m = 1–100 μM)-affinity glutamate uptake exist in neurons and glial cells (Logan and Snyder, 1971;Schousboe, 1981). The transporters involved have long been known to be electrogenic (Brew and Attwell, 1987) and have thus been studied by electrophysiological means (Arriza et al., 1994; Kanai et al., 1994;Klochner et al., 1994; Wadiche et al., 1995a). An initial stoichiometry was determined as follows: 2 Na+and 1 glutamate− cotransported in exchange of 1 K+ and 1 OH− (Bouvier et al., 1992; Kanai et al., 1995b).
During the postcloning era, SKDGTs were expressed in Xenopusoocytes and transfected cell lines, which allowed the demonstration that EAAT-1 to -5 can transport l-glutamate as well as l- and d-aspartate with a high affinity (Table 3). In addition, using the reversal potential of EAAT-3, Zerangue and Kavanaugh (1996) could establish a more definitive stoichiometry for this transporter as follows: 3 Na+ and 1 H+ cotransported with 1 glutamate− for 1 K+countertransported. This stoichiometry was recently extended to EAAT-2 (Levy et al., 1998).
In 1995, Fairman et al. demonstrated that EAAT-4 has an intrinsic Cl− conductance gated by glutamate, and subsequent studies showed that this property is shared with the other SKDGT, EAAT-1–3 (Wadiche et al., 1995; Billups et al., 1996). In fact, EAAT-1 to -5 behave as true glutamate carriers, contaminated by a Cl− conductance (Wadiche et al., 1995; Otis and Jahr, 1998). In other words, the uphill transport of glutamate by EAAT1–5 is Cl−-independent whereas Cl− permeation is linked to particular steps of the glutamate transport cycle (Wadiche et al., 1995; Sonders and Amara, 1996; Otis and Jahr, 1998). Interestingly, EAAT-4 (in the cerebellum) and EAAT-5 (in the retina) are characterized by a relatively large glutamate-elicited Cl−conductance (33 Cl−/glutamate transported) (Arriza et al., 1997; Eliasof et al., 1998) when compared with the one induced by EAAT-1, -2, and -3 (1–2 Cl−/glutamate transported) (Arriza et al., 1994;Wadiche et al., 1995; Sonders and Amara, 1996).
Consequently, EAAT-4 and -5 are bifunctional proteins, each acting as both a transporter and an ion channel. However, the actual functional significance of the channel activity is still unknown.
3. Cellular and Subcellular Localization.
Northern blot studies showed that EAAT-1 and -2 are expressed exclusively in the brain (Pines et al., 1992; Storck et al., 1992; Nakayama et al., 1996). EAAT-3 mRNA is found in the intestine, kidney, heart, liver, and brain (Kanai and Hediger, 1992; Nakayama et al., 1996). EAAT-4 is present in the brain and placenta (Fairman et al., 1995), and the fifth SKDGT, EAAT-5, is expressed in the retina and, at lower levels, in the liver and brain (Arriza et al., 1997). Specific antibodies have been raised against the first four subtypes of glutamate transporters: EAAT-1 (Rothstein et al., 1994; Lehre et al., 1995; Wahle and Stoffel, 1996;Schmitt et al., 1997), EAAT-2 (Danbolt et al., 1992; Rothstein et al., 1994; Lehre et al., 1995), EAAT-3 (Rothstein et al., 1994; Shashidharan et al., 1997) and EAAT-4 (Yamada et al., 1996; Furuta et al., 1997;Nagao et al., 1997). Immunohistochemical labeling with anti-EAAT-1 and anti-EAAT-2 antibodies was observed throughout the brain, but with variable intensity from one region to another. Thus, EAAT-1 is especially abundant in the molecular layer of the cerebellar cortex (Chaudhry et al., 1995; Lehre et al., 1995; Shibata et al., 1996), whereas the areas containing the highest levels of EAAT-2 are the cerebral cortex, hippocampus, lateral septum, thalamus, striatum, nucleus accumbens, and cerebellum. In these regions, EAAT-1 and -2 are found primarily in the plasma membrane of astrocytes and Bergman glia (in the cerebellum) (Chaudhry et al., 1995; Lehre et al., 1995; Schmitt et al., 1996, 1997; Torp et al., 1997). In addition, EAAT-1 is also present in ependymal cells bordering ventricles (Schmitt et al., 1996,1997; Torp et al., 1997), and EAAT-2 is expressed in subsets of hippocampal and cortical neurons (Schmitt et al., 1996; Torp et al., 1997).
In the brain, EAAT-3 and -4 are present only in neurons (Kanai and Hediger, 1992; Rothstein et al., 1994; Kanai et al., 1995a;Velaz-Faircloth et al., 1996; Yamada et al., 1996; Furuta et al., 1997;Nagao et al., 1997; J. Tanaka et al., 1997; Torp et al., 1997). Interestingly, EAAT-3 is found in both glutamatergic (such as granule cells in the dentate gyrus and pyramidal cells in the hippocampus and cerebral cortex) and GABAergic (such as Purkinje cells in the cerebellum and medium spiny neurons in the striatum) systems. At the ultrastructural level, EAAT-3 immunoreactivity is observed in the plasma membrane of the somas and dendrites of these neurons (Coco et al., 1997). However, a different targeting was noted in the deep cerebellar nuclei because EAAT-3 is locally associated with the axon terminals of GABAergic Purkinje cells (Rothstein et al., 1994; Furuta et al., 1997). In contrast, such a distribution has never been found in case of glutamatergic neurons. Thus, axotomy of glutamatergic pathways (i.e., the corticostriatal and the fimbria-fornix projections) does not decrease EAAT-3 in projection areas (Ginsberg et al., 1995, 1996). Indeed, EAAT-3 is a postsynaptic transporter at glutamatergic synapses.
EAAT-4, the other neuronal SKDGT, is almost entirely restricted to the GABAergic Purkinje cells in the cerebellum. Thus, the levels of EAAT-4 are ∼30-fold lower in the hippocampus than in the cerebellum (Furuta et al., 1997). Interestingly, EAAT-4-immunoreactive material exhibits a parasagittal compartmentation which is perpendicular to the Purkinje cell layer in the cerebellum (Nagao et al., 1997). At the electron microscopic level, EAAT-4 is never associated with axon terminals or Bergman glia, but is exclusively confined to the plasma membrane of Purkinje cell soma and dendritic spines (Yamada et al., 1996; Furuta et al., 1997; Nagao et al., 1997). Indeed, both EAAT-3 and -4 are colocalized in dendritic spines of Purkinje cells (Furuta et al., 1997), whereas EAAT-1 and -2 are present in Bergman glia surrounding these cells. For the Purkinje cells, the two neuronal transporters EAAT-3 and EAAT-4 play a triple role: 1) by taking up glutamate, they decrease the local extracellular concentration of this excitatory aa; 2) their functioning produces Cl−influx (cf. I. Cellular and Subcellular Localization), which leads to local hyperpolarization, thereby preventing excessive excitation by extracellular glutamate (Fig. 7); and 3) by accumulating the latter aa into the cells, they provide the substrate for the neosynthesis of GABA (Furuta et al., 1997).

Schematic diagram illustrating the functional roles of neuronal EAAT. Glutamate released by an excitatory bouton acts at postsynaptic glutamatergic receptors (EAAR); in most cases, this results in postsynaptic membrane depolarization. The removal of glutamate from the synaptic cleft is carried out by the transporter EAAT-4 in the plasma membrane of the postsynaptic neuron. In addition, EAAT-4 also acts as a Cl− channel, leading to local hyperpolarization.
In summary, SKDGTs are strategically distributed to control the extracellular levels of glutamate in brain. Emphasis has to be put on the fact that although excitatory aa uptake by hippocampal glutamatergic terminals has been demonstrated (Gundersen et al., 1993,1995), no “presynaptic” glutamate transporter has yet been isolated to date.
4. Regulation of SKDGT Activity and Expression.
The primary sequences of SKDGTs contain several PKA and PKC consensus phosphorylation sites (Kanai et al., 1993; Gegelashvili et al., 1997). For example, a conserved PKC site is present in the first intracellular loop of EAAT-1 to -5, and one PKA site is found just before the large conserved hydrophobic domain of EAAT-1–4 (Arriza et al., 1997). Indeed, numerous studies showed that glutamate transporters are regulated by protein kinases and phosphatases (Casado et al., 1991,1993).
EAAT-3 (but not EAAT-1 and -2) is endogenously expressed in a subline of glioma cells (C6), where its activity can be stimulated by phorbol-myristate-13-acetate (PMA) but not forskolin (Dowd et al., 1996). This effect is associated with a 2.5-fold increase in itsV max. Furthermore, Davis et al. (1998)have recently established by confocal microscopy that PMA triggers both an increase in the number of EAAT-3 molecules and their clustering at the cell surface. The rapid onset of PMA-induced stimulation of glutamate uptake is compatible with a direct PKC-mediated phosphorylation of EAAT-3. Indeed, this effect is abolished when Ser113 in the EAAT-3 sequence has been mutated into an asparagine residue (Casado et al., 1993).
On the other hand, short-term PKC-dependent down-regulation of EAAT-1/GLAST has been described in transfected HEK-293 cells andXenopus oocytes (Conradt and Stoffel, 1997). Immunofluorescence experiments allowed the demonstration that the resulting PKC-mediated inhibition of glutamate transport is not due to an altered targeting of the transporter at the cell membrane. Although its amplitude is proportional to the amount of32P incorporated into EAAT-1 (Conradt and Stoffel, 1997), PMA-induced inhibition of glutamate transport persists when the three PKC consensus sites of EAAT-1 have all been removed by site-directed mutagenesis (Conradt and Stoffel, 1997). It can therefore be hypothesized that the decreased activity of EAAT-1 upon PKC activation involves other phosphorylated protein(s) possibly interacting with the transporter.
EAAT-1 is the only SKDGT expressed in undifferentiated astrocyte monocultures (Swanson et al., 1997). However, when astrocytes are cultured on a neuronal layer, their morphology changes and they express EAAT-2 in addition to EAAT-1 (Swanson et al., 1997). Indeed, EAAT-2 mRNA and protein are found in astrocytes cultured in media conditioned by cortical neurons (Gegelashvili et al., 1997). These data suggest that soluble factors released by neurons are able to trigger the transcription of the EAAT-2 gene in astrocytes (Gegelashvili et al., 1997). Interestingly, long-term treatment of astrocytes with dibutyryl-cAMP resulted in an increased expression of EAAT-1 and -2 as well as an enhancement ofd-[3H]aspartate uptake (Gegelashvili et al., 1996; Swanson et al., 1997). In addition, glutamate receptor activation by glutamate itself or kainate, but not α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid or trans-(±)-1-amino-1,3-cyclopentane-dicarboxylic acid, also results in an up-regulation of both EAAT-1 expression andd-[3H]aspartate uptake (Gegelashvili et al., 1996).
In summary, phosphorylation by PKC activates EAAT-3 (and also EAAT-2; see Casado et al. (1991, 1993) but inhibits EAAT-1. On the other hand, EAAT-1 and -2 can be up-regulated at the transcriptional level by neuronal soluble factors including glutamate itself (possibly acting at kainate receptors) and dibutyryl cAMP. However, the physiological relevance of this complex pattern of short- and long-term regulation of glutamate reuptake remains to be established.
5. Pharmacological and Functional Aspects.
Biochemical and pharmacological studies performed on brain preparations provided the first evidence in favor of the existence of several subtypes of SKDGTs (Ferkany and Coyle, 1986; Robinson et al., 1993). The pharmacological profile of each subtype could then be precisely determined using cell lines transfected with the corresponding cDNAs (Kanai and Hediger, 1992; Pines et al., 1992; Storck et al., 1992; Arriza et al., 1994,1997; Tanaka, 1994; Fairman et al., 1995; Dowd et al., 1996). However, additional SKDGT subtypes are probably still to be discovered (Dowd et al., 1996). To date, only a few compounds are able to discriminate between the different subtypes. For example, kainate and cysteine inhibit preferentially EAAT-2 and EAAT-3, respectively (Vandenberg et al., 1997). All the drugs acting at the EAAT that are currently available are competitive inhibitors. However, many of them also act at glutamate receptors.
It has long been known that neuronal cells can be destroyed by sustained exposure to glutamate (Olney and Sharpe, 1969). The neurotoxicity of this aa is due to overstimulation of its ion channel receptors leading to excessive intracellular level of Ca2+. Concentrations of glutamate in the micromolar range are needed to stimulate excitatory receptors in the CNS (Fonnum, 1984). Consequently, to prevent neurotoxicity, glutamate levels have to be maintained below this range in the extracellular space. It has often been claimed that the termination of glutamate transmission occurs primarily via the action of SKDGTs. However, as shown in Table 3, the K m values of the human SKDGT range between 30 and 97 μM and are consequently at least 1.5 order of magnitude higher than the resting levels of glutamate in the extracellular space (∼1 μM). Thus, it can be surmised that, because of their high K m values, SKDGTs must also have high V max values to efficiently take up extracellular glutamate. Alternatively, mechanisms other than reuptake might be in charge of the termination of glutamatergic transmission. Indeed, ionotropic EAAT receptors, like all ligand-gated ion channel receptors, desensitize very rapidly. If receptor desensitization is the primary mechanism of glutamate inactivation, then SKDGTs may play a critical role in the resensitization of glutamate receptors.
A large body of evidence supports the idea that elevated concentrations of glutamate in the extracellular space are implicated in the pathophysiology of various neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS), Huntington’s chorea, and Alzheimer’s disease (Appel, 1993; Portera-Caillau et al., 1995; Lesch et al., 1996; Kanai, 1997). Moreover, glutamate has been shown to induce severe damage during trauma, stroke, ischemia, anoxia, and status epilepticus (Benveniste et al., 1984; Sandberg et al., 1986;Sherwin et al., 1988; Storm-Mathisen et al., 1992; During and Spencer, 1993; Attwell and Mobbs, 1994; Szatkowski and Attwell, 1994; Kanai et al., 1995).
A loss of EAAT-2, but not of other SKDGTs, has been reported in Huntington’s chorea (Arzberger et al., 1997), Alzheimer’s disease (Scott et al., 1995; Masliah et al., 1996; Li et al., 1997), and ALS (Rothstein et al., 1992, 1995; Glenn Lin et al., 1998). For example, in postmortem brains of patients who died with Huntington’s chorea, the decrease in EAAT-2 mRNA was shown to be proportional to the severity of the illness (Arzberger et al., 1997). In contrast, EAAT-2 mRNA levels are not altered in postmortem brain and spinal cord of patients suffering from Alzheimer’s disease or ALS. Therefore, the 30 to 90% reduction of the EAAT-2 protein (Rothstein et al., 1992, 1995; Li et al., 1997) in the latter neurodegenerative diseases (as compared to normal controls) probably results from multiple aberrant processing of its mRNAs (Glenn Lin et al., 1998). On the other hand, a direct inhibitory effect of the amyloid precursor protein on EAAT-2 also probably contributes to the reduction of excitatory aa reuptake in Alzheimer’s disease (Keller et al., 1997; Li et al., 1997).
Glutamatergic neurotransmission has also been shown to be implicated in the pathophysiology of epilepsy. Alteration of EAAT has notably been reported in amygdala-kindled rats (Akbar et al., 1997; Prince Miller et al., 1997). In this model, EAAT-1 amounts are decreased (−60%) in the piriform cortex with no change in other limbic areas (Prince Miller et al., 1997). In contrast, EAAT-2 mRNA and protein are unchanged in limbic areas (amygdala, piriform cortex, and hippocampus). Finally, the levels of the neuronal subtype EAAT-3 are augmented in the piriform cortex (+40%) and hippocampus (+28%) of these kindled animals (Prince Miller et al., 1997).
Interestingly, typical and atypical neuroleptics have recently been shown to affect glutamatergic transmission (Meshul et al., 1996). In particular, chronic treatment with clozapine or haloperidol was found to reduce the striatal levels of EAAT-2 mRNA in rats (Schneider et al., 1998). In line with these observations, a deficit in EAA reuptake has been described in the basal ganglia of schizophrenic patients (Simpson et al., 1992). Accordingly, not only neurological disorders but also psychiatric diseases might well be associated with altered functioning of SKDGTs, especially EAAT-2.
Transient down-regulation of EAAT-1, -2, and -3 has been achieved through the chronic i.c.v. infusion of antisense oligonucleotides in rats (Rothstein et al., 1996). Thus, the brain levels of EAAT-1, -2, and -3 could be reduced by −85, −60, and −78%, respectively (Rothstein et al., 1996). Interestingly, the down-regulation of both astroglial transporters, EAAT-1 and -2, was found to be associated with a dramatic increase in the extracellular concentration of glutamate, by 13- and 32-fold, respectively. In contrast, an almost complete loss of EAAT-3 had no affect on the extracellular levels of glutamate (Rothstein et al., 1996). That EAAT-2 plays a key role in glutamate reuptake has been additionally evidenced by Tanaka et al. (1997) who showed that the knockout of its encoding gene results in a 94% decrease in this process. In contrast, no changes in glutamate levels and uptake could be detected in knockout mice that do not express EAAT-3 (Peghini et al., 1997). Altogether, these data led to the following order of SKDGT efficiency for the clearance of glutamate in brain: EAAT-2 ≫ EAAT-1 ≫ EAAT-3. Neuronal degeneration and epilepsy are observed in mice with deficits in the glial transporters, EAAT-1 and -2, but not in those with low levels of EAAT-3 (Rothstein et al., 1996; Peghini et al., 1997; Tanaka et al., 1997). Nevertheless, behavioral studies showed that the loss of EAAT-1, -2, and -3 results in locomotor impairment (Rothstein et al., 1996; Peghini et al., 1997;Tanaka et al., 1997).
In conclusion, EAAT-2 seems to play a pivotal role in the control of glutamatergic transmission and in neurodegenerative processes. The physiological relevance of EAAT-1 and -3 seems to be more subtle and remains to be further evaluated. Because null-mutant mice have proven to be valuable models for the assessment of the respective roles of EAAT-2 and -3 in EAA neurotransmission, the knockout of EAAT-1 and -4 are also eagerly expected to gain clear-cut information on the physiological implications of these transporters.
III. Vesicular Neurotransmitter Transporters (VNTs)
Neurotransmitters are synthesized in neurons, where they are concentrated in vesicles for their subsequent Ca2+-dependent release (see Fig. 1). Vesicular transport has been demonstrated for several classical neurotransmitters including acetylcholine (Toll and Howard, 1980), monoamines (Njus et al., 1986), glutamate (Disbrow et al., 1982; Shioi et al., 1989; Tabb et al., 1992), GABA (Fykse and Fonnum, 1988), glycine (Kish et al., 1989; Burger et al., 1991), and ATP (Luqmani, 1981). The accumulation of intraneuronal neurotransmitters into storage vesicles acts as an amplification step for the overall process of Na+-dependent uptake of these molecules from the extracellular space, since it controls their concentration gradient across the plasma membrane. Moreover, vesicular accumulation of neurotransmitters protects these molecules from leakage and/or intraneuronal metabolism. Finally, this storage process also prevents the possible toxic effects of neurotransmitters that could occur when their cytoplasmic concentration exceeds a critical level.
A. Cloning of VNTs
1. The Vesicular Monoamine Transporter (VMAT)/Vesicular Acetylcholine Transporter (VAChT) Family.
In 1992, two groups successfully isolated cDNAs encoding the vesicular monoamine (serotonin, DA, norepinephrine, epinephrine, and histamine) transporters (VMATs, see Fig.8) in the rat. On the one hand, Erickson et al. (1992) isolated a cDNA from mRNA of rat basophilic leukemia cells. This cDNA was shown to promote 5-HT vesicular accumulation in transfected and permeabilized CV-1 cells (Erickson et al., 1992). On the other hand, Y. Liu et al. (1992) used an expression cloning strategy based on the observation that PC12 cells (derived from pheochromocytoma) are resistant to the neurotoxic agent MPP+. Thus, CHO cells were transfected with PC12 cDNA library and MPP+-resistant clones were isolated. In fact, resistance to MPP+ could be attributed to the vesicular accumulation of this agent, notably because reserpine, a potent inhibitor of vesicular transport, restored its toxicity in transfected CHO cells (Y. Liu et al., 1992).

Alignment of the aa sequences of the vesicular transporters. Vesicular transporters are rVMAT-1 (Liu et al., 1992b), rVMAT-2 (Erickson et al., 1992; Liu et al., 1992b), and rVAChT (Erickson et al., 1994; Roghani et al., 1994). Putative membrane spanning domains (I-XII) are indicated by bars. Conserved residues are shaded.
The two clones were initially named CGAT, for chromaffin granule amine transporter (Y. Liu et al., 1992), and SVAT or MAT for synaptic vesicle amine transporter or monoamine transporter (Erickson et al., 1992; Y.Liu et al., 1992). Then, because SVAT expression appeared not to be restricted to synaptic vesicles, a new nomenclature was adopted: CGAT was renamed VMAT-1, and SVAT/MAT, VMAT-2 (Fig. 8; Table4).
Mammalian vesicular neurotransmitter transporter family
Using a homology cloning strategy, VMAT-1 and VMAT-2 were then successfully isolated in bovine (Krejci et al., 1993; Howell et al., 1994) and humans (Erickson and Eiden, 1993; Lesch et al., 1993; Surratt et al., 1993; Erickson et al., 1996) (see Table 4). VMAT-1 is primarily present in endocrine and paracrine cells of peripheral organs; on the other hand, VMAT-2 is the predominant monoamine vesicular transporter in the CNS, and it is also found in histaminergic cells of the stomach, adrenal medulla, and blood cells (Mahata et al., 1993; Peter et al., 1995; Weihe et al., 1995; Erickson et al., 1996). Finally, it has to be noted that VMAT-1 and -2 are mutually exclusive except in human and rat chromaffin cells where they are coexpressed.
Hydropathic analysis of the primary sequences of all these clones predicts 12 putative transmembrane segments (Fig.9). In addition, a large hydrophilic loop with three to five potential sites of N-linked glycosylation probably exists between transmembrane domains 1 and 2. This loop has been proposed to be inside the lumen of the vesicle, whereas both the N and C termini should be in the cytoplasm. Major sequence divergences between the different clones occur at this large luminal loop and at the N- and C-terminal domains (Fig. 9). Interestingly, VMATs show some sequence homology with several drug resistance H+antiporters in bacteria (Schuldiner, 1994).

Hypothetical topologies of the vesicular transporters VMAT-1–2/VAChT A and rVIAAT B. The predictions of secondary structures propose 12 α-helical membrane-spanning domains for the VMAT-1 and -2 and the VAChT transporters (A). In contrast, only 10 α-helical membrane-spanning domains can be inferred from the aa sequence of rVIAAT (B), further supporting the idea that it belongs to another VNT family.
Extensive pharmacological studies have shown that both VMAT isoforms have rather similar properties (except for tetrabenazine binding to the human proteins, Erickson et al., 1996). However, characterization of a VMAT-1/VMAT-2 chimera revealed that the domains located between TM6 and TM11 could account for the better affinity of VMAT-2 than VMAT-1 for histamine (Erickson, 1998). Furthermore, TM1 and TM11 to TM12 appeared to contain residues critical for the specific binding of tetrabenazine (Peter et al., 1996; Erickson, 1998). The same conclusion was reached independently by microsequencing proteolytic peptides from VMAT-2 covalently modified by photoactivable tetrabenazine or ketanserin derivatives (Sagné et al., 1997b; Sievert and Ruoho, 1997).
Site-directed mutagenesis also contributed to the identification of residues involved in VMAT functioning. Thus, Asp33 in TM1 and the triplicate Ser180, 181, and 182 in TM3 are probably implicated in substrate recognition (Merickel et al., 1995). Asp404 in TM10 and Asp431 in TM11 probably bind protons (Steiner-Mordoch et al., 1996), and His419 in the fifth cytosolic loop appears to participate in the energetic coupling of the antiport (Shirvan et al., 1994). Recently,Finn and Edwards (1997) identified several residues between TM9 and TM12 which are required for the high-affinity interactions of VMAT-2 with multiple ligands and also account for the preferential recognition of serotonin over DA by this transporter.
Successful cloning of the vesicular transporter for acetylcholine, VAChT, initially derived from studies in invertebrates. In a first step, homology screening with a probe from unc-17, a gene responsible for the integrity of neuromuscular function in the nematodeCaenorhabditis elegans (Alfonso et al., 1993), allowed the isolation of DNA clones from Torpedo marmorata andTorpedo ocellata (Varoqui et al., 1994). The homology of these clones with VMAT-1 and VMAT-2 (43% identity) led to their classification in the same gene family (Fig. 8). Indeed, the corresponding proteins expressed in fibroblasts were able to bind vesamicol and to transport acetylcholine (Varoqui et al., 1994). Mammalian VAChTs (human and rat) were subsequently isolated (Erickson et al., 1994; Varoqui and Erickson, 1996) (see Table 4).
2. Vesicular Inhibitory Amino Acid Transporter.
Several genes have been identified in the nematode C. elegans, the mutations of which cause defects in GABAergic transmission (McIntire et al., 1993). In particular, the defect observed in unc-47mutant suggested that it was due to a loss of GABA transport into synaptic vesicles. Subsequent studies based on this hypothesis actually led to the isolation of the vesicular GABA (and glycine) transporter (McIntire et al., 1997; Sagné et al., 1997a).
McIntire et al. (1997) successfully rescued the unc-47mutant phenotype with injection of cosmids having the unc-47gene. The rescuing activity was precisely localized, and a cDNA encoding the vesicular GABA uptake could be isolated. The rat cDNA homolog, designated VGAT, was then used for the transfection of PC12 cells. Uptake measurement using vesicles isolated from these cells demonstrated that VGAT is endowed with the capacity for accumulating GABA (McIntire et al., 1997).
On the other hand, Sagné et al. (1997a) used another strategy based on the screening of genome databases. They mapped the region spanning the unc-47 gene and selected a cDNA possibly encoding the C. elegans vesicular GABA/glycine transporter. Indeed, the selected sequence corresponded to a protein with the expected secondary structure (i.e., more than six putative transmembrane domains) and size. This candidate was used for homology screening in mouse and human genome expressed sequence tag databases, and the putative mouse vesicular GABA/glycine transporter could then be isolated from corresponding expressed sequence tag clones. Indeed, in situ hybridization and expression experiments subsequently demonstrated that this transporter actually corresponds to the vesicular GABA/glycine transporter in the mouse brain (Sagné et al., 1997a).
Comparison of the respective sequences of vesicular GABA transporter (VGAT) and vesicular inhibitory aa transporter (VIAAT) that were cloned independently by McIntire et al. (1997) and Sagné et al. (1997a), respectively, clearly showed that they correspond to the same protein.
Further comparison of VGAT/VIAAT with VMAT and VAChT allowed the conclusion that they did not belong to the same gene family (McIntire et al., 1997; Sagné et al., 1997a). Indeed, VGAT/VIAAT is the first member of a new neurotransmitter transporter family. Its putative structure predicts 10 TMs, a long NH2- and a short COOH-intracytoplasmic termini, and a large intraluminal loop between TM1 and TM2 (Fig. 9B).
B. Regulation of Transport
VNT expression and activity can be controlled by both long- and short-term regulatory processes, as shown notably in studies on VMAT-2 in chromaffin cells which endogenously express this transporter (Desnos et al., 1992, 1995; Krejci et al., 1993; Mahata et al., 1993; Laslop et al., 1994). Thus, chronic stimulation (for several days) of these cells by a high extracellular concentration of K+ was shown to produce a marked increase in VMAT-2 synthesis, as assessed by measurement of [3H]tetrabenazine-specific binding (Desnos et al., 1992, 1995). This suggests that a functional link exists among cell stimulation, catecholamine secretion, and the synthesis of VMAT-2 in chromaffin cells (Krejci et al., 1993; Desnos et al., 1995).
Numerous consensus sites for protein kinases (PKA and PKC) are present in intracytoplasmic portions of VMAT-1 and -2, suggesting that second messengers are able to play a role in post-translational regulation of VMAT activity. Relevant investigations performed on PC12 cells have shown that activation of the cAMP pathway leads to a decrease in vesicular monoamine uptake (Nakanishi et al., 1995). Additonal studies on transfected cells (CHO, PC12, and COS) demonstrated that VMAT-2 is constitutively phosphorylated at two consensus sites (Ser512 and 514) for casein kinase II (Krantz et al., 1997). On the other hand, no phosphorylation of VMAT-1 could be detected, suggesting the existence of different regulatory mechanisms for the two subtypes (Krantz et al., 1997). Finally, it has to be emphasized that directed mutagenesis of Thr154 in rat VMAT (rVMAT)-2, a potential site for phosphorylation by PKC, does not affect monoamine transport function (Merickel et al., 1995). Indeed, whether PKA- and/or PKC-mediated modulations of VMATs transport function result from the phosphorylation of the transporters themselves or of other interacting protein(s) is still an unsolved question.
The VAChT gene is contained within the first intron of the gene encoding choline acetyltransferase (ChAT, the key enzyme for acetylcholine synthesis). Moreover, these two genes are transcribed in the same orientation (Bejanin et al., 1994; Erickson et al., 1994). Several factors (nerve growth factor, retinoic acid, trophic factors, cholinergic differentiation factor, cAMP) were initially reported to affect the levels of ChAT mRNA (Usdin et al., 1995), and it was of interest to examine whether they could also influence VAChT transcription. Indeed, coregulation of both ChAT andVAChT gene transcription by retinoic acid and differentiation factor/leukemia inhibitory factor was actually demonstrated in cultured sympathetic neurons and septal cells (Berrard et al., 1995; Berse and Blusztajn, 1995).
Thus, the VNTs appear to be regulated at the transcriptional, translational, and post-translational levels in the various cell types where they are expressed.
C. Ionic Dependence and Electrogenic Properties
The vesicular transport system consists of two components: an ATP-driven H+ pump that, on the one hand, acidifies the organelle lumen (ΔpH) and, on the other hand, generates a potential gradient (Δψ), and a transporter that exchanges internal H+ ions with a given substrate (Schuldiner et al., 1978; Johnson and Scarpa, 1979). Studies on the electrochemical components of the vesicular transport allowed stoichiometric calculations and showed that the steady-state monoamine concentration gradient depends both on Δψ and on 2 × ΔpH (Njus et al., 1986; Rottenberg, 1986; Johnson, 1988; Nguyen et al., 1998). In particular, these investigations clearly established that the monoamine and the acetylcholine vesicular transporters exchange two protons with one molecule of respective substrate. Interestingly, nigericin, which exchanges K+ for H+, reduced VMAT-2 activity by 65% but rVGAT activity by only 40% (McIntire et al., 1997). This observation supports the idea that the transport activity of the three VNTs is differentially sensitive to the ΔpH and/or the Δψ. In particular, VMAT-2 appears to be more dependent than VGAT on ΔpH (McIntire et al., 1997).
D. Pharmacological Properties
It is well established that VMAT-1 and -2 recognize each monoamine (5-HT, adrenaline, DA, histamine, noradrenaline) with high affinity (Johnson, 1988; Erickson et al., 1992, 1996; Peter et al., 1994;Merickel and Edwards, 1995). However, VMAT-2 has a higher affinity than VMAT-1 for some monoamine substrates, notably histamine (Merickel and Edwards, 1995). In addition, VMAT-2 is approximately 10-fold more sensitive to the inhibitor tetrabenazine than VMAT-1. Metamphetamine also inhibits preferentially VMAT-2, apparently by competing at the site of amine recognition (Peter et al., 1994; Erickson et al., 1996). However, reserpine has a similar affinity for both VMATs (Peter et al., 1994; Erickson et al., 1996).
Numerous acetylcholine (ACh) derivatives are recognized by VAChT (Parsons et al., 1993), some of which being actively transported, just as well as ACh itself (Clarkson et al., 1992). Vesamicol has been identified as a selective high-affinity inhibitor of the vesicular accumulation of ACh (Erickson et al., 1994; Varoqui and Erickson, 1996).
Experiments using vesicular fractions from PC12 cells stably transfected with the rVGAT-encoding sequence showed that γ-vinyl-GABA inhibits the vesicular GABA transport but with low potency (K m = 5 mM) (McIntire et al., 1997). Thus, high-affinity-selective VGAT inhibitors are eagerly expected.
Studies with native brain synaptic vesicles suggested that a common transporter is responsible for the vesicular accumulation of both GABA and glycine (Christensen and Fonnum, 1991). Direct demonstration of this hypothesis was recently provided by Sagné et al. (1997a), who showed that COS-7 cells transfected by the VIAAT-encoding sequence actively accumulated both aas, and that GABA and glycine interacted competitively with the same transport mechanism. These findings accounted for the name chosen for this vesicular transporter: VIAAT.
E. Cellular and Subcellular Localization
VMATs and VAChTs are selective markers of monoaminergic and cholinergic neurons, respectively (Schäfer et al., 1994; McIntire et al., 1997; Nirenberg et al., 1997; Sagné et al., 1997a). Thus,Efange et al. (1997) proposed the use of selective VAChT ligands as probes for assessing the loss of cholinergic projections in Alzheimer-type dementias.
The localization of these transporters on the secretory vesicles in axon terminals determines their prime role, i.e., to accumulate the neurotransmitter for subsequent quantal release at the synapse. VAChT is localized in small, clear synaptic vesicles of axon terminals that make symmetric contacts with dendrites (Gilmor et al., 1996). VMAT-2 is also expressed in small synaptic vesicles, but its main location in axon terminals is on large dense core vesicles (Nirenberg et al., 1997). This peculiar subcellular targeting of VMAT-2 addresses the question of the role of large dense core vesicles in the storage and release processes for classical neurotransmitters such as monoamines.
Somatic and dendritic expressions have also been observed for both VAChT and VMAT-2. The somatic localization probably represents newly synthesized proteins associated with the Golgi complex and the endoplasmic reticulum (Nirenberg et al., 1995; Gilmor et al., 1996). At the level of dendrites, expression of VMAT-2 and VAChT supports the proposal that DA and ACh can be stored in and released from these neurites (Arvidsson et al., 1997; Nirenberg et al., 1997).
In situ hybridization experiments showed that VIAAT/VGAT is synthesized in GABAergic neurons (McIntire et al., 1997; Sagné et al., 1997a). Furthermore, Sagné et al. (1997a) provided evidence that, in some cells, VIAAT can be coexpressed with GLYT2, a specific marker of glycinergic neurons. These data further support the idea that VIAAT/VGAT is a neuronal vesicular transporter for both GABA and glycine. Indeed, the protein has been observed in GABAergic as well as glycinergic terminal boutons (Chaudhry et al., 1998; Dumoulin et al., 1999). At the ultrastructural level, this VIAAT/VGAT is restricted to small synaptic vesicles. Because this transporter: 1) can accumulate glycine and GABA in synaptic vesicles and 2) is present in both types of nerve endings, the name VIAAT should be adopted. Interestingly, some GABAergic as well as glycinergic terminals are devoid of immunoreactive VIAAT/VGAT (Chaudhry et al., 1998; Dumoulin et al., 1999). This suggests that other member(s) of this vesicular transporter subfamily are still to be cloned.
F. Dysfunctioning—Models for Neurodegenerative Disorders or Drug Abuse
Vesicular sequestering seems to be crucial for protecting neurons from potential toxic effects of neurotransmitters and/or intraneuronal metabolites. In this context, possible implications of vesicular transporters in pathogenic mechanisms have been the subject of numerous studies.
Reserpine, an inhibitor of vesicular amine transport, has been used in the treatment of hypertension because it potently reduces blood pressure. However, high dosages frequently produce a disabling effect which resembles that observed in depressed patients (Frize, 1954). Accordingly, VMATs may also be implicated in psychiatric disorders. Indeed, the hVMAT-2 gene is localized in the vicinity of chromosomal breaks that have been identified in some mentally ill patients with cutis verticis gyrata (Surratt et al., 1993).
MPP+ is a substrate for both the plasmic DA transporter and VMATs. This neurotoxin has been used in numerous studies aimed at generating relevant animal models of parkinsonism (Y.Liu et al., 1992, Liu et al., 1994; Stern-Bach et al., 1992; Edwards, 1993). In light of the neuroprotective effect resulting from the sequestration of MPP+ in synaptic vesicles, it can be hypothesized that alterations in the activity of VMAT-2 may contribute to the peculiar vulnerability of nigrostriatal dopaminergic neurons in case of drug-induced and/or idiopathic Parkinson’s disease (Tanner and Langston, 1990). Attempts to enhance the vesicular accumulation of DA and/or neurotoxic compounds may well be of therapeutic interest for (early-stage) parkinsonian patients.
Mutant mice lacking VMAT-2 have recently been of great help to further assess the physiological importance of this protein (Fon et al., 1997;Takahashi et al., 1997; Wang et al., 1997). Indeed, VMAT-2 (−/−) animals exhibit marked deficits in locomotor activity and feeding behavior, and die shortly after birth. However, their brains present no obvious morphological changes, and monoaminergic pathways do not seem to be altered by the mutation (Fon et al., 1997). Nevertheless, biochemical measurements showed marked reductions in the levels of monoamines in hetero- and homozygous mutants (Fon et al., 1997;Takahashi et al., 1997; Wang et al., 1997). In addition, the catalytic activity of tyrosine and tryptophan hydroxylases is augmented in homozygous animals (Wang et al., 1997), possibly as a compensatory adaptation to the accelerated degradation of monoamines. Interestingly, behavioral studies showed that the increased locomotor activity evoked by acute treatment with amphetamine, cocaine, and apomorphine was significantly higher in heterozygous VMAT-2(±) mice than in wild-type animals (Fon et al., 1997; Takahashi et al., 1997; Wang et al., 1997). A larger release of DA through both the blockade of vesicular monoamine transport and the reverse functioning of the plasma membrane transporter (DAT) by amphetamine probably accounted for the differences in the behavioral response to this drug in the mutant versus the wild-type mice. In line with this interpretation, Fon et al. (1997)reported that amphetamine was more efficient to trigger DA release in primary cultures of midbrain dopaminergic neurones from knockout mice than from wild-type animals. On the other hand, the increased behavioral response to apomorphine is in favor of the presence of supersensitive DA receptors in heterozygous VMAT-2(±) mice. However, direct investigations of D1- and D2-dopaminergic receptors, as well as of α1A- and β-adrenergic receptors and 5-HT1A and 5-HT2serotoninergic receptors revealed no changes in the mutant mice as compared to wild-type animals (Takahashi et al., 1997).
With regard to cholinergic neurons, the coordinate regulation of the genes encoding VAChT and ChAT (Berrard et al., 1995) may play a key role, especially during development in the central and peripheral nervous systems. In addition, functional alterations of cholinergic neurotransmission in Alzheimer-type dementias might also concern both of these genes and their regulation.
IV. Conclusion
Recent progress in molecular cloning and the elucidation of carriers’ structures allowed the identification of several gene families encoding neurotransmitter transporters. In particular, two families of plasma, as well as two families of vesicular, membrane neurotransmitter transporter genes were isolated. These proteins are Na+/Cl−-, Na+/K+-, or H+-dependent. Their pharmacological profiles, regulatory properties, regional and cellular localizations, and implications in neuropathologies begin to be elucidated but a lot of questions still have to be addressed. Clearly, the knowledge of the precise ultrastructural localization of these transporters should contribute to the understanding of their implications in the physiological and pathophysiological functioning of specific synapses. On the other hand, addition, alteration, or deletion of genes encoding the transporters allow the generation of animal models that are of considerable interest for assessing the actual role of these proteins, notably in relevant psychiatric and neurological diseases.
Exploration of neurotransmitter transporters as cell-specific markers should also be a key for progressing in the knowledge of brain functioning. At present, antibodies specific of neurotransmitter synthesizing enzymes, such as tyrosine hydroxylase for catecholaminergic neurons and glutamic acid decarboxylase for GABAergic neurons, are generally used as markers of specific neuronal populations. However, neurons using excitatory aas as neurotransmitters cannot be specifically labeled by such probes. Antibodies raised against transporter(s) selectively expressed in these neurons should constitute alternative tools for their specific labeling and visualization.
Acknowledgments
We thank C. Sais for excellent secretarial assistance. We are also extremely grateful to Dr. S Supplisson for expert scientific advice and support.
Footnotes
-
↵1 Address for correspondence: S. El Mestikawy, INSERM U288, Faculté de Médecine Pitié-Salpêtrière, 91 Boulevard de l’Hôpital, 75634 Paris Cedex 13, France. E mail: elmesti{at}ext.jussieu.fr
Abbreviations
- CNS
- central nervous system
- GABA
- γ-aminobutyric acid
- SCDNT
- Na+/Cl−-dependent neurotransmitter transporter
- GAT
- GABA transporter
- ALS
- amyotrophic lateral sclerosis
- aa
- amino acid
- 5-HT
- 5-hydroxytryptamine, serotonin
- CHO
- Chinese hamster ovary
- ChAT
- choline acetyltransferase
- ACh
- acetylcholine
- PKC
- protein kinase C
- PKA
- protein kinase A
- DAT
- dopamine transporter
- DA
- dopamine
- EAAT
- excitatory amino acid transporter
- GLYT
- glycine transporter
- PMA
- phorbol 12-myristate 13-acetate
- SERT
- serotonin transporter
- TM
- α-helical transmembrane domain
- PROT
- proline transporter
- SKDGT
- Na+/K+-dependent glutamate transporter
- VAChT
- vesicular acetylcholine transporter
- VGAT
- vesicular GABA transporter
- VIAAT
- vesicular inhibitory amino acid transporter
- VMAT
- vesicular monoamine transporter
- VNT
- vesicular neurotransmitter transporter
- rVMAT
- rat VMAT
- MPP+
- 1-methyl-4-phenylpyridinium
- NET
- norepinephrine transporter
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
{kind=link}
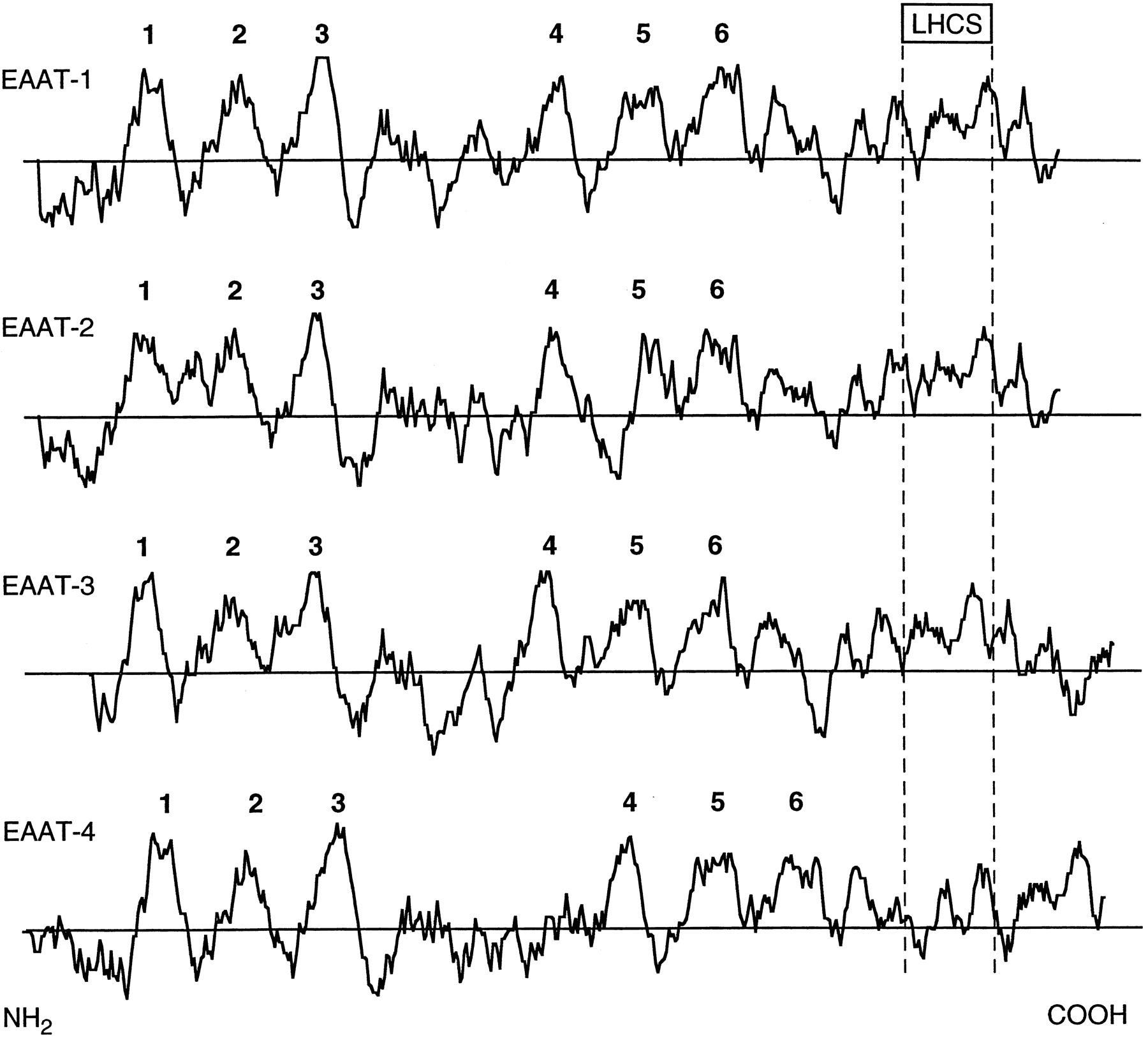{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
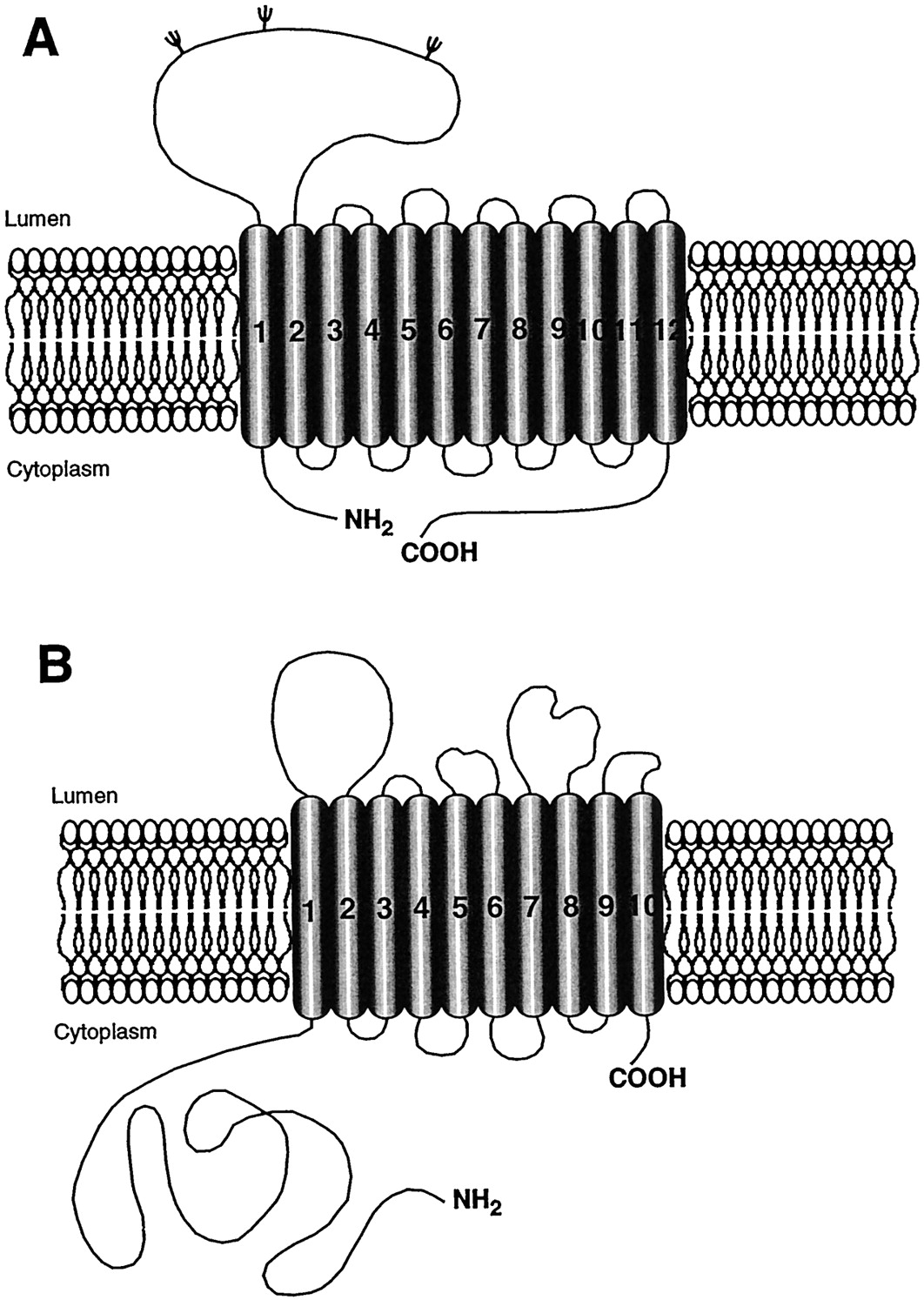{kind=link}