

Abstract
Protease-mediated signaling is an important modulator of the nervous system. However, identifying the specific signaling substrates of such proteases is limited by the rapidity with which intermediate substrate forms are cleaved and released. Here, a screening method to detect noncleaved enzyme-bound forms was developed and used to identify a novel neuropsin/neuregulin-1 (NRG-1) proteolytic signaling system, which is specifically localized in the microdomain of synaptic cleft, in the mouse hippocampus. The extracellular protease, neuropsin, cleaved mature NRG-1 (comprising the extracellular domain of the NRG-1) at three newly identified sites to remove the heparin-binding domain of NRG-1. This released the ligand moiety from the matrix-glycosaminoglycan pool and enabled it to trigger the phosphorylation of NRG-1 receptor, p185 (ErbB4). Proteolysis of mature NRG-1 by neuropsin led to colocalization of the processed NRG-1 with ErbB4 in parvalbumin-positive hippocampal interneurons and consequent phosphorylation of tyrosine residues of proteins in the cells. Moreover, neuropsin knock-out mice exhibited impairments in Schaffer collateral early phase long-term potentiation, and application of the recombinant NRG-1 lacking heparin-binding activity reversed the effects through the activation of ErbB4 and GABAA receptors. Thus, ErbB4 signaling induced by neuropsin-dependent processing of NRG-1 contributes to the modulation of synaptic plasticity via regulation of GABAergic transmission. This signaling system may be involved in human cognition and mental disorders, such as schizophrenia and bipolar disorder, by its dysfunction.
Introduction
Neural activity-dependent processing of extracellular proteins, particularly in the synaptic cleft, is of pivotal importance for the function of molecules such as neurotrophic factors, matrix proteins, cell adhesion molecules, and receptor extracellular domains, and has implications for cell migration, neurite outgrowth, synaptogenesis, neural plasticity, and neurite retraction (Lu, 2003; Reiss and Saftig, 2009; Dityatev et al., 2010; Shiosaka and Ishikawa, 2011). However, the target molecules for proteases remain to be identified, primarily due to difficulties in determining specific substrate-enzyme interactions (López-Otín and Overall, 2002). Enzyme-bound intermediate forms of substrates are cleaved and released by the enzyme too quickly to be isolated as noncleaved Michaelis–Menten intermediate complexes.
Neuropsin (also referred to as KLK8) is a kallikrein-related protease that displays extracellular serine protease activity and is expressed focally in the limbic system including the hippocampus, amygdala, prefrontal cortex, septum, and Mynert nucleus (Chen et al., 1995). Human neuropsin is thought to influence cognitive brain function, as single nucleotide polymorphisms (SNPs) are associated with attention/concentration and verbal IQ disorders and with bipolar disorder (Izumi et al., 2008). In animal experiments, neuropsin gene deficiency causes severe impairments in the early phase (E-) of long-term potentiation (LTP), in late-associativity (comprising processes of cross-synaptic interventions within several minutes after induction late phase of LTP), and in spatial working memory (Tamura et al., 2006; Ishikawa et al., 2008). However, the downstream signaling mechanism underlying neuropsin that is responsible for synaptic plasticity is still poorly understood.
Because efforts to identify protease substrates are limited by nonpersistent Michaelis–Menten intermediate complex formation during the rapid proteolysis and release reaction, fixing such intermediates in bound form may permit the identification of specific neuropsin substrates. Here, we developed a new unbiased screening method for substrates (including protease inhibitors) to detect noncleaved stable forms. The strong binding of noncleaved mutant proteases enabled the screening of candidate substrates using a proteomics approach. Applying this novel approach to mouse hippocampal tissue, we identified several substrate molecules that are directly cleaved by neuropsin. Serine protease inhibitors were also observed as specific bound molecules. The present study focused predominantly on neuregulin-1 (NRG-1), a receptor tyrosine kinase ligand, which was identified as a major modulator of neuropsin-dependent E-LTP. Perturbation of this novel neuropsin-NRG-1 signaling system may contribute to symptoms associated with certain psychiatric disorders, particularly working memory defects.
Materials and Methods
Animals.
A total of 36 pregnant ICR female mice (SLC), 63 male C57BL/6J mice (3–8 weeks old; SLC), and 101 neuropsin knock-out mice and corresponding wild-type C57BL/6J mice (3–4 weeks old) were used in the present study. Neuropsin knock-out mice were produced as previously described (Hirata et al., 2001). Mice were maintained under a 12 h light/dark cycle and given food and water ad libitum. Animal husbandry and all aspects of experimental manipulation of animals were in accordance with National Institutes of Health guidelines and approved by the Nara Institute of Science and Technology Animal Care and Use Committee. All efforts were made to minimize animal suffering and the number of animals used.
Plasmid construction.
All constructs were produced using standard methods and confirmed by DNA sequencing (Oka et al., 2002). To obtain mutants disrupted in the protease-active pocket, Val was substituted for Asp206 at the S1-specific pocket (D206V) and Ala for Ser212 in the catalytic triad (S212A). Ser was substituted for Cys208 (SS6) to disrupt the disulfide bond (C208S). Ser87-Leu-Gln-Ser-Arg-Asp-Gln-Pro was deleted to disrupt loop C (ΔS87LQSRDQP). NcoI-NotI fragments of full-length preproneuropsin and preproneuropsin mutant cDNAs were amplified from pED1-neuropsin (Oka et al., 2002) by PCR using the forward primer 5′-CCA CCA TGG GAC GCC CCC CAC-3′ and reverse primer 5′-GGT GCG GCC GCG TCC CTG TTG TCC ATT GTC-3′ and introduced into the pGEM-T Easy vector (Promega). The numerals in the clone names indicate the amino acid position, counting from the start codon. preneuropsins were created by PCR using the forward primer 5′-ATC CTG GAA GGT CGA GAG TGT ATA-3′ and reverse primer 5′-AGC TCT GGT GAG CCC TGC CCA CGC-3′, with pGEM-T-preproneuropsin as the template. Mutant preneuropsins were prepared by replacing the ScaI79Y insert fragment of pGEM-T-preneuropsin with those of pGEM-T-prepromutants. The EcoRI-NotI fragments of all neuropsins inserted into the pGEM-T Easy vector were introduced into the EcoRI-NotI site of pEF1-Myc-His vector (Invitrogen) containing the human elongation factor 1α-subunit promoter. preproneuropsin-, preneuropsin-, and preneuropsinC208S-Myc-His were cloned into the adenoviral expression vector pLP-Adeno-X-CMV-E3-ZsGreen1 (Clontech).
Mouse Nrg-1 was amplified from mouse brain first-strand cDNA (Genostaff) using primers 5′-ATG TCT GAG CGC AAA GAA GG-3′ and 5′-TAC AGC AAT AGG GTC TTG GTT AG-3′ and cloned into pEGFP-N1 (Clontech). Mutations were made in each target sequence using overlapping PCR. The mutated Nrg-1 target sequences were as follows: R18A (5′-GAC GCG GGA TCC CGC GGG AAG CC-3′), R67A (5′-GCA TTC AAA TGG TTC AAG AAC GGG AAT GAG C-3′) and R98A (5′-GCA ATC AAC AAA GCG TCC CTG GCT G-3′).
Primary hippocampal neuron cultures.
Mouse hippocampi were dissected from embryos at embryonic day 17–18 and treated with papain (90 U; Worthington), l-cysteine (1 mm; Nacalai Tesque), 0.02% bovine serum albumin (BSA; Sigma-Aldrich), glucose (28 mm; Wako), and 0.01% DNase- (Sigma-Aldrich) in 0.1 m PBS, pH 7.4, for 30 min at 37°C. Neurons were then triturated and suspended in Neurobasal medium (Invitrogen) supplemented with 2% B27 (Invitrogen) and GlutaMAX (Invitrogen). The cell suspension was poured through a 70 μm cell strainer (BD Biosciences) into a 50 ml tube. For immunostaining, neurons were plated on 13 mm coverslips (Matsunami) coated with poly-l-lysine (0.1 mg/ml; Sigma-Aldrich) in flat-bottomed 24-well tissue culture plates at a density of 1 × 105 cells per coverslip. For immunoblot analysis, neurons were seeded on poly-l-lysine-coated 24-well plates at high density (4–5 × 105/well). Neuron cultures were incubated at 37°C in a humidified atmosphere in the presence of 5% CO2, 95% air. Transient transfection of hippocampal neurons was performed at day 9 in vitro using Lipofectamine 2000 (Invitrogen).
Surface immunocytochemistry.
Transfected neurons were incubated at 4°C for 1 h in the presence of rabbit anti-neuropsin polyclonal antibody (1:1000; a gift from Dr. H. Castro, Universidade Federal Fluminense, Brazil). The neurons were then washed with ice-cold PBS to remove the unbound antibody, fixed for 15 min with 4% paraformaldehyde/4% sucrose, and then incubated with an anti-rabbit secondary antibody coupled to Alexa Fluor 546 (1:1000; Invitrogen) for 1.5 h under nonpermeabilized conditions. For immunostaining using anti-PSD-95 antibody or anti-NRG-1 antibody, the neurons were preincubated in a solution of PBS containing 5% BSA and 0.3% Triton X-100 for 1 h at room temperature and incubated overnight with mouse anti-PSD-95 antibody (1:2500; Millipore Biotechnology) or rabbit polyclonal NRG-1 N-terminal extracellular domain (N-NRG-1) antibody (1:50; Santa Cruz Biotechnology). Immunoreactivity was detected with an Alexa Fluor 647-coupled anti-mouse (1:1000; Invitrogen) or rabbit (1:1000; Invitrogen) secondary antibody. Neurons were examined with a Zeiss confocal microscope. The confocal micrographs shown here are digital composites of a Z-series scan of 3–4 optical sections through a depth of 0.5 μm. Final images were constructed with Zeiss software (ZEN 2009). Quantification of the neuropsin-immunoreactive signal or neuropsin colocalized with NRG-1 was measured by ImageJ software (National Institutes of Health). Coexistence of neuropsin and NRG-1 in transfected neurons is expressed as the ratio between the mean of neuropsin and NRG-1 colocalized area and the mean neuropsin area ± SEM as percentages.
Detection of high-molecular-weight complex.
Cell lysates (1% detergent-solubilized) were prepared in native PAGE sample buffer (4×) (Invitrogen), 1% protease inhibitor cocktail (Sigma-Aldrich), 1 U benzonase, 2 mm MgCl2, and NativePAGE 5% G-250 Sample Additive (Invitrogen) and subjected to 3–12% BlueNative gel analysis and immunoblotting according to the manufacturer's protocol (Invitrogen). Protein concentrations were determined using the BCA Protein Assay (Thermo Fisher Scientific). After electrophoresis, the gel was transferred to a polyvinylidene difluoride (PVDF; Bio-Rad) membrane and then incubated in 8% acetic acid for 15 min to fix the proteins. The membrane was then rinsed with deionized water and air-dried. The membrane was placed in Tris-buffered saline blocking solution containing 5% skim milk and 0.1% Tween 20 for 30 min and then incubated with mouse monoclonal anti-Myc antibody (9B11; 1:2500, Cell Signaling Technology) or rabbit polyclonal anti-neuropsin antibody (1:7000) for 1 h at room temperature. The membrane was incubated with donkey anti-mouse (1:40,000; Jackson ImmunoResearch Laboratories) or rabbit (1:10,000; Jackson ImmunoResearch) IgG coupled to peroxidase and exposed to X-blue film (Fujifilm) after treatment with chemiluminescence reagents (Immobilon Western; Millipore). For SDS-PAGE, transfected cells were lysed with CelLytic M Cell Lysis Reagent (Sigma-Aldrich), separated on a 10% SDS-PAGE gel, and then transferred to a PDVF membrane. The membrane was analyzed by immunodetection as described above.
Immunoprecipitation of high-molecular-weight complex.
Transfected cell lysates were incubated with anti-neuropsin antibody overnight at 4°C. Immobilized Protein G Sepharose beads (GE Healthcare) were added to immunocomplexes and incubated for 4 h at 4°C. The precipitate was collected after centrifugation at 12,000 × g and washed five times in wash buffer (50 mm HEPES, pH 7.4, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100). In some experiments, immunoprecipitates were treated with heparitinase (Seikagaku) in 50 mm HEPES, pH 7.4, 150 mm NaCl, 5 mm CaCl2, and 1% Triton X-100 overnight at 37°C. The beads were eluted with loading buffer, and the eluted materials were resolved by SDS-PAGE and immunoblotted with anti-Myc antibody (9B11; 1:2500, Cell Signaling Technology).
Peptide sequencing by mass spectrometry.
Immunoprecipitated samples were electrophoresed on a reducing SDS-polyacrylamide gel, visualized by silver staining (Wako), excised and, following in-gel digestion, identified by mass spectrometry. Nanoscale capillary liquid chromatography-tandem mass spectrometric analyses of in-gel digests were performed using an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) combined with a Paradigm MS4 HPLC system (AMR).
Cleavage of mNRG-1, fibronectin, and vitronectin by neuropsin.
Recombinant neuropsin was produced as previously described using a baculovirus-insect cell expression system (Shimizu et al., 1998). After activation by lysyl endopeptidase (Wako), the enzymatic activity of neuropsin was measured with Boc-VPR-4-methylcoumaryl-7-amide (Boc-VPR-MCA), one unit being defined as the amount required to hydrolyze 1 μmol/min of this substrate at 30°C. Neuropsin was incubated with human NRG-1-β1 extracellular domain (mNRG-1; amino acids 2–246; R&D Systems), bovine fibronectin (Trevigen), and human vitronectin (amino acids 20–478; R&D Systems) at 37°C in buffer containing 0.5 mm CaCl2 and 50 mm Tris-HCl, pH 8.0. The products of mNRG-1 were separated on 15% SDS-PAGE gels. Following transfer, nitrocellulose membranes were probed with rabbit polyclonal N-NRG-1 antibody (1:400; Santa Cruz Biotechnology). Silver staining was used to detect the products of fibronectin and vitronectin after electrophoretic separation on 6% and 10% polyacrylamide gels, respectively.
Hippocampal tissues were homogenized in cold modified RIPA buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mm EDTA, 5 mm sodium fluoride, and 1 mm sodium orthovanadate. The lysates were incubated with neuropsin (10 mU/ml) or an equal dose of proneuropsin for 1 h at 37°C, resolved by SDS-PAGE, and immunoblotted with anti-N-NRG-1 antibody (1:400; Santa Cruz Biotechnology).
Coimmunoprecipitation assays.
Transfected cells were lysed with CelLytic M Cell Lysis Reagent containing 1% protease inhibitor cocktail. The cell lysates were incubated with anti-neuropsin antibody or anti-N-NRG-1 antibody overnight at 4°C. Immobilized Protein G (GE Healthcare) was added to immunocomplexes and incubated for 4 h at 4°C. After washing, the beads were eluted with loading buffer, and the eluted materials were resolved by SDS-PAGE and immunoblotted with anti-N-NRG-1 antibody (1:400; Santa Cruz Biotechnology).
N-terminal peptide sequence analysis.
The N-terminal sequences of the NRG-1 fragments identified by SDS-PAGE after coincubation of recombinant neuropsin and NRG-1 were determined. The fragments were transferred to a PVDF membrane after SDS-PAGE. The membrane was stained with Coomassie Brilliant Blue (CBB), and the bands corresponding to fragments of 32, 28, 19, 15, and 6 kDa were excised and used for N-terminal amino acid sequence analysis. The amino acid sequence of the transferred fragments was determined using the Edman method with a Procise 492 cLC Sequencer (Applied Biosystems).
Enzyme activity and kinetic parameters assay.
Recombinant neuropsin amidolytic activities and kinetic parameters were determined at 30°C in 50 mm Tris-HCl buffer, pH 8.0, for various MCA substrates. The release of the fluorophore 7-amino-4-methyl coumarin (AMC) was measured with a microplate reader (Mithras LB940, Berthold Technologies) using an excitation wavelength of 355 nm and emission wavelength of 460 nm. The reaction rate [V] of neuropsin depending on enzyme concentration [E] was determined using a Hanes–Woolf plot, but those of neuropsin for Ac-KER-MCA and Ac-DVR-MCA were fitted to a sigmoidal curve using the equation V = VmaxEh/(Eh + Kmh), where Vmax, Km, and h indicate the maximal activity, enzyme concentration at which the reaction rate is half of Vmax, and Hill coefficient, respectively. Note that h values >1 indicate positively cooperative AMC synthesis.
Glycosaminoglycan inhibition of mNRG-1-induced receptor phosphorylation.
Preparations of recombinant human NRG-1-β1 extracellular domain (amino acids 2–246; R&D Systems) were diluted in 10 μl Tris buffer (50 mm Tris-HCl, pH 8.0, 1 mm CaCl2, and 1 mg/ml BSA). Then, 20 min after addition of neuropsin or vehicle, glycosaminoglycans (Sigma-Aldrich) were added to give the final indicated concentrations and incubated at room temperature with mNRG-1 for 10 min, after which the mixture was incubated with MCF-7 cells (human breast cancer, JCRB0134, established by Soule H. D. et al., Health Science Research Resources Bank, Osaka, Japan). Phosphorylation of p185 tyrosine residues was measured as described previously (Loeb and Fischbach, 1995). After a 20 min treatment of the cells with mNRG-1, the medium was discarded. The cells were solubilized in lysis buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 1% Triton X-100, 1% CHAPS, 5 mm EDTA, 50 mm sodium fluoride, 1 mm PMSF, 10 mm sodium pyrophosphate, 1 mm sodium orthovanadate, and 1% protease inhibitor cocktail) and then centrifuged at 12,000 × g to remove debris. Phosphotyrosine on p185 was detected by immunoblot analysis using a monoclonal antibody (4G10; 1:1000, Millipore) after resolution on 7.5% reducing SDS-polyacrylamide gels. The blots were developed using a donkey anti-mouse IgG linked to peroxidase (1:10,000; Jackson ImmunoResearch) and chemiluminescence as described above. Horseradish peroxidase-conjugated anti-β-actin antibody (1:2000; Abcam) was used as loading control.
Recombinant neuropsin-induced release of cell surface-localized NRG-1.
COS-7 and MCF-7 cells were plated on 24-well plates 1 d before transfection in high-glucose DMEM with l-glutamine (Sigma-Aldrich), supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. COS-7 cells were transfected for 20 h with the pEGFP-N1 vector encoding NRG-1-EGFP and then treated with recombinant neuropsin or heparitinase (10 mU/ml, Seikagaku). The medium was applied to MCF-7 cells for 20 min at 37°C and then discarded. Cells were solubilized in lysis buffer and then boiled in SDS sample buffer for 5 min. Phosphorylated forms of the ErbB receptor (p185) were detected by immunoblot analysis as described above. Band densities were quantified using ImageJ software (National Institutes of Health) and normalized to those of β-actin.
Biotinylation of mNRG-1, cleavage by neuropsin, and injection.
To identify the binding site for processed NRG-1 (pNRG-1) in the hippocampus, human recombinant mNRG-1 (R&D Systems) was biotinylated and cleaved by neuropsin according to the following procedure. Briefly, mNRG-1 was incubated with EZ-Link Sulfo-NHS-LC-Biotin (Pierce) at a molar ratio of 1:20 for 2 h on ice. The reaction mixture was dialyzed for 1.5 h to remove uncoupled biotinylation reagent (Slide-A-Lyzer MINI Dialysis Unit, 2K, Thermo Fisher Scientific). Biotinylated mNRG-1 was incubated with neuropsin for 20 min at 37°C and then confirmed by immunoblot analysis using anti-biotin antibody (1:10,000; Sigma-Aldrich) and anti-N-NRG-1 antibody (1:400; Santa Cruz Biotechnology). Adult mice were anesthetized with urethane (1.5 g/kg; i.p.; Wako), and then biotinylated pNRG-1 or PBS was injected (2.06 mm posterior and 1.5 mm lateral to bregma; 1.5 mm ventral from the dural surface) at a flow rate of 0.05 μl/min for 15 min using a Hamilton syringe driven by syringe pump (CFV-2100; Nihon Kohden). A 1.5% solution of the biological dye Chicago Sky Blue 6B (Sigma-Aldrich), dissolved in PBS, was used to confirm the injection site as well as the diffusion into the hippocampus.
For immunoprecipitation of ErbB4, pNRG-1-injected dorsal hippocampus was homogenized in RIPA buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 5 mm EDTA, 10 mm sodium fluoride, 10 mm sodium pyrophosphate, 1 mm sodium orthovanadate, and 1% protease inhibitor cocktail). The lysates (1.2 mg of protein) were incubated with anti-ErbB4 antibody (1.2 μg; Santa Cruz Biotechnology) overnight at 4°C. The immunocomplexes were recovered with Protein G (GE Healthcare) and subjected to immunoblotting with anti-phosphotyrosine (4G10; 1:1000, Millipore). The antibodies were stripped with 62.5 mm Tris-HCl, pH6.8, 2% SDS, and 100 mm 2-mercaptoethanol at 60°C for 30 min, after which the membrane was reblocked and reprobed with anti-ErbB4 antibody (1:500; Santa Cruz Biotechnology).
Immunohistochemistry.
Mice deeply anesthetized with urethane (1.5 g kg−1; i.p.; Wako) were transcardially perfused with saline followed by 4% paraformaldehyde in PBS. Brains were removed, postfixed overnight in the same fixative at 4°C, rinsed twice with PBS, and placed in 30% sucrose in PBS for 2 d at 4°C. The brains were then frozen quickly and cut into serial coronal sections (40 μm thickness) on a cryostat. Sections were collected as free-floating sections in PBS, incubated in blocking solution (5% BSA and 0.3% Triton X-100) for 1 h at room temperature and then incubated overnight at 4°C with the following antibodies: rabbit anti-NRG1 epidermal growth factor (EGF)-like domain (1:1000; Abcam), chicken anti-MAP2 (1:5000; Abcam), mouse anti-PSD-95 (1:2500; Millipore), mouse anti-synaptophysin (1:2500; Millipore Bioscience Research Reagents), mouse anti-parvalbumin (1:2000; Sigma-Aldrich), goat anti-biotin (1:2500; Sigma-Aldrich), rabbit anti-ErbB4 (1:500; Santa Cruz Biotechnology), and mouse anti-phosphotyrosine (4G10; 1:1000, Millipore). Sections were then washed and incubated overnight at 4°C with donkey anti-rabbit Alexa Fluor 488 (1:1000; Invitrogen) and 647 (1:500; Invitrogen), goat anti-rabbit Alexa Fluor 594 (1:1000; Invitrogen), goat anti-chicken DyLight 594 (1:1000; Jackson ImmunoResearch), goat anti-mouse Alexa Fluor 647 (1:1000; Invitrogen), donkey anti-mouse Alexa Fluor 594 (1:1000; Invitrogen), and donkey anti-goat Alexa Fluor 488 (1:1000; Invitrogen) secondary antibodies. Following a 10 min incubation with Hoechst 33342 (1:1000; Wako), sections were washed and mounted on microscope slides with ProLong Gold antifade reagent (Invitrogen). Images were captured using a confocal laser scanning microscope (LSM710; Carl Zeiss).
Subcellular fractionation of mouse brain extracts.
Subcellular fractions of adult mouse brain were prepared as described previously (Matsumoto-Miyai et al., 2009). Briefly, mouse hippocampi were homogenized in 0.32 m sucrose, 0.5 mm EDTA, 5 mm HEPES, pH 7.4, and 1% protease inhibitor cocktail. The homogenate was centrifuged at 1000 × g for 5 min to remove nuclei (pellet P1). The supernatant (S1) was centrifuged at 12,000 × g for 20 min, yielding a pellet (P2) and supernatant (S2). Pellet P2 was resuspended in homogenization buffer and represented the crude synaptosome fraction. The P2 fraction was loaded onto a 7.5%/12% Ficoll gradient and centrifuged at 68,000 × g for 1 h. The material remaining above the 7.5% Ficoll layer was collected as the myelin fraction (My). Material concentrated between 7.5% and 12% Ficoll was collected as the synaptosomal fraction (SS). The SS was lysed by osmotic shock by adding 10 volumes of ice-cold water and homogenized. The synaptic membrane fraction (SM) was collected from this homogenate by centrifugation at 68,000 × g for 20 min. Equal amounts of protein from each fraction were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and analyzed by immunoblotting. To identify the subcellular localization of NRG-1, the membrane was probed with a NRG-1 cytoplasmic tail (C-NRG-1) polyclonal antibody (1:400; Santa Cruz Biotechnology) and anti-N-NRG-1 antibody (1:400; Santa Cruz Biotechnology). The synaptic fractions were confirmed by immunoblotting with an antibody against the synaptic marker protein PSD-95 (1:6000; Millipore).
Electrophysiological methods.
Electrophysiological analysis was performed as described previously (Bjarnadottir et al., 2007). Mice were anesthetized with urethane (1.5 g/kg; i.p.; Wako) and transcardially perfused with ice-cold artificial CSF (ACSF). After decapitation, the brain was rapidly removed and placed in cold oxygenated (95% O2/5% CO2) ACSF. Slices (400 μm) were cut with a Lineaslicer PRO7 (Dosaka EM) and maintained at room temperature for at least 90 min in a holding chamber filled with oxygenated ACSF. After recovery, a single slice was transferred to a recording chamber where it was held on nylon nets and continuously perfused with oxygenated ACSF (31–32°C) at a 2 ml/min flow rate. The same ACSF was used for both perfusion and cutting and contained the following (in mm): 120 NaCl, 3.5 KCl, 0.7 CaCl2, 4.0 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. The same ACSF was used for both incubation and recording, and contained the following (in mm): 120 NaCl, 3.5 KCl, 2.6 CaCl2, 1.3 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose.
For extracellular recording in the hippocampal CA1 region, a bipolar stainless stimulating electrode was placed in the Schaffer collateral axons to elicit field population responses. The population spikes or field EPSPs (fEPSPs) were recorded via a glass micropipette filled with ACSF (1–3 MΩ) placed in the stratum pyramidale or radiatum. The population spikes were evaluated by calculating the voltage difference between onset and peak. The fEPSP responses were quantified as the slope of field potentials. Data were acquired using a PowerLab system (ADInstruments) and analyzed with LabChart software (ADInstruments). Stimuli (80 μs duration) were delivered every 60 s. Baseline responses were collected at 0.017 Hz with a stimulation intensity that yielded a 30–40% maximal response. Paired pulses were applied at interpulse intervals of 10 ms. The paired-pulse inhibition was defined as the second evoked response expressed as a percentage decrease of the first evoked response and described by the following equation: % paired-pulse inhibition = [(First response − Second response)/(First response)] × 100. LTP was induced by a single tetanic stimulation, which consisted of 100 stimuli delivered at 100 Hz at the test intensity. Recombinant human NRG-1 EGF-like domain [NRG-1177–246, a functional NRG-1 (Bjarnadottir et al., 2007; Chen et al., 2010; Pitcher et al., 2011); the EGF-like domain consists exactly of amino acid residues 177–246] (Peprotech) was applied to slices at 0.5 or 10 nm for 20 or 35 min after a stable baseline had been attained. ACSF was also supplemented as indicated with AG1478 (5 μm; LC Laboratories) or bicuculline methiodide (Bicu) (10 μm; Santa Cruz Biotechnology). In experiments using Bicu, the CA3 region of the hippocampal slice was removed by a microsurgical cut to prevent the propagation of epileptiform activity in the recording of LTP. Responses (mean ± SEM) are expressed as percentage of baseline response.
Statistical analysis.
Statistically significant differences were determined by Student's t test or one-way ANOVA with Scheffe's post hoc test. All data are presented as the mean ± SEM, the number of experiments is indicated by “n,” and differences were considered significant at p < 0.05.
Results
Neuropsin mutated in loop G loses peptidolytic activity but retains synthetic peptide-binding ability
Our previous studies on the crystal structure and conformation of neuropsin (Kishi et al., 1997, 1999) identified a unique loop G conformation for arginine and lysine surrounding the catalytic triad (Fig. 1A). Further analysis confirmed that loop G was necessary for the catalytic efficiency of neuropsin: disruption of loop G resulted in complete loss of cleavage activity, although both the protease activity pocket and the catalytic triad were intact (Oka et al., 2002). These observations led us to develop a new methodology to directly screen native substrates from tissues or cultured cells.

NeuropsinC208S forms a high-molecular-weight complex on the surface of hippocampal neurons. A, Neuropsin structure (Protein Data Bank code 1NPM). The catalytic triad (marked in yellow) consists of His73, Asp120, and Ser212. The disulfide bond-forming cysteines are marked SS1–SS6 (blue lines). Asp206 (green) in the S1-specific pocket and Cys208 (red) are indicated. Neuropsin forms eight loops and has one N-glycosylated site (GlcNac). Loops C and G are designated in the structure. B, Neuropsin domains and mutation sites used in this study. The signal sequence, activity-masking amino acid sequence (Q29GSK), Myc-tag, and His-tag are indicated. For modifications, Ser was substituted for Cys208 in loop G, or Ser87-Leu-Gln-Ser-Arg-Asp-Gln-Pro in loop C was deleted (ΔS87LQSRDQP). To modify the protease activity pocket, Val was substituted for Asp206 in the S1-specific pocket and Ala for Ser212 in the catalytic triad. C, Quantitative analysis of neuropsin-immunoreactive signal under the conditions specified in D. Bar diagram represents the average values (AU) obtained for different cultures (n = 5 different cultures, 2 neurons per cultures in each condition). *p < 0.05; **p < 0.01. Error bars indicate the SEM. D, Extracellular protein complex bound to mutant neuropsin. Immunostained images are merged with those showing intracellular EGFP fluorescence (green). Neuropsin was detected using an anti-neuropsin antibody (red) under nonpermeabilized conditions. Hippocampal neurons were transfected with preneuropsin, preneuropsinC208S, preproneuropsinC208S, preneuropsinD206V, or preneuropsinS212A. Scale bar, 10 μm. Bottom, High-magnification images of the rectangles in the top. Scale bar, 2 μm. E, Double immunostaining of preneuropsinC208S-transfected hippocampal neurons with an anti-neuropsin antibody under nonpermeabilized conditions (red; top) and an anti-PSD-95 antibody under permeabilized conditions (blue; middle), and their superimposed images (bottom). Neuropsin immunoreactivity was localized to the surface of dendrites in close apposition with intracellular PSD-95. Scale bar, 1 μm. F, BlueNative-PAGE of preproneuropsin-, preneuropsin-, preproneuropsinC208S-, or preneuropsinC208S-transfected cell lysate, immunoblotted and probed using an anti-neuropsin antibody. An ∼1-MDa high-molecular-weight protein complex was detected in the preneuropsinC208S cell lysate (arrow) (n = 3). G, SDS-PAGE of preneuropsinC208S-transfected cell lysates under nonreducing (preneuropsinC208S), boiling (+boil) or reducing conditions (+DTT). Although the high-molecular-weight band (arrow) was weaker or disappeared after boiling or DTT pretreatment, a monomeric neuropsin protein band was apparent in both cases (arrowhead) (n = 3). H, Top, Cell lysates transfected with preneuropsinC208S immunoprecipitated with an anti-neuropsin antibody. Immunoprecipitates were treated with heparitinase to degrade glycosaminoglycans and immunoblotted with an anti-Myc antibody. Note that the high-molecular-weight complex (black arrow) was weaker after heparitinase pretreatment, and an ∼150 kDa band (red arrow) appeared (n = 3). IP, Immunoprecipitation; IB, immunoblotting. Bottom, The high-molecular-weight band (arrow) was weaker after neuropsin pretreatment (n = 3).
We constructed mutant mouse neuropsin genes to yield preneuropsin bearing a Myc-tag and His-tag tail at the C terminus, and a deletion of prosequence (ΔQ29GSK in preproneuropsin; shown in pink in Fig. 1B) to render the mutant neuropsins easily accessible to potential substrates immediately after signal sequence removal and secretion (Shimizu et al., 1998). Further, we constructed four mutant neuropsin genes harboring single point mutations: D206V (preneuropsinD206V) in the protease activity pocket, C208S (preneuropsinC208S) in loop G, S212A (preneuropsinS212A) in the catalytic triad, and deletion of eight amino acids (ΔS87LQSRDQP) (preneuropsinΔS87LQSRDQP) in loop C near the protease activity domain (Fig. 1B). We also constructed a mutant preproneuropsin gene harboring the C208S point mutation (preproneuropsinC208S) (Fig. 1B). Recombinant proteins were immunopurified from the supernatants of cells transfected with one of the above mutant genes and assayed for potential binding or cleavage activity against the synthetic peptide substrate, VPR-MCA. Mutant neuropsins expressed from genes encoding a genetically modified S1 pocket, catalytic triad, or loops G or C, showed no (or negligible) peptidolytic activity. Supernatants from cells transfected with preneuropsinD206V, preneuropsinS212A, preneuropsinC208S, and preneuropsinΔS87LQSRDQP showed mean activities of 4.4, 6.1, 17.0, and 7.8 mU/ml, respectively, whereas the mean activity in supernatants from preneuropsin-transfected cells was 3160 mU/ml. The kinetic parameters associated with recombinant mutant proteins derived from the preneuropsinD206V, preneuropsinS212A, and preneuropsinC208S genes (kcat = 0.14, 0.04 and 0.51 s−1, respectively) were 200–2500-fold lower than that of normal neuropsin (100 s−1). Km values for each of these proteins (82, 86, and 100 μm, respectively) were ∼3-fold lower than that of wild-type neuropsin (280 μm). By contrast, disruption of loop C (preneuropsinΔS87LQSRDQP) induced an increase in Km (to 970 μm). Thus, the peptidolytic activity of neuropsinC208S was markedly decreased, although it maintained a strong binding affinity for the synthetic substrate, indicating that substrates that are bound to the catalytic domain of neuropsinC208S are not cleaved and form complexes with neuropsinC208S.
NeuropsinC208S is localized at discrete locations along dendritic and somatic membranes
Since mutant neuropsins exhibited impaired proteolytic activity while maintaining their substrate-binding ability, in situ immunodetection using these mutants was performed in cultured hippocampal neurons. Primary cultured neurons transfected with preneuropsinC208S or preneuropsin secreted approximately equal amounts (0.16 and 0.18 μg/ml, respectively) of recombinant proteins into the culture medium after 36 h of cultivation. Numerous mutant neuropsin-immunoreactive puncta [preneuropsinC208S: 9.1 ± 0.9 arbitrary units (AU), n = 10, one-way ANOVA, p < 0.01 vs preneuropsin; Fig. 1C,D, red fluorescence] were observed surrounding neuronal cell bodies and processes (Fig. 1D, green fluorescence) in cultures transfected with the preneuropsinC208S gene and were examined by surface immunocytochemistry (see Materials and Methods). A few puncta were also observed in preneuropsinD206V-transfected cultures (preneuropsinD206V: 3.2 ± 0.3 AU, n = 10, one-way ANOVA, p < 0.05 vs preneuropsin; Fig. 1C,D). However, no (or negligible) puncta were observed in cultures transfected with the genes producing enzymatically active neuropsin (preneuropsin: 0.88 ± 0.04 AU, n = 10), the nonprocessed precursor form (preproneuropsinC208S: 0.18 ± 0.06 AU, n = 10, one-way ANOVA, p > 0.8 vs preneuropsin), or the catalytic triad-lesion form (preneuropsinS212A: 1.3 ± 0.3 AU, n = 10, one-way ANOVA, p > 0.9 vs preneuropsin; Fig. 1C,D). We interpret these results as reflecting strong complex formation by a substrate-mutant neuropsin intermediate.
Higher-magnification photographs indicated that C208S and D206V neuropsin mutants assembled at the surface of cell bodies (Fig. 1D) and dendrites (Fig. 1E). The puncta were partially colocalized with postsynaptic density protein 95 (PSD-95) (Fig. 1E), an intracellular postsynaptic scaffolding protein. A similar staining pattern was also observed using an antibody against the C-terminal Myc-tag tail (data not shown), confirming the concentration of neuropsin protein at the neuronal cell surface. Thus, the recombinant C208S neuropsin mutant appears to bind to substrate/inhibitor candidate(s) localized outside the postsynaptic membrane in both soma and dendrites.
A high-molecular-weight complex is formed in cultured hippocampal neurons expressing neuropsinC208S
Because the results of histological analysis suggested that the C208S neuropsin mutant formed complexes with intermediate substrates/inhibitors, preneuropsinC208S-transfected cell lysates were electrophoresed in a BlueNative-polyacrylamide gel to analyze the size of the complexes. A high-molecular-weight band of ∼1 MDa (Fig. 1F, arrow) (far larger than the molecular weight of neuropsinC208S; ∼33 kDa) was detected when preneuropsinC208S-transfected cell lysates were transblotted and probed with anti-Myc antibody (Fig. 1F) or anti-neuropsin antiserum (data not shown). No high-molecular-weight band was observed in the lysates of cells transfected with preproneuropsin, preneuropsin, or preproneuropsinC208S when they were run in a native polyacrylamide gel (Fig. 1F) This indicates that the gene products of preproneuropsin, preneuropsin, and preproneuropsinC208S do not form, or stably maintain, the intermediate complex (see also Fig. 1D). The high-molecular-weight band detected after neuropsinC208S expression was stable in SDS gels, but was dissociated from the mutant neuropsin by boiling or pretreatment of cell lysates with dithiothreitol (DTT) (Fig. 1G).
Because the neuropsin protein interacts with glycosaminoglycans (Kishi et al., 1997), we investigated whether glycosaminoglycans associated with neuropsin and potential substrates/inhibitors in the high-molecular-weight band. Heparitinase treatment of the immunoprecipitated sample (to degrade glycosaminoglycans) resulted in a new band of ∼150 kDa (Fig. 1H, top, red arrow). However, monomeric neuropsin (∼33 kDa) was not observed. The high-molecular-weight band is, therefore, composed of mutant neuropsin, unidentified substrate/inhibitor candidates, and glycosaminoglycans to form a large SDS-stable complex. Moreover, mutant neuropsin and the substrate/inhibitor candidates may directly couple within the 150 kDa complex. When lysates from cells transfected with preneuropsinC208S were treated with recombinant (active) neuropsin, the band intensity weakened considerably (Fig. 1H, bottom, arrow), supporting our hypothesis that the band may contain substrate candidates that interact directly with mutant neuropsin.
Analysis of substrate candidates by LC-MS/MS
The high-molecular-weight band was immunopurified from a cell lysate of preneuropsinC208S-transfected cultured neurons using anti-neuropsin antiserum. After digestion with trypsin, the high-molecular-weight band was analyzed by liquid chromatography/tandem mass spectrometry (LC-MS/MS). The peptide sequences of protein fragments derived from the band were determined, and the band was found to contain several proteins other than neuropsin, including the extracellular domain of NRG-1 (the mature form or mNRG-1), fibronectin, vitronectin, and protease inhibitors of neuropsin as the major components, some of which were identified in earlier studies (Kato et al., 2001; Scott et al., 2007). Thus, the screening method appeared to be appropriate for detecting specific substrates/inhibitors of neuropsin. Peptide fragments identified in these experiments are listed in Table 1.
Peptide fragment sequences determined from candidate proteins that associated with mutant neuropsin
To further investigate whether the obtained substrate candidates were indeed specific substrates for neuropsin, mNRG-1 was coincubated with recombinant neuropsin at various concentrations in vitro. The mature form was used in this experiment because pro-NRG-1 (full-length; membrane-associated) is thought to be cleaved by type I transmembrane proteases, such as tumor necrosis factor-α converting enzyme (TACE), β-site amyloid precursor protein cleaving enzyme 1 (BACE1), and meltrin β for maturation, and the released mNRG-1 has the potential to bind to a receptor (Mei and Xiong, 2008). Immunoblot analysis using an antibody against mNRG-1 and silver-staining for products derived from fibronectin and vitronectin (Fig. 2A–C) revealed the progressive appearance of truncated fragments. Thus, the substrate candidates identified in the intermediate complex are true and effective substrates for neuropsin in vitro.

Neuropsin preferentially cleaves mNRG-1. Activity response and time course of neuropsin-dependent cleavage of mNRG-1 (A), fibronectin (B), or vitronectin (C), visualized in the top panel by immunoblotting for mNRG-1 using anti-N-NRG-1 polyclonal antibody (A) or silver staining (B, C). The band densities were quantified by densitometric scanning, with densities in the vehicle only condition (0 mU/ml) or sampled just after application of neuropsin at 4°C (0 min) set to 100%; relative densities (vertical axis) were plotted for various activities of applied neuropsin (top graphs) or time points after application of neuropsin (bottom graphs; horizontal axis) (n = 3 in each case). The band intensities of the indicated products (blue or red squares) and full-length proteins (black squares) are plotted. Time course reactions included neuropsin at 10 mU/ml. Data represent the mean ± SEM.
Neuropsin efficiently and quickly cleaved mNRG-1, as determined by comparison with serial dose responses and coincubation time courses obtained using recombinant neuropsin (Fig. 2A). As shown in Figure 2A, after only 15 min, two major immunoreactive fragments (32 and 19 kDa) approached their plateau levels (Fig. 2A, see also Fig. 4B). Endogenous hippocampal mNRG-1 was also efficiently degraded by neuropsin (Fig. 3A).

Endogenous mNRG-1 is cleaved by neuropsin and is associated with mutant neuropsin. A, Treatment with neuropsin causes complete loss of mNRG-1. Immnoblot analysis of hippocampal homogenates treated with vehicle, recombinant proneuropsin, or neuropsin with an anti-N-NRG-1 antibody (n = 3). B, Mutant neuropsin associates with mNRG-1 in hippocampal neurons. Hippocampal neurons were infected with adenovirus containing ZsGreen1 (mock), preproneuropsin, or preneuropsinC208S at day 9 in vitro. After 2 d, cell lysates were immunoprecipitated with an anti-neuropsin antibody or anti-N-NRG-1 antibody. Precipitated materials were immunoblotted with an anti-N-NRG-1 antibody or anti-neuropsin antibody (n = 3 in each case). The asterisk (*) denotes a nonspecific band. C, Double immunocytochemical staining for NRG-1 (green) and neuropsinC208S (red) in the hippocampal neurons. The immunoreactivity for neuropsinC208S and NRG-1 partially colocalized in the surface of dendrites of the hippocampal neurons (arrowhead) (n = 3 cultures, 2 neurons per cultures). Merge, A superimposed image of NRG-1, neuropsinC208S, and EGFP. Scale bar, 2 μm.
To confirm that neuropsinC208S interacts with mNRG-1 in vivo, we performed coimmunoprecipitation experiments using cultured hippocampal neurons. Incubation of neuropsinC208S immunoprecipitates with an NRG-1 antibody directed against its N-terminal moiety or anti-neuropsin antibody revealed that endogenous mNRG-1 or neuropsinC208S coimmunoprecipitated with transfected neuropsinC208S from cultured hippocampal neurons (Fig. 3B). Moreover, double-immunolabeling of NRG-1 and neuropsinC208S revealed that both molecules are distributed primarily as discrete puncta along dendrites (77.5 ± 5.2% surface colocalized signal) (Fig. 3C). These experiments suggest that the punctuate staining shown in Figure 1E and the intermediate complex shown in Figure 1H are, at least partly, due to binding of mutant neuropsin to mNRG-1.
Neuropsin cleaves mNRG-1 at three sites
Immunoblot analysis using an antibody against mNRG-1 (34 kDa) demonstrated that neuropsin cleaved mNRG-1 into two bands of 32 kDa and 19 kDa (Fig. 2A). Next, an in vitro coincubation assay was conducted to detect all the sites in recombinant human mNRG-1 cleaved by recombinant human His-tagged neuropsin (37 kDa) followed by CBB staining. Three fragments of 32 kDa (G19SGKKP…), 28 kDa (F68KWFKN…), and 19 kDa (I99NKASL… ; Fig. 4A,B) were observed and subsequently characterized by N-terminal peptide sequencing. Comparison of the cleavage sites with the sequence data identified one site located between the Ig and heparin-binding (HB) domains and two sites within the Ig domain (Fig. 4B). Among these fragments, the most major 32 kDa fragment was defined as pNRG-1 (see also Fig. 6A). In another experiment, additional 15 and 6 kDa fragments were detected on Tris-tricine gels (data not shown), both having the same N-terminal sequence (S2ERKEGRGKGKGKKK…) as the HB domain. We interpret these 15 and 6 kDa fragments as cleaved counterparts of the 19 and 28 kDa fragments, respectively. Collectively, these data indicate that neuropsin cleaves mNRG-1 at three distinct cleavage sites: KER18/GSG, SLR67/FKW, and ELR98/INK (Fig. 4B).

Neuropsin cleavage of mNRG-1. A, Recombinant human mNRG-1 was incubated at 37°C in the absence (control) or presence (+neuropsin) of recombinant human neuropsin. The samples were separated by SDS-PAGE, transferred to a PVDF membrane, and then stained with CBB. Neuropsin cleaves mNRG-1 to generate 32, 28, and 19 kDa fragments (n = 3). The corresponding bands were subjected to N-terminal amino acid sequence analysis. B, Domain structure of mNRG-1. HB, Heparin-binding domain; Ig, Ig domain; EGF, epidermal growth factor-like domain. Neuropsin cleavage sites in mNRG-1 are indicated by arrows and, below, by red bars in the six-residue amino acid sequence spanning each site. C, Heparin does not block receptor phosphorylation by neuropsin-cleaved mNRG-1. mNRG-1, containing the HB, Ig, and EGF-like domains (3 nm final concentration) was treated with 10 mU/ml neuropsin in 50 mm Tris-HCl, pH 8.0, for 20 min at 37°C. Both treated and undigested mNRG-1 were mixed with or without 3 mg/ml heparin and added to MCF-7 cells for 20 min. p185 receptor phosphorylation was measured by immunoblot analysis with an anti-phosphotyrosine antibody (n = 4). D1, Activity-response effect of neuropsin on the cleavage of mNRG-1 and effect of neuropsin-treated medium on p185 tyrosine phosphorylation. COS-7 cells were incubated for 20 min with the neuropsin at the indicated activity or for different times with 10 mU/ml neuropsin. The supernatant from each of the experimental conditions was added to MCF-7 cells, and p185 phosphorylation levels were measured by immunoblot analysis (n = 4). As control experiment, MCF-7 cells were treated with the medium collected from COS-7 cells transfected with empty vector after application of neuropsin (mock). D2, Quantification of p185 phosphorylation levels under the conditions specified in D1 (black line) and of the band density ratio of 32 kDa/34 kDa NRG-1 from Figure 2A (red line). The profiles are similar. D3, The dashed lines represent regression lines for p185 phosphorylation levels under the conditions specified in D1 (black) and the ratio of 32 kDa/34 kDa NRG-1 (red); correlation coefficients are 0.9865 and 0.9696, respectively. E, Heparitinase treatment enhances p185 phosphorylation to almost the same level of neuropsin. COS-7 cells were incubated with 10 mU/ml neuropsin or heparitinase, and the culture media were then applied to MCF-7 cells to analyze p185 phosphorylation activity (n = 8–9). *p < 0.05; **p < 0.01. Error bars indicate the SEM. F, Supernatant from mutated mNRG-1-expressing cells after treatment with 10 mU/ml neuropsin does not enhance p185 phosphorylation. Mutant mNRG-1, in which Arg18, Arg67, and Arg98 were each replaced by Ala, was expressed in COS-7 cells, which were then treated with 10 mU/ml neuropsin for 60 min. Supernatant samples were added to MCF-7 cells. The MCF-7 cell lysates were then subjected to immunoblot analysis with the anti-phosphotyrosine antibody, 4G10 (n = 8).
Using synthetic substrates spanning the newly identified cleavage sites, we determined accurate enzyme kinetics for neuropsin. We produced synthetic MCA fluorogenic substrates (KER-MCA, SLR-MCA, and ELR-MCA from human NRG-1, and KDR-MCA from amino acid residues in mouse NRG-1 corresponding to the human NRG-1 amino acid sequence, K16ER) and measured their kinetic parameters (Km and kcat; Table 2). VPR-MCA, a preferential substrate for α-thrombin, was also included as a positive control (Shimizu et al., 1998). The cleavage sites KER-MCA (human) and KDR-MCA (mouse) are consistent with formation of the 32 kDa fragment (which initially appeared after coincubation with recombinant neuropsin; Fig. 2A), and these substrates were digested efficiently by neuropsin. SLR-MCA, corresponding to the first cleavage site in the Ig domain of NRG-1 (Fig. 4B), was also effectively digested. However, recombinant neuropsin demonstrated low amidolytic activity for ELR-MCA, the cleavage site of which is consistent with formation of the 19 kDa fragment; the affinity of neuropsin for this fragment may be low because it was cleaved only by a high concentration (>10 mU/ml) of recombinant neuropsin (Fig. 2A). The amino acid sequences in NRG-1 susceptible to the nucleophilic activity of neuropsin were, thus, confirmed to be the three sites: KER, SLR, and ELR (Fig. 4B, arrows). In additional experiments, we detected only negligible cleavage activity by recombinant neuropsin at DVR-MCA and YGR-MCA, which correspond to reported neuropsin cleavage sites in fibronectin and EphB2, respectively (Shimizu et al., 1998; Attwood et al., 2011). Neuropsin-mediated cleavage of fibronectin and EphB2 may occur at sites other than those suggested by these two studies; thus, the cleavage system for these proteins should be further investigated.
Enzyme activities and kinetic parameters of neuropsin as measured on MCA-containing synthetic substrates
Cleaved mNRG-1 fragments trigger phosphorylation of p185
Since mNRG-1 appears to be the preferred substrate for neuropsin, its cleavage product(s) should retain functional activity for the signaling system downstream of mNRG-1. Therefore, we tested the functional significance of mNRG-1 cleavage by neuropsin. Because the HB domain consensus motif interacts with heparan sulfate proteoglycans (HSPGs), which are components of the extracellular matrix (Dreyfuss et al., 2009), we hypothesized that removal of the HB domain of mNRG-1 by neuropsin would be critical to NRG-1-driven signaling; the first step of which involves phosphorylation of the NRG-1 receptor p185 (ErbB). Since heparin-bound mNRG-1 does not trigger the phosphorylation of p185 in cultured MCF-7 human breast carcinoma cells that express the endogenous NRG-1 receptor, p185 (Loeb and Fischbach, 1995), the extent of p185 tyrosine phosphorylation reflects the extent of functional release of the activity domain of mNRG-1 from the heparin reservoir in the synaptic cleft. Thus, receptor interaction can be assessed by measuring p185 tyrosine phosphorylation using immunoblot analysis (Loeb and Fischbach, 1995).
Experiments using an antibody against tyrosine-phosphorylated p185 showed that p185 was highly phosphorylated following mNRG-1 application in absence of heparin, though it was clearly abolished in presence of heparin. When recombinant neuropsin was pretreated to mNRG-1, p185 was phosphorylated in both the absence and presence of heparin (Fig. 4C). Since treatment with neuropsin can, thus, lead to phosphorylation of p185 in the presence of heparin, we concluded that neuropsin-dependent proteolytic cleavage of mNRG-1 inactivates its HB activity by removing the HB domain. A dose-dependent increase in p185 tyrosine phosphorylation in MCF-7 cell lysates following the addition of neuropsin (0.3, 1.0, 3.2, 10, and 32 mU/ml) was observed (Fig. 4D1,D2). Both the extent of p185 phosphorylation by 10 mU/ml neuropsin, and the cleavage ratio of 32 kDa/34 kDa NRG-1 by neuropsin, increased linearly between 0 and 60 min (Fig. 4D1,D3). Phosphorylation of p185 increased markedly upon treatment with neuropsin at <3.2 mU/ml (Fig. 4D2, black line), and this increase was paralleled by the cleavage ratio of 32 kDa/34 kDa NRG-1 (Fig. 4D2, red line). Treatment of MCF-7 cells with the medium collected from COS-7 cells transfected with empty pEF1-Myc-His vector after application of neuropsin did not yield detectable phosphorylation of p185 (Fig. 4D1, mock), indicating that the tyrosine-phosphorylated p185 was triggered by the released NRG-1 fragments. Therefore, cleavage of the HB domain of mNRG-1 by neuropsin releases a functional NRG-1 domain.
To confirm the confinement of mNRG-1 by glycosaminoglycans and its release by neuropsin, heparitinase or recombinant neuropsin was applied to MCF-7 cells, and p185 phosphorylation was examined. Heparitinase and recombinant neuropsin induced p185 phosphorylation to a similar extent (heparitinase: 134.91 ± 12.6% of control, n = 9, one-way ANOVA, p < 0.05 vs control; neuropsin: 176.6 ± 17.3% of control, n = 8, p < 0.01 vs control; Fig. 4E). Thus, degradation of glycosaminoglycans and neuropsin-related cleavage of mNRG-1 show comparable stimulatory effects on NRG-1 signaling.
A mutant mNRG-1 (R18A/R67A/R98A) was constructed to eliminate the neuropsin cleavage sites identified above. Application of recombinant neuropsin to mutant-mNRG-1-expressing cells caused no increase in the level of p185 phosphorylation compared with that in vehicle-treated controls (phosphorylation of p185 by neuropsin in cells expressing mutant mNRG-1:91.1 ± 13.4% of phosphorylation of p185 in control cells, n = 8, t test, p > 0.5 vs control; Fig. 4F), confirming the importance of neuropsin-mediated mNRG-1 cleavage for p185 phosphorylation. Together, these results indicate that mNRG-1 cleavage by neuropsin removes the HB domain, thereby releasing the functional EGF-like domain of mNRG-1 (pNRG-1) from the heparin reservoir so that it can trigger phosphorylation of p185.
Localization of mNRG-1 and pNRG-1 in adult mouse brain
Consistent with previous in situ hybridization analysis (Woo et al., 2007), numerous NRG-1 immunoreactive neurons were localized in the granular layer of the dentate gyrus and in the pyramidal layer of the CA1–3 subfield (Fig. 5A), and in all the layers, in layers V with high density and in layers II, III, IV, and VI with moderate density (Fig. 5D). The anti-NRG-1 antibody specifically recognized NRG-1 because its immunoreactivity was diminished if the antibody was preabsorbed with blocking peptide (Fig. 5G), and the antibody reacted with the apparent molecular mass NRG-1 (Frenzel and Falls, 2001) in Western blots of mouse hippocampal extracts (Fig. 5H). Majority of the immunoreactive neurons are considered excitatory neurons by their morphology stained with immunoreactive MAP-2 (Fig. 5B,E). In addition, there were a number of NRG-1 immunoreactive interneurons coexpressing parvalbumin (Fig. 5B,E). Thus, NRG-1 is expressed both in excitatory and inhibitory neurons of the hippocampus and the prefrontal cortex. In higher magnification observation, the small fluorescent punctates, majority of which colocalized with PSD-95, surrounded pyramidal cell bodies and dendrites of the pyramidal cells (Fig. 5C, left, arrowheads). However, NRG-1-immunoreactivity did not colocalize with synaptophysin, a presynaptic vesicle-related protein (Fig. 5C, right). Collectively, NRG-1 is extracellularly localized in the synaptic elements. Subcellular fractionation showed that both of pro-NRG-1 and mNRG-1 are present in the synaptic membrane and synaptosomal fraction (Fig. 5F). The mNRG-1 is present in synaptosomal fraction rather than synaptic membrane fraction (Fig. 5F). Therefore, mNRG-1 is predominantly localized to the synaptic cleft, presumably in association with glycosaminoglycans.

NRG-1 immunoreactivity in the hippocampus and prefrontal cortex. A, D, Coronal sections of hippocampus and prefrontal cortex from adult mice were stained with anti-NRG-1 antibody. NRG-1 immunoreactivity was detected in the pyramidal cell layer (CA1–3) throughout Ammon's horn, the granular cell layer of the dentate gyrus (DG), and in layers II–VI (particularly layer V) of the prefrontal cortex. Scale bar, 200 μm. B, E, Superimposed images of triple immunohistochemical staining for Hoechst (blue), NRG-1 (red), and MAP2 (green), or Hoechst (blue), NRG-1 (red), and parvalbumin (green) in the hippocampus and prefrontal cortex. Arrows indicate parvalbumin-positive cells (right images). NRG-1 was expressed in both excitatory and inhibitory neurons of the hippocampus and prefrontal cortex. Scale bar, 10 μm. C, Superimposed images of double immunohistochemical staining for NRG-1 (red) and PSD-95 (green) or NRG-1 (red) and synaptophysin (green) in the hippocampus. Higher-magnification views of the white rectangles, to the right of each main image, show that NRG-1 colocalized with PSD-95 (in yellow; arrowheads, left), but not with synaptophysin, a presynaptic vesicle-related protein (right). Scale bars: 10 μm (main images), 2 μm (higher-magnification images). F, Representative immunoblots of SS and SM preparations from mouse hippocampus (n = 3). PSD-95 was abundant in the SS and SM fractions and served as a control for the quality of the preparations. Pro-NRG-1 was found in the SS and SM fractions. The majority of mNRG-1 was detected in the SS fraction. H, Homogenate; S1, supernatant 1; P1, pellet 1; S2, supernatant 2; P2, pellet 2; My, myelin fraction. G, Coronal sections of hippocampus were incubated with the anti-NRG-1 antibody or antibody preabsorbed with blocking peptide (n = 3). Scale bar, 20 μm. H, Specificity of anti-NRG-1 antibody used for immunohistochemistry in the mouse bran was analyzed with Western blot (n = 3).
To analyze the target cells for pNRG-1 produced by neuropsin proteolysis, we injected biotin-labeled recombinant pNRG-1 into the hippocampus (Fig. 6A,B) and performed immunohistochemical analysis using an antibody against biotin. Triple staining for biotin (exogenous pNRG-1), ErbB4, and parvalbumin clearly showed that pNRG-1 was directed to parvalbumin- (and ErbB4-) positive interneurons within the CA1 region (Fig. 6C). In addition, triple staining for biotin, ErbB4, and phosphotyrosine showed that biotin-positive pNRG-1-labeled cell somata containing ErbB4 immunoreactivity were highly phosphorylated, as indicated by phosphotyrosine immunoreactivity (Fig. 7A, top). Furthermore, we found that ErbB4 was tyrosine-phosphorylated in the pNRG-1-injected hippocampus: ErbB4 immunoprecipitated from hippocampal extracts was recognized by an anti-phosphotyrosine antibody 1 h after injection of pNRG-1 (Fig. 7B). Notably, no pyramidal neurons were labeled by pNRG-1, consistent with previous reports (Vullhorst et al., 2009). Immunoreactive phosphotyrosine was never observed after injection of vehicle alone into the mouse hippocampus; therefore, the high levels of phosphorylated cellular proteins in ErbB4-positive neurons were due to the activity of exogenously applied pNRG-1 on ErbB4-containing fast-spiking neurons (Fig. 7A, bottom). Collectively, these data show that processing of mNRG-1 by neuropsin regulates GABAergic fast-spiking neurons via NRG-1-ErbB4 signaling.

pNRG-1 and ErbB4 colocalize in parvalbumin-positive interneuron of the hippocampus. A, Biotinylated-mNRG-1 was pretreated without or with neuropsin (10 mU/ml) and then subjected to immunoblotting (IB) with anti-biotin or anti-N-NRG-1 antibodies (n = 3). B, Both pNRG-1 and Chicago Sky Blue 6B were injected in the CA1 region of the hippocampus, and the sections were visualized by Nissl staining (n = 3 independent experiments). Scale bar, 0.5 mm. C, Coronal sections of hippocampus from adult mice injected with biotinylated-pNRG-1 were stained with Hoechst 33342 (blue) and anti-biotin (red), anti-ErbB4 (green), and anti-parvalbumin (purple) antibodies. Biotinylated-pNRG-1 immunoreactivity was detected on parvalbumin-positive interneurons (n = 3 independent experiments). Arrows, Colocalization of biotinylated-pNRG-1, ErbB4, and parvalbumin. Insets show higher-magnification views of another biotinylated-pNRG-1-immunoreactive neuron in the stratum pyramidale of the CA1 subfield. Scale bar, 10 μm.

Activation of ErbB4 immunoreactive neurons triggered by hippocampal injection of pNRG-1. A, mNRG-1 was biotinylated and cleaved with neuropsin to produce biotinylated-pNRG-1. Coronal sections of hippocampus from adult mice injected with biotinylated-pNRG-1 (top) or vehicle (bottom) were stained with Hoechst 33342 (blue) and anti-biotin (red), anti-ErbB4 (green), and anti-phosphotyrosine (purple) antibodies. Injected pNRG-1 was labeled in a highly phosphorylated ErbB4-immunoreactive neuron in the hippocampus (top, arrows) (n = 3 independent experiments). Insets show higher-magnification views of another ErbB4-immunoreactive neuron in the stratum pyramidale of the CA1 subfield. Scale bar, 10 μm. No phosphorylation of ErbB4-positive neurons was observed in the vehicle-injected hippocampus (bottom, arrowheads) (n = 3 independent experiments). B, The hippocampal lysates were prepared at 0, 1, 3, and 6 h after hippocampal injection of biotinylated-pNRG-1 to adult mice, immunoprecipitated (IP) with the anti-ErbB4 antibody, and subjected to immunoblotting (IB) with anti-phosphotyrosine (anti-pY) or anti-ErbB4 antibodies (n = 4).
Impairment of GABAergic inhibition in neuropsin knock-out mice
We further analyzed how neuropsin-NRG-1-ErbB4 signaling functions in hippocampal plasticity using electrophysiological techniques. GABA mediates the paired-pulse inhibition of population spikes at postsynaptic GABAA receptors (Steffensen and Henriksen, 1991). When two population spikes were applied to the pyramidal layer of the hippocampus 10 ms apart, clear inhibition was observed in wild-type mice as a result of GABA transmission (64.2 ± 6.7% of initial stimulus, n = 9; Fig. 8A,B). However, the degree of inhibition significantly decreased in neuropsin knock-out slices compared with wild-type slices (neuropsin (−/−), 41.7 ± 8.2% of initial stimulus, n = 7, t test, p < 0.05 vs wild-type; Fig. 8A,B). This effect was reversed by acute bath application (20 min) of 0.5 nm recombinant NRG-1177–246 (neuropsin (−/−) + NRG-1177–246, 57.3 ± 5.3%, n = 7; t test, p < 0.05 vs neuropsin (−/−); Fig. 8A,B). Combined with the histological data, these results indicate that neuropsin regulates GABAergic transmission via pNRG-1 binding to fast-spiking neurons.

Effects of neuropsin gene deletion on paired-pulse inhibition. A, Representative traces from paired-pulse recordings of wild-type, neuropsin knock-out, and 0.5 nm NRG-1177–246-treated neuropsin knock-out hippocampal slices are shown. Paired stimuli were applied through the same stimulation electrode at a 10 ms interpulse interval. Calibration: 2 mV (vertical), 10 ms (horizontal). B, Population spike response to the second stimulus expressed as a percentage decrease of initial stimulus, [(First response − Second response)/(First response)] × 100%, is plotted. The data indicate that the degree of paired-pulse inhibition of neuropsin knock-out mice was decreased compared with that of wild-type mice (n = 7–8 slices).
Rescue of E-LTP in neuropsin knock-out mice by NRG-1177–246
We next investigated whether the impaired hippocampal Schaffer-collateral E-LTP in neuropsin-deficient mice (Tamura et al., 2006; Ishikawa et al., 2008) was rescued by bath application of NRG-1177–246 to hippocampal slices. Bath application of low-dose (0.5 nm) NRG-1177–246 in neuropsin knock-out slices with a single tetanic stimulation resulted in restoration of a similar E-LTP level to that in wild-type slices [30–45 min after tetanus: neuropsin (−/−), 130.1 ± 5.9% of baseline, n = 6; wild-type, 153.7 ± 4.3% of baseline, n = 6; one-way ANOVA, p < 0.05; red triangles, neuropsin (−/−) + NRG-1177–246 (0.5 nm), 154.2 ± 4.1% of baseline, n = 7; one-way ANOVA, p < 0.05 vs neuropsin (−/−); Fig. 9A–C], although a higher dose of NRG-1177–246 (10 nm) cancelled this effect [30–45 min after titanic stimulation: neuropsin (−/−) + NRG-1177–246 (10 nm), 120.9 ± 5.0% of baseline, n = 5; one-way ANOVA, p < 0.01 vs neuropsin (−/−) + NRG-1177–246 (0.5 nm); Fig. 9A–C]. These dose-dependent effects of NRG-1177–246 on E-LTP were consistent with the reverse U-shaped dose–response effects of previous studies (Bjarnadottir et al., 2007; Role and Talmage, 2007). In wild-type hippocampal slices, where sufficient endogenous pNRG-1 content may be present, application of NRG-1177–246 (0.5 nm) markedly suppressed E-LTP [30–45 min after tetanus: wild-type + NRG-1177–246 (0.5 nm), 112.4 ± 5.1% of baseline, n = 6; one-way ANOVA, p < 0.01 vs wild-type; Fig. 9A–C]. This observation is consistent with previous reports demonstrating a suppressive effect of mNRG-1 on LTP (Huang et al., 2000; Ma et al., 2003; Iyengar and Mott, 2008; Pitcher et al., 2008; Chen et al., 2010; Pitcher et al., 2011). By contrast, bath application of recombinant NRG-1177–246 had no effect on basal synaptic strength in wild-type or neuropsin knock-out slices (see Fig. 11A).

Effects of bath-applied NRG-1177–246 on LTP in neuropsin knock-out hippocampal slices. A, Representative waveforms of fEPSP slopes recorded before (1) or after (2) tetanization in wild-type (open circles in B), neuropsin knock-out [neuropsin (−/−), red triangles in B], and 0.5 or 10 nm NRG-1177–246-treated neuropsin knock-out [neuropsin (−/−) + NRG-1177–246 (0.5 nm), blue triangles in B; neuropsin (−/−) + NRG-1177–246 (10 nm), green triangles in B] hippocampal slices are shown. Calibration: 0.5 mV (vertical), 10 ms (horizontal). B, Normalized fEPSP slopes from control and NRG-1177–246-treated hippocampal slices. ACSF or NRG-1177–246 was applied during the period indicated by the bar. The arrow denotes the point of tetanic stimulation, and numbers (1 and 2) indicate waveforms in A. C, Bar diagram shows mean values expressed as percentage change in fEPSP slope from 30 to 45 min after tetanus in slices from wild-type and neuropsin knock-out mice without and with NRG-1177–246 treatment at a concentration of 0.5 or 10 nm. *p < 0.05; **p < 0.01. The suppression of E-LTP in neuropsin knock-out mice was alleviated by a low concentration (0.5 nm) of NRG-1177–246; this effect was reversed when the higher concentration of 10 nm was applied (n = 5–7 slices). Error bars indicate SEM.
We further analyzed whether cellular modulation (phosphorylation of ErbB4 and GABAergic transmission) via pNRG-1 binding was responsible for the potentiation effect of neuropsin on E-LTP using an ErbB4 inhibitor, AG1478. The rescue of E-LTP in neuropsin-deficient mouse slices by bath application of 0.5 nm NRG-1177–246 was completely cancelled out by bath-applied AG1478 [45–60 min after tetanus: NRG-1177–246 (0.5 nm) + AG1478, 118.4 ± 2.1% of baseline, n = 6; NRG-1177–246 (0.5 nm), 148.9 ± 16.2% of baseline, n = 6; one-way ANOVA, p < 0.01; Fig. 10A–C]. Moreover, rescue of E-LTP by bath application of 0.5 nm NRG-1177–246 was also cancelled by bath application of a GABAA inhibitor, Bicu [45–60 min after tetanic stimulation: wild-type + Bicu, 134.6 ± 4.5% of baseline, n = 6; neuropsin (−/−) + Bicu, 130.3 ± 3.5% of baseline, n = 7; neuropsin (−/−) + NRG-1177–246 (0.5 nm) + Bicu, 134.5 ± 3.8% of baseline, n = 8; Fig. 10D–F]. Since subsequent high-frequency stimulation following NRG-1177–246-treated LTP induced additional LTP, the blockade effect of NRG-1177–246 is not a ceiling effect that could mask potential enhancement of NRG-1177–246 (Fig. 11B).

Effects of bath-applied NRG-1177–246 on LTP rescue are cancelled by ErbB4 inhibitors or GABAA antagonists. A–C, E-LTP rescue by bath application of NRG-1177–246 requires ErbB4 activation in neuropsin knock-out mice. Slices were treated without or with AG1478 15 min before the application of 0.5 nm NRG-1177–246. Representative waveforms of fEPSP slopes recorded before (1) or after (2) tetanization in the presence of vehicle (control; red triangles in B), NRG-1177–246 (blue triangles in B), or NRG-1177–246 + AG1478 (gray triangles in B) are shown at the top. D–F, Bicu blocks E-LTP rescue by NRG-1177–246. Bicu was bath-applied 35 min before the application of NRG-1177–246. Representative waveforms of fEPSP slopes recorded before (1) or after (2) tetanization in wild-type (filled circles in E) or neuropsin knock-out slices in the presence of vehicle (red triangles in E) or NRG-1177–246 (blue triangles in E) are shown at the top. NRG-1177–246 (0.5 nm) was applied during the period indicated by the bar. Arrows denote the point of tetanus. Values in B and E are expressed as a percentage of the baseline. Calibration: 0.5 mV, 10 ms. On the left, mean values expressed as percentage change in fEPSP slope from 45 to 60 min after tetanus are represented in bar-diagram form (n = 6–8 slices). **p < 0.01. Error bars indicate SEM.

The blockade effect of NRG-1177–246 by GABAA antagonists is not ceiling effect. A, Basal transmission is not altered in NRG-1177–246-treated hippocampal slice (n = 4 slices). B, Subsequent high-frequency stimulation following NRG-1177–246-treated LTP induced additional LTP in neuropsin knock-out slice. Normalized fEPSP slopes from NRG-1177–246-treated wild-type (open circles in A) and neuropsin knock-out (blue triangles in A, B) hippocampal slices (n = 5 slices in each case). NRG-1177–246 was applied during the period indicated by the bar. Bicu was bath applied 35 min before the application of NRG-1177–246. NRG-1177–246 (0.5 nm) was applied during the period indicated by the bar. Arrows denote the point of tetanus. Values are expressed as a percentage of the baseline. Symbols indicate the mean ± SEM.
Together, the present data show that the effect of neuropsin on E-LTP requires pNRG-1 binding to ErbB4 followed by GABAA receptor-dependent transmission. Thus, the neural activity-dependent processing of mNRG-1 into the NRG-1 ligand domain (pNRG-1) triggers activation of ErbB4 in fast-spiking GABAergic neurons and leads to a modulation of Schaffer-collateral E-LTP (Fig. 12A,B).

Model of signaling molecules accumulated within microdomain of synaptic cleft and neuronal circuitry in the hippocampus depicting the role of neuropsin in modulating GABAergic transmission. A, Signaling molecules are accumulated and associated with HSPGs in the synaptic cleft. Neuropsin cleaves directly mNRG-1 at three sites. This cleavage removed the heparin-binding domain of NRG-1, releasing the ligand moiety from the matrix-glycosaminoglycan pool and enabling it to trigger the phosphorylation of ErbB4. B, Basket interneurons control pyramidal cell output by forming inhibitory synapses on the soma of pyramidal cells. In wild-type animals, neuropsin cleaves mNRG-1 and causes its ligand domain release, thus stimulating GABAergic neurons via activation of ErbB4. Therefore, parvalbumin-positive interneurons could regulate the excitability of pyramidal cells and contribute to their synchronization. Loss of neuropsin causes a decrease in inhibitory inputs to the soma of the pyramidal cells because impairment of the cleavage of mNRG-1 results in attenuation of the GABAergic neurons activity. This attenuation leads to reduced synchronization of pyramidal cells, followed by prevention of the induction of NMDA receptor-dependent LTP.
Discussion
The present study developed a novel method to detect a direct protease-substrate interaction. Using this method, we identified mNRG-1, the major signaling ligand protein for ErbB4 receptor kinase and a risk factor for psychiatric diseases, as a novel native substrate for neuropsin. mNRG-1 is thought not to transmit signals into postsynapses via the ErbB4 receptor until it has been processed by an unidentified protease (Loeb and Fischbach, 1995). We clearly showed that neuropsin removes the HB domain of mNRG-1 to release functional pNRG-1. This activity-dependent processing of mNRG-1 is responsible for downstream signaling through the ErbB4 receptor and consequently, E-LTP.
Furthermore, we identified parvalbumin-immunoreactive neurons as the target of pNRG-1, processed and released as an active domain of mNRG-1. Parvalbumin-positive neurons are fast-spiking GABAergic neurons and are anatomically categorized into basket and chandelier cells, both of which project inhibitory synapses into hippocampal pyramidal neurons (Markram et al., 2004). When ErbB4 is activated via pNRG-1, the GABAergic neurons send signals to pyramidal neurons via GABAA receptor-mediated transmission. Impairment in GABAergic transmission leads to excessive postsynaptic excitation, thereby preventing the induction of NMDA receptor-dependent LTP (Moore et al., 1993; Barr et al., 1997). In neuropsin-deficient mice, GABAergic input to the pyramidal neurons is impaired, as shown in the paired-pulse inhibition experiments. Consistent with this, neuropsin-deficient mice demonstrate behavioral and neuronal phenotypes of hyperexcitability, with a compensatory increase of parvalbumin-immunoreactive neurons in the hippocampus, although the total number of glutamate decarboxylase 67 (GAD67)-immunoreactive cells was unchanged (Davies et al., 2001; Hirata et al., 2001). Neuropsin was therefore considered to be responsible, at least in part, for inhibitory neurotransmission, particularly to fast-spiking chandelier and/or basket neurons. Thus, reduction of ErbB4 signaling in parvalbumin-positive neurons caused by neuropsin deficiency produces an impairment of E-LTP via GABAergic transmission.
Recombinant NRG-1 has been reported to suppress protein synthesis-independent Schaffer-collateral LTP (E-LTP) in CA1 hippocampal synapses in the mouse brain (Huang et al., 2000; Ma et al., 2003; Kwon et al., 2005; Iyengar and Mott, 2008; Pitcher et al., 2008; Chen et al., 2010; Pitcher et al., 2011). Interestingly, previous studies have contradicted this suppressive effect and reported that NRG-1 potentiates tetanus-induced LTP in heterozygous mice deficient for the EGF domain of NRG-1, Nrg1(ΔEGF)+/−, although it had a similar suppressive effect on E-LTP in normal mouse hippocampal slices (Bjarnadottir et al., 2007; Role and Talmage, 2007). Thus, the effect of NRG-1 (EGF-like domain) on LTP may depend on endogenous NRG-1 expression levels. Consistently, a low concentration of recombinant NRG-1177–246 consisting of EGF-like domain had a potentiating effect on E-LTP and rescued an impairment of E-LTP in neuropsin-deficient mice to wild-type levels. By contrast, applying a higher concentration of recombinant NRG-1177–246 to neuropsin-deficient mouse hippocampal slices had a suppressive effect. Similarly, in wild-type hippocampal slices, where endogenous NRG-1 levels are normal, application of recombinant NRG-1 suppressed E-LTP. The potentiation effect of bath-applied recombinant neuropsin on E-LTP took the form of an inverted U curve between concentrations of 1 and 170 nm, with the strongest effect observed at 2.5 nm (Komai et al., 2000; Tamura et al., 2006). Taking these observations together, the modulatory effect of neuropsin on E-LTP appears to reflect an inverted U relationship between downstream NRG-1 signaling and LTP.
Loeb et al. (1999) reported that HSPGs within the brain extracellular matrix are a reservoir for mNRG-1. Taking this finding into account, our present data suggest that HSPGs are involved in the regulation of synaptic plasticity by acting as a supplier of mNRG-1. In fact, heparan sulfates (e.g., N-syndecan), which constitute a group of glycosaminoglycans, were reported to be involved in 100 Hz/1 s tetanus-induced Schaffer-collateral LTP (E-LTP), with both heparitinase preincubation and heparin application, in hippocampal slices leading to a marked reduction in LTP (Lauri et al., 1999). Glypican, a glycosaminoglycan, is an another candidate reservoir for mNRG-1 (Malavé et al., 2003) and is involved in neurite extension (Coles et al., 2011). These findings indicate that heparan sulfates are critical for establishing LTP and neural plasticity. Consistently, the present study identified a high-molecular-weight complex (∼1 MDa) that was coimmunoprecipitated by mutant neuropsinC208S and was cleaved by heparitinase to yield a lower molecular-weight complex of 150 kDa. Based on our present data, the high-molecular-weight complex may contain HSPGs in addition to neuropsinC208S, mNRG-1, fibronectin, vitronectin, and protease inhibitors. Further, neuropsin cleaved the HB domain of mNRG-1, leading to activation of fast-spiking GABAergic neurons. Inconsistent with the report described above (Lauri et al., 1999), adult N-syndecan-deficient mice show enhanced LTP in area CA1 of the hippocampus and, contradictorily, impaired hippocampus-dependent memory performance (Kaksonen et al., 2002). Interestingly, mutant mice were not responsive to N-syndecan ligand heparin-binding growth-associated molecule (HB-GAM). Therefore, a possible interpretation for the enhancement of LTP in mutant mice might be that a shortage of storage capacity for mNRG-1 in the synaptic area in the N-syndecan-null mutant brain causes uncontrolled GABAergic transmission regardless of neural activity-dependent neuropsin function. However, further studies are necessary to clarify the involvement of HSPGs in neural plasticity.
Finally, impairment in GABA neurotransmission results in altered gamma oscillations and weakened working memory (Lewis et al., 2012). Recent findings indicate that schizophrenia is associated with abnormalities in parvalbumin-positive basket cells, which weakens their inhibitory control of pyramidal cells (Cho et al., 2006; Minzenberg et al., 2010). Interestingly, neuropsin knock-out mice represent a similar phenotype, with spatial working memory impairment (Tamura et al., 2006) and intact reference memory, as well as increased anxiety behavior (Horii et al., 2008). Linkage analysis in humans has identified Nrg-1 as a susceptibility gene for bipolar disorder and schizophrenia (Stefansson et al., 2002; Green et al., 2005) and the neuropsin gene as a susceptibility gene for bipolar disorder (Izumi et al., 2008). Although linkage of the neuropsin gene to schizophrenia was not significant in Japanese populations, a Val286Ile missense mutation in exon 6 of klk8 (SNP22) was detected in a rare case of severe schizophrenia (Izumi et al., 2008). Our study provides a new insight into a psychiatry-related signaling system: the neuropsin-NRG-1-ErbB4-GABAergic connection. Neuropsin-dependent GABAergic regulation may play important roles in animal and human cognition, and its dysfunction may contribute to mental disorders such as schizophrenia and bipolar disorder.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research (No. 22700396 to H.T. and No. 20300128 to S.S.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, the CREST program of the Japan Science and Technology Agency, and the Global Centers of Excellence Program at the Nara Institute of Science and Technology (Frontier Biosciences: strategies for survival and adaptation in a changing global environment), MEXT, Japan. We thank Rie Kurata [Nara Institute of Science and Technology (NAIST)] for LS-MS/MS and Edman sequencing, Yoshie Tawara (NAIST) for plasmid construction, Dr. Honda Naoki (Kyoto University) for analyzing the kinetic parameters, and Dr. Ian Smith (NAIST) and Dr. Robert Pawlak (University of Leicester) for their valuable comments.
The authors declare no competing financial interests.
- Correspondence should be addressed to either Hideki Tamura or Sadao Shiosaka, Laboratory of Functional Neuroscience, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0192, Japan. h-tamura{at}bs.naist.jp or sshiosak{at}bs.naist.jp





{kind=link}
{kind=link}
{kind=link}
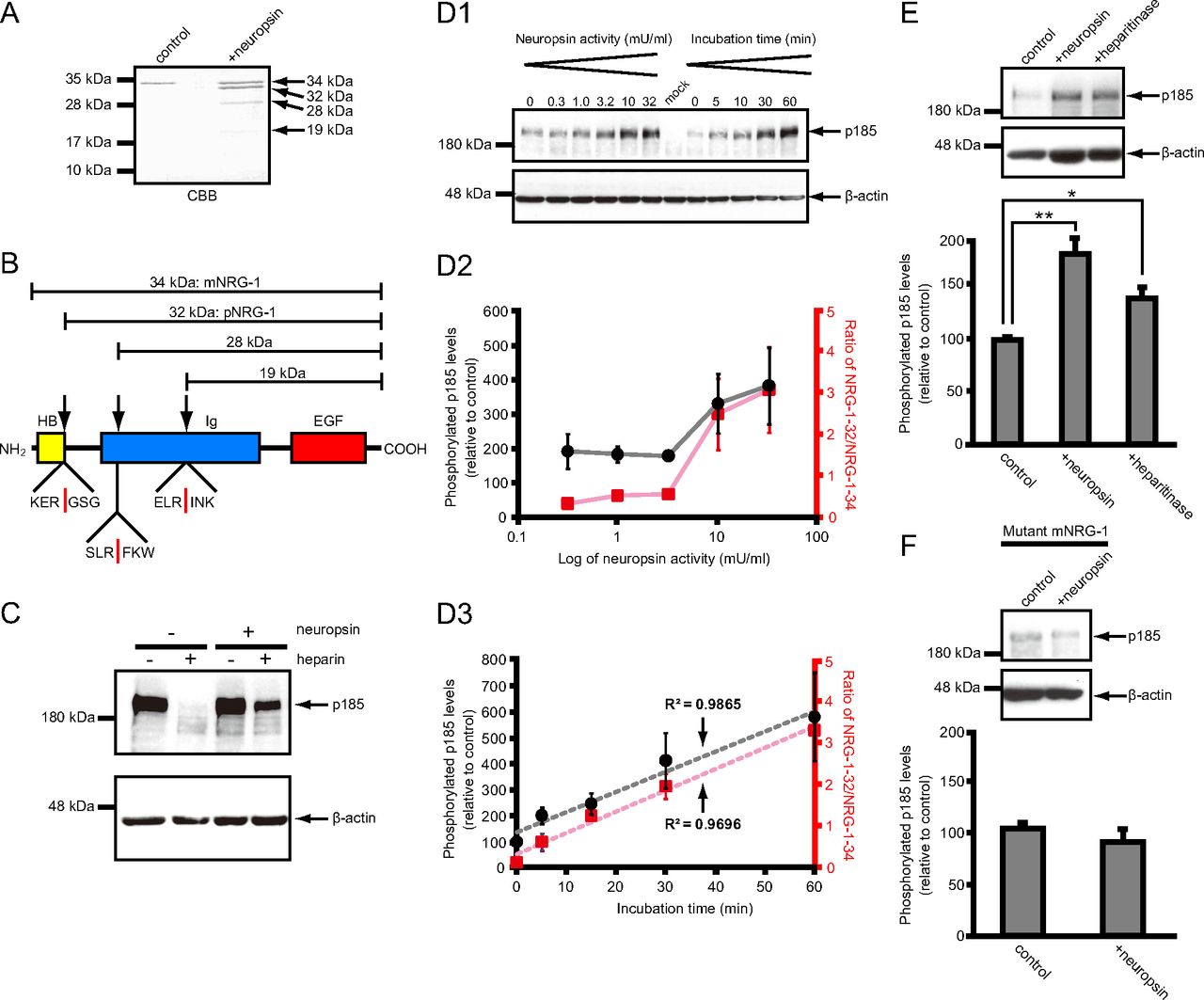{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}