

Abstract
The muscle-specific kinase MuSK is one of the key molecules orchestrating neuromuscular junction (NMJ) formation. MuSK interacts with the Wnt morphogens, through its Frizzled-like domain (cysteine-rich domain [CRD]). Dysfunction of MuSK CRD in patients has been recently associated with the onset of myasthenia, common neuromuscular disorders mainly characterized by fatigable muscle weakness. However, the physiological role of Wnt-MuSK interaction in NMJ formation and function remains to be elucidated. Here, we demonstrate that the CRD deletion of MuSK in mice caused profound defects of both muscle prepatterning, the first step of NMJ formation, and synapse differentiation associated with a drastic deficit in AChR clusters and excessive growth of motor axons that bypass AChR clusters. Moreover, adult MuSKΔCRD mice developed signs of congenital myasthenia, including severe NMJs dismantlement, muscle weakness, and fatigability. We also report, for the first time, the beneficial effects of lithium chloride, a reversible inhibitor of the glycogen synthase kinase-3, that rescued NMJ defects in MuSKΔCRD mice and therefore constitutes a novel therapeutic reagent for the treatment of neuromuscular disorders linked to Wnt-MuSK signaling pathway deficiency. Together, our data reveal that MuSK CRD is critical for NMJ formation and plays an unsuspected role in NMJ maintenance in adulthood.
Introduction
The formation of the neuromuscular junction (NMJ) relies upon the accurate apposition and contact between presynaptic nerve terminals and postsynaptic muscle targets. The muscle-specific tyrosine kinase receptor MuSK and its coreceptor Lrp4 (low density lipoprotein receptor) constitute the central hub orchestrating all steps of NMJ formation and maintenance (Wu et al., 2010; Burden et al., 2013). MuSK contains in its extracellular region a Frizzled-like domain (cysteine-rich domain [CRD]) mediating its interaction with several Wnts molecules, including Wnt4, Wnt11, and Wnt9a in vitro (Jing et al., 2009; Strochlic et al., 2012; Zhang et al., 2012). Activation of the MuSK-Lrp4 complex regulates the prepatterning step, before muscle innervation, during which AChRs begin to aggregate in a broad central and prospective synaptic region of the muscle (Lin et al., 2001; Yang et al., 2001; Arber et al., 2002; Weatherbee et al., 2006; Kim and Burden, 2008). Moreover, in vivo knockdown of Wnt4 and Wnt11 affects muscle prepatterning and axon guidance, indicating a role for Wnt signaling in this process (Jing et al., 2009; Gordon et al., 2012; Strochlic et al., 2012). Upon innervation, the MuSK-Lrp4 complex is further stimulated by neural agrin, which induces multiple signaling pathways leading to clustering and remodeling of aneural AChR clusters (Kim et al., 2008; Zhang et al., 2008; Wu et al., 2010). In addition to their role in prepatterning, Wnt proteins have been shown to regulate agrin-induced AChR clustering in vitro (Henriquez et al., 2008; Wang et al., 2008). However, the general role of Wnt-induced mammalian NMJ formation and maintenance in adult life mediated by MuSK remains largely unknown.
Mutations in genes or autoantibodies directed against proteins critical for NMJ formation or function are responsible for myasthenia, respectively congenital (CMS) or acquired (myasthenia gravis), mainly characterized by fatigable muscle weakness resulting from defective neuromuscular transmission (Hantaï et al., 2013; Berrih-Aknin et al., 2014). Recently, several cases of CMS linked to mutations in the CRD of MuSK as well as autoantibodies against the CRD and Ig1/2 domains of MuSK in anti-AChR negative myasthenia gravis patients were identified, indicating that dysfunction of MuSK CRD is associated with the onset of myasthenia (Mihaylova et al., 2009; Takamori, 2012, 2013).
In the present study, to get insight into the functional role of Wnt-MuSK-induced NMJ formation and maintenance in vivo, we generated knock-out mice in which MuSK CRD (MuSKΔCRD) was deleted. Analysis of the NMJ phenotype of MuSKΔCRD embryos revealed profound defects in both muscle prepatterning and synapse differentiation. More surprisingly, we found that MuSKΔCRD adult mice exhibited dislocation of NMJs and fatigable muscle weakness characteristic of CMS-like symptoms. We also demonstrated, for the first time, that lithium chloride (LiCl), a well-known reversible inhibitor of the glycogen synthase kinase-3 (Gsk3) rescued NMJ defects in MuSKΔCRD embryos and adult mice.
Collectively, our data demonstrate that the absence of MuSK CRD affects NMJ formation from the prepatterning step to NMJ maintenance in adult, leading to a pathogenic phenotype. Importantly, the biological benefits of LiCl treatment on NMJ formation in MuSKΔCRD mice suggest the use of Gsk3 inhibitors as potential therapeutic reagents to counteract neuromuscular disorders linked to Wnt/MuSK pathway deficiency.
Materials and Methods
All experiments on mice were performed in accordance with European Community guidelines legislation and reviewed by the local ethical committee of the Paris Descartes University (CEEA34.LS.030.12). The investigators had valid licenses (A-75-1970) to perform experiments on live vertebrates delivered by Direction des Services Vétérinaires (Préfecture de Police, Paris). The mutant line was established on the C57BL/6 genetic background, and experimental procedures were performed on mutant males and wild-type (WT) littermates.
Generation of MuSKΔCRD mutant mice and genotyping
The MuSKΔCRD/ΔCRD mutant mouse line, lacking MuSK 315–478 amino acids corresponding to the CRD, was established at the Mouse Clinical Institute using proprietary vector containing floxed neomycin resistance cassette and protamine-Cre cassette (Illkirch, France; http://www-mci.u-strasbg.fr). The use of protamine cassette in the construction vector offers an efficient solution for auto-excision of the floxed region when chimaera mice were bred with Cre-expressing mice. The targeting vector was constructed by successive cloning of PCR products and contained a 5.5 kb fragment (corresponding to the 5′ homology arm), a 4 kb floxed fragment including protamine Cre and neomycin selection cassettes, and a 5 kb fragment (corresponding to the 3′ homology arms). Two LoxP sequences delimiting the floxed fragment were located upstream of exon 9 and downstream of exon 11. The linearized construct was electroporated in BALB/CN mouse embryonic stem (ES) cells. Targeted ES clones were screened by 5′ external and Protamine Cre (inside the targeting vector) long-range PCR and by external and internal Southern blot. Two positive ES clones were injected into C57BL/6N blastocysts, and derived male chimaeras gave germline transmission. The resulting lines were crossed with a Cre deleter mouse generated on a pure inbred C57BL/6N background, in which the CRE gene is driven by the chicken β-actin promoter, and showing high and stable recombination efficiency to induce deletion of the floxed region (Birling et al., 2012). The following primers were used for PCR validation of the injected clones: 5′external PCR, Efl (forward) 5′ATTTGAAATGCTGATTTGGATCCAT3′ and Nr (reverse) 5′GCGGCCGGAGAACCTGCGTGCAATC3′; protamine Cre PCR, Cref (forward) 5′GGTAACTAAACTGGTCGAGCGATGG3′ and Er (reverse) 5′ATGGATCCAAATCA GCATTTCAAAT3′. A 3′ external probe (forward, 5′GGCGTAATGGAAGAAATAATCCT CT3′; reverse, 5′GACTGCAGAATCAGAAGGC3′) with Drd1 restriction enzyme and a neo probe with ApaL1, EcoR1, Drd1, and Xcm1 restriction enzymes were used for external and internal Southern blot analyses. Genotyping was performed in a single PCR reaction with a three primer set to detect both WT and mutated alleles. The following primers were used for genotyping: for the WT allele, Ef (forward) 5′CTC TTC TCC CTT CTG CCC ACC GAT3′ and Wr (reverse) 5′CCC TGG GAA TAT GGT TTC TCA TTG CT3′; for the MuSKΔCRD allele, Ef (forward) and Lxr (reverse) 5′AGT TAT ACT AGA GCG GCC GTT CAC CG3′. The WT amplified sequence was 234 bp long, whereas the knock-out amplification was 200 bp long.
Antibodies
The following antibodies were used: polyclonal and monoclonal Alexa Fluor 488-conjugated (Invitrogen, 1/1000), polyclonal Cy3 conjugated (Jackson ImmunoResearch Laboratories, 1/1000), monoclonal and polyclonal peroxidase-conjugated (GE Healthcare, 1/10,000), rabbit monoclonal anti-synaptophysin (Syn) (Invitrogen, 1/5), polyclonal anti-neurofilament (NF) 68 kDa (Millipore Bioscience Research Reagents, 1/1000), polyclonal anti-NF 165 kDa (DSHB, 1/750), polyclonal anti-HA (1/2500; Abcam), and monoclonal anti-β-catenin (Invitrogen, 1/500). Polyclonal anti-MuSK (Abcam, 1/200), monoclonal anti-transferrin receptor (Invitrogen, 1/500), and anti-α-tubulin (Sigma-Aldrich, 1/6000) were used for Western blot. Polyclonal anti-MuSK (1/500) used for immunohistochemistry and immunoprecipitation is a gift from M. Ruegg (Basel, Switzerland). DAPI (1/20,000) and α-BTX Alexa Fluor 488 conjugate (1/1000) were purchased from Euromedex and Invitrogen, respectively.
Plasmids
The rat MuSK-HA and MuSKΔCRD-HA cDNA plasmids have been previously described (Cartaud et al., 2004; Strochlic et al., 2012).
Biotinylation of cell surface MuSK and MuSKΔCRD, immunoprecipitation, and Western blot analyses
HEK293T cells (ATCC) were cultured in DMEM supplemented with 10% FBS, 2 mm glutamine, and 2%penicillin/streptomycin (500 U) at 37°C in 5% CO2. Cells were grown to 70% confluence and transfected (2 μg of plasmids) using Fugen (Promega) transfection technique. Forty-eight hours after transfection, biotinylation of cell surface proteins and Western blot were performed as described previously (Sigoillot et al., 2010). The membrane transferrin receptor was used as a loading control to normalize the results. Relative signal intensity of total and cell surface MuSK-HA or MuSKΔCRD-HA proteins was measured using ImageJ software. The levels of MuSK-HA and MuSKΔCRD-HA in total extracts were normalized to α-tubulin signals.
MuSK immunoprecipitation was performed from mutant or heterozygote E18.5 hindlimb muscles (N = 4 for each genotype) as described previously (Punga et al., 2011).
Muscle primary cultures
Muscle cells were isolated from P7-P10 hindlimb tibialis anterior (TA) and gastrocnemius muscles from MuSKΔCRD or WT mice. Briefly, muscle tissues were excised, separated from connective tissue, minced in dissecting medium (DMEM-F12 medium containing 2 mm glutamine, 2% penicillin/streptomycin (500 U), 2% Fungizone), and dissociated in dissecting medium containing 0.2% Type I collagenase (Invitrogen) for 90 min at 37°C. Cells were centrifugated, filtered, and resuspended in proliferating medium (dissecting medium supplemented with 20% horse serum and 2% Ultroser G, Pall). After overnight preplating, cells were expanded on Matrigel-coated dishes (Corning) for 3–5 d and differentiated in differentiating medium (dissecting medium supplemented with 2% horse serum) for 5 d. When indicated, myotubes were treated with recombinant agrin (0.4 μg/ml, R&D Systems), Wnt11 (10 ng/ml, R&D Systems), or LiCl (2.5 mm, Sigma) for 16 h.
CT scan analysis and measurement of the kyphotic index (KI)
Micro-CT scan analysis was performed in collaboration with the imaging platform PIPA installed in the imaging laboratory of EA 2496 (Montrouge). P90 WT and MuSKΔCRD mice were sedated using 1.5% isoflurane in air (TEC 3, Anestéo France). Entire body in dorsal and ventral decubitus of each animal was scanned by Quantum FX PerkinElmer micro-CT device (Caliper Rikagu) in dynamic mode. A tube voltage of 90 kV and a tube current of 160 μA were selected. Total scan time was 2 × 17 s per total animal scan. The scan field of view was 2 × 60 mm with a spatial resolution of 118 micros (voxel size). Each scan was achieved by optimizing the resolution/dose ratio. Therefore, for the resolution selected, each animal was exposed to a low dose of 26 mGy. Image reconstructions and measurements were performed by the Osirix software version 5.6. The KI was determined from the direct multiplanar reconstructions as previously described (Laws and Hoey, 2004). Briefly, the distance (D1) from the seventh cervical vertebra (C7) to the sixth lumbar vertebra (L6) and then the perpendicular distance (D2) from D1 to the point of maximum vertebra curvature were measured. KI corresponds to the ratio D1/D2. KI is inversely proportional to kyphosis.
Rail grip and grip strength measurements
Animals were placed on a suspended metal rail, and their latency to release the rail was recorded. Each mouse (N = 6 for each genotype) was subjected to five trials with at least 10 min rest period between tests. The same group of mice was tested for the grip test assay at different time points. The grip strength was measured using a grip force tensiometer (Bioseb) according to the TREAT-NMD guidelines. Forelimb and hind limb traction strength was recorded following the manufacturer's instructions. Three measurements were performed per animal.
Ex vivo isometric tension analyses
P120 WT and MuSKΔCRD mice were killed by dislocation of the cervical vertebrae followed by immediate exsanguination. Left hemidiaphragm muscles with their respective associated phrenic nerves were mounted in a silicone-lined bath filled with Krebs–Ringer solution of the following composition: 154 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 11 mm glucose, and 5 mm HEPES (buffered at pH 7.4 with NaOH), continuously perfused with O2 at 23.1 ± 0.4°C. One of the hemidiaphragm tendons (at the rib side) was securely anchored onto the silicone-coated bath via stainless steel pins while the other tendon was tied with silk thread, via an adjustable stainless steel hook, to an FT03 isometric force transducer (Grass Instruments). Muscle twitches and tetanic contractions were evoked by stimulating the motor nerve via a suction microelectrode adapted to the diameter of the nerve, with supramaximal current pulses of 0.15 ms duration, at frequencies indicated in the text. For each preparation investigated, the resting tension was adjusted at the beginning of the experiment (to obtain maximal contractile responses) and was monitored during the whole duration of the experiment. Signals from the isometric transducer were amplified, collected, and digitized with the aid of a computer equipped with an analog to digital interface board (Digidata 1200, Molecular Devices) using Axoscope 9 software (Molecular Devices).
Electron microscopy
P120 WT and MuSKΔCRD mice were sedated using 1.5% isoflurane in air (Minerve Equipement vétérinaire). TA muscles were then dissected and immediately fixed in 2% glutaraldehyde and 2% PFA in PBS for 1 h at room temperature and overnight at 4°C. TA muscles were rinsed in water, and AChE staining following Koelle's protocol was performed. The endplate-containing tissue blocks were cut in small pieces. Subsequently, tissue samples were washed three times in 0.1 m sodium phosphate buffer, incubated 30 min at 4°C in 1% osmic acid in sodium phosphate buffer, and dehydrated in graded ethanol solutions (2 × 10 min in 70%, 2 × 10 min in 90% and 2 × 10 min in 100% ethanol). Samples were then incubated twice in propylene for 1 min, 10 min in 50%–50% Epon-50% propylene oxide, then embedded in Epon and incubated for polymerization 24 h at 60°C. The 90 nm sections were cut on a Reichert Ultracut S and laid on a grid for staining 10 min with 2% uranyl acetate and 4 min with lead citrate at room temperature. The observations were performed with a JEOL 1011 transmission electron microscope and the images recorded at 80 kV with a GATAN Erlangshen 1000 camera.
Biochemical analyses of acetylcholinesterase activity
Frozen diaphragm and soleus muscles from P90 WT and MuSKΔCRD mice were homogenized in a protein extraction buffer (50 mm phosphate buffer, pH 7.6, 1% chaps, 0.8 m NaCl, and 10 mm EDTA) completed with a protease inhibitors mixture (Roche). Total AChE activity in the extracts was assayed using 0.7 mm acetylthiocholine and 0.5 mm 5,5-dithiobis(2-nitrobenzoic acid) (DTNB) in the presence of 50 μm tetra(monoisopropyl)-pyrophosphortetramide (Sigma). The extracts were first incubated in the absence of acetylthiocholine for at least 20 min to block butyrylcholinesterase and saturate the free sulfhydryl groups that interact with DTNB. The change in optical density was measured at 414 nm.
Immunohistochemistry
Whole-mount diaphragm staining.
Diaphragm muscles were dissected and fixed (4% PFA in PBS) for 1 h at room temperature and further fixed (1% formaldehyde in PBS) overnight at 4°C. Muscles were washed three times for 15 min in PBS, incubated for 15 min with 100 mm glycine in PBS, and rinsed in PBS. Muscles were permeabilized (0.5% Triton X-100 in PBS) for 1 h and blocked for 4 h in blocking buffer (3% BSA, 5% goat serum and 0.5% Triton X-100 in PBS). Muscles were incubated overnight at 4°C with rabbit polyclonal antibodies against NF and Syn in blocking solution. After three 1 h washes in PBS, muscles were incubated overnight at 4°C with Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 488-conjugated α-BTX in blocking solution. After three 1 h washes in PBS, muscles were flat-mounted in Vectashield (Vector Laboratories) mounting medium.
Tissue section staining.
Dissected TA muscles from P90 adult mice were fixed (1% PFA in PBS) for 1 h at 4°C, rinsed twice at 4°C in PBS, cryoprotected (30% sucrose in PBS) overnight at 4°C, embedded in TissueTek (Sakura), and quickly frozen in isopentane cooled in liquid nitrogen. Cryostat cross sections (12 μm) were permeabilized with 0.5% Triton X-100 in PBS for 10 min and labeled with various antibodies as for whole-mount immunostaining. Incubation times were the following: 1 h for blocking, overnight at 4°C for incubation with primary antibodies and 1 h for incubation with secondary antibodies.
Isolated muscle fiber staining.
Dissected and isolated TA muscle fibers from P20, P40, and P60 adult mice were fixed (4% PFA in PBS) for 30 min at 4°C and rinsed with PBS at room temperature. Isolated muscle fibers were labeled with antibodies as for whole-mount immunostaining. Incubation times were the following: 4 h for blocking, overnight at 4°C for incubation sucessively with primary or secondary antibodies.
Images acquisition and processing
All images were collected on a microscope (model BX61; Olympus) equipped with a Fast 1394 Digital CCD FireWire camera (model Retiga 2000r; Qimaging) and a 20× objective or on a confocal laser scanning microscope (Zeiss, LSM-710) equipped with a 20× objective and 63× oil objective. Collected Z-stack confocal images (5–20 stacks with 1–1.5 μm (20×) z-steps) and image capture were made using LSM Image Browser. The same laser power and parameter setting were applied to ensure reliable comparison between WT and MuSKΔCRD muscles. Multiple tile scanned images were taken to cover the size of the whole diaphragm. Confocal images presented are single-projected image derived from overlaying each set of stacks. For quantification of the AChR cluster number, volume, and intensity, image stacks were quantified using the ImageJ (version 1.46m) plugin “3D object counter” (Bolte and Cordelières, 2006). The threshold intensity was set by visual inspection of AChR clusters, being the same between WT and MuSKΔCRD images. The endplate band width was defined by the distance between the two farthest AChR clusters from the main nerve trunk. Approximately 100 measurements regularly spaced and covering the entire diaphragm were taken. At least 4 diaphragms or 50 isolated muscle fibers of each genotype were analyzed and quantified. To evaluate β-catenin translocation to subsynaptic nuclei in isolated muscle fibers, image stacks corresponding to nuclei were used for quantification using the ImageJ intensity plot profile measuring the intensity of β-catenin and DAPI fluorescence within the segmented line.
Neonatal and pregnant mice intraperitoneal injections
E12 pregnant mice or P10 adult mice were intraperitoneally injected with LiCl (Sigma, 600 mm 10 μl/g body weight) or placebo NaCl solution (0,0009% 10 μl/g body weight). Daily injections were made from E12 to E18 or from P10 to P60. After embryos genotyping, E18.5 or P60 diaphragms of LiCl-treated MuSKΔCRD mice were analyzed and compared with WT or NaCl-treated MuSKΔCRD.
Statistical analysis
Data are expressed as mean ± SEM. Statistical analyses and graphs were performed with Prism 6.0 (Graphpad) software. All data were analyzed using the Mann–Whitney U test or two-way ANOVA, wherever appropriate (p < 0.05 considered significant). Each experiment was conducted a minimum of three times.
Results
Generation of MuSKΔCRD transgenic mice
We generated a mouse line deleted from the MuSK CRD by homologous recombination (Fig. 1A). To construct the MuSK-CRD floxed allele, we created a targeting vector consisting of the MuSK gene flanked by two loxP sites upstream of exon 9 and downstream of exon 11. The construct was electroporated in BALB/CN mouse ES cells, and the injected clones were controlled by PCR (5′external and Protamine Cre PCR) followed by internal (neo probe with ApaL1 or Drd1) and external (3′ external probe with Drd1 restriction enzyme) Southern blot analyses to confirm 5′ and 3′ correct integration and absence of second random integration (Fig. 1B). Two positive and independent ES cell clones were injected into C57BL/6N blastocysts to generate two independent MuSKflox(CRD) mice. These mice were then crossed with Cre deleter mice to generate offspring with MuSK CRD deletion (see Material and Methods; Fig. 1C for genotyping). A single band corresponding to MuSK deleted from its CRD (85 kDa) was detected in E18.5 MuSKΔCRD hindlimb muscles after MuSK immunoprecipitation confirming that the CRD deletion occurred in the mutant mice (Fig. 1D). Both heterozygous MuSK+/ΔCRD and homozygous MuSKΔCRD/ΔCRD (MuSKΔCRD) mutant mice were indistinguishable from WT mice. Although 5% of the MuSKΔCRD mice died a few days after birth, being smaller in weight and size, exhibiting respiratory failure as well as limb motor deficiency, most of them were viable, able to suck milk, and developed up to adulthood with normal fertility. In addition, no difference in the weight curve between MuSKΔCRD and WT mice could be detected (data not shown).

Generation of MuSKΔCRD transgenic mice. A, Schematic representation of the KO strategy. First panel, Genomic structure of the MuSK gene encompassing MuSK CRD. Second panel, Targeting vector. Third panel, MuSK-targeted allele (MuSKflox(CRD)). Fourth panel, Recombinant MuSK allele (MuSKΔCRD). Ef, Wr, and Lxr (black arrowheads), primers for PCR genotyping; Efl, Nr, Cref, and Er, primers for PCR validation of the ES clones (blue arrowheads for 5′ external and violet arrowheads for protamine Cre PCR). Pink line indicates 3′ external Southern blot probe. Green line indicates neo internal Southern blot probe. A, ApaL1; D, Drd1; X, Xcm1. B, Examples of 5′ external and protamine Cre PCR validation and internal and external Southern blot hybridization analyses of two injected ES mutant clones (lanes 1 and 2). Amplified bands of the expected size for PCR (8.2 and 6 kb) and for 5′ internal (ApaL1, targeted band, 11.1 kb), 3′ internal (Drd1, targeted band, 9.5 kb), and 3′ external (Drd1; WT band, 7.4 kb; targeted band, 9.5 kb) Southern blots were obtained. C, Genotyping by PCR using a three primer set. The 234 and 200 bp bands represent WT and MuSKΔCRD alleles, respectively. D, Western blot of MuSK or MuSKΔCRD using MuSK antibodies in E18.5 MuSK+/ΔCRD or MuSKΔCRD hindlimb muscle after MuSK immunoprecipitation. E, Western blot of cell surface and total MuSK-HA and MuSKΔCRD-HA in HEK293T cells transfected with MuSK-HA or MuSKΔCRD-HA. Transferrin receptor (Tfr) and α-tubulin were used as a loading control for biotinylated proteins and input, respectively. Transfection of MuSK-WT-HA or MuSKΔCRD-HA was performed in duplicate. F, Confocal images of P60 WT or MuSKΔCRD TA muscle cross sections stained with MuSK antibody (red) together with α-BTX (AChR, green). G, Quantification of WT and mutated MuSK signal intensities at the synapse. H, Koelle's histochemical staining of AChE performed on isolated TA muscle fibers from P90 WT and MuSKΔCRD. I, Quantification of AChE activity extracted from P90 WT and MuSKΔCRD soleus and diaphragm. J, Examples of myotubes isolated from WT or MuSKΔCRD primary cultures, treated or not with recombinant agrin and stained with α-BTX. K, Quantitative analysis of the number of AChR clusters in WT and MuSKΔCRD myotubes. Data are mean ± SEM. ***p < 0.001. ns, Not significant. N = 3 independent experiments. Scale bar: F (merged image), J, 20 μm; H, 50 μm.
Given that deletion of MuSK CRD could lead to a deficit of the mutated MuSK expression at the cell membrane, we quantified the level of membrane WT or mutated MuSK using in vitro biotinylation experiments and tissue immunohistochemistry. Labeling of surface proteins by biotinylation in HEK293T cells expressing MuSK-HA or MuSKΔCRD-HA showed similar levels of MuSK and MuSKΔCRD at the plasma membrane (Fig. 1E). Moreover, both WT and mutated MuSK colocalized with membrane AChRs clusters labeled with α-BTX in P60 WT and MuSKΔCRD TA, indicating that deletion of MuSK CRD does not disrupt the mutated MuSK membrane localization at the NMJ (Fig. 1F). Quantification of the level of WT and mutated MuSK signal intensity at the synapse showed an increase of 70% in MuSKΔCRD compared with WT mice (Fig. 1G). In addition, because MuSK localization at the synapse is required for anchoring AChE in the postsynaptic membrane (Cartaud et al., 2004), we asked whether AChE localization and activity were disturbed in MuSKΔCRD muscles. Histochemical staining revealed that AChE was accumulated in the postsynaptic membrane of both WT and MuSKΔCRD P90 TA muscles (Fig. 1H). No difference in AChE activity could be detected between WT and MuSKΔCRD muscles (Fig. 1I).
To test whether deletion of MuSK CRD could disturb agrin-Lrp4-MuSK signaling, we quantified agrin-induced AChR clusters in WT and MuSKΔCRD primary muscle cultures. No difference in the number of AChR clusters following agrin treatment was detected in MuSKΔCRD compared with WT primary myotubes, indicating that MuSK CRD deletion does not perturb agrin-induced AChR clustering. These data suggest that agrin is able to interact with Lrp4 in the Lrp4/MuSKΔCRD complex and that MuSKΔCRD/Lrp4 interaction can transduce downstream signaling (Fig. 1J,K).
Deletion of MuSK CRD affects NMJ formation
Given that Wnts are known to play a role during the early steps of NMJ formation through binding to MuSK CRD (Jing et al., 2009; Strochlic et al., 2012), we tested whether deletion of MuSK CRD could lead to NMJ defects during development. First, we compared NMJ phenotype in E14 WT and MuSKΔCRD embryos (Fig. 2). Whole-mount diaphragms were stained with α-BTX to detect AChR clusters and with a mixture of antibodies against neurofilament (NF) and synaptophysin (Syn) to label axonal branches and nerve terminals, respectively. Both dorsal and ventral portions of each hemidiaphragm were innervated, indicating that axonal extension is fully developed in MuSKΔCRD embryos (Fig. 2A). Nonetheless, the neurites were increased in length by 82% (Fig. 2B,C). In addition to this presynaptic defect, AChR clustering was also affected in MuSKΔCRD embryos. In WT embryos, AChR clusters were concentrated in the central zone of the muscle as expected during prepatterning. In contrast, in mutant embryos, AChR clusters were almost undetectable and were distributed in a twofold wider muscle area (Fig. 2B,D, arrowheads). Quantitative analysis revealed a mean 64% decrease in the number of MuSKΔCRD AChR clusters (Fig. 2E). Moreover, MuSKΔCRD AChR clusters volume and intensity were reduced by 70% and 65%, respectively, and non-innervated AChR clusters were increased by 30% in MuSKΔCRD compared to WT embryos (Fig. 2F–H). These results suggest that deletion of MuSK CRD leads to an early developmental defect of the NMJ with a severely reduced AChR prepatterning.

Impaired muscle prepatterning in MuSKΔCRD embryos. A, B, Confocal images of whole-mount left hemidiaphragms from E14 WT and MuSKΔCRD embryos stained with NF (red) and Syn (red) antibodies (A, B) together with α-BTX (AChRs, green, B). Arrowheads indicate AChR clusters. White dashed lines indicate the synaptic endplate band and include most AChR clusters. C–H, Quantitative analysis of the mean neurite length (C), the endplate band width (D), the AChR cluster number (E), volume (F), intensity (G), and non-innervated AChR clusters (H). Number of AChR clusters analyzed: 790 in WT and 461 in MuSKΔCRD. Data are mean ± SEM. *p < 0.05 (Mann–Whitney U test). **p < 0.01 (Mann–Whitney U test). ***p < 0.001 (Mann–Whitney U test). N = 4 embryos per genotype. Scale bar: A, 300 μm; B (merged image), 50 μm.
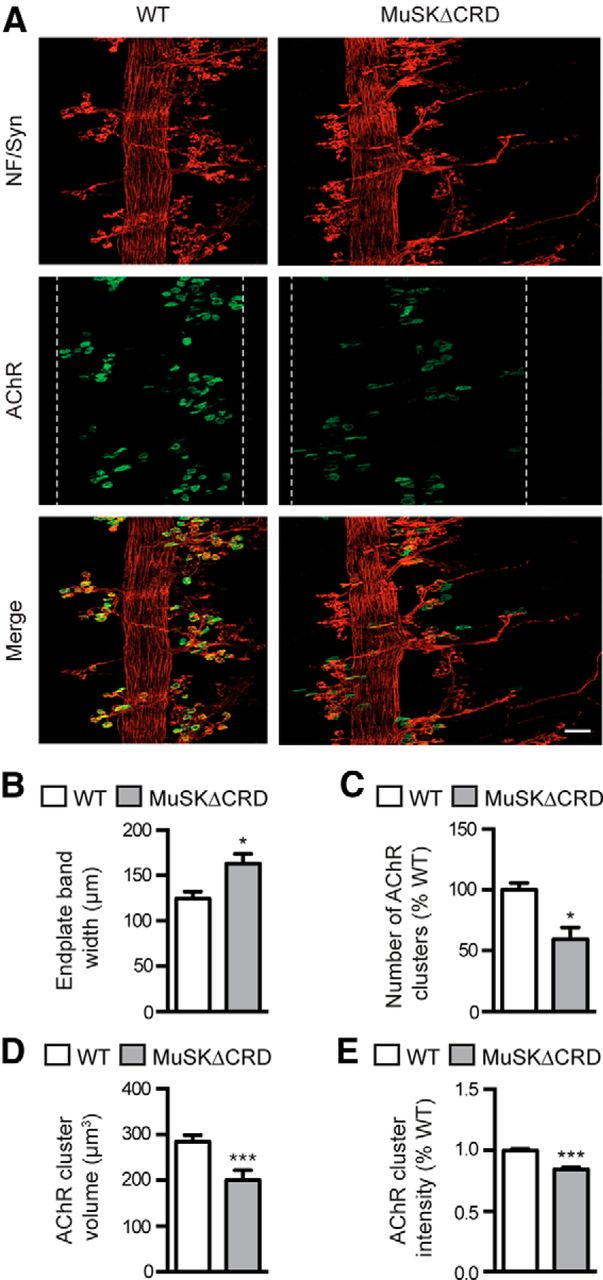
We further analyzed NMJ morphology later during development in E18.5 MuSKΔCRD diaphragms (Fig. 3). Whereas AChR clusters were tightly restricted to a thin endplate band in WT hemidiaphragms, the endplate band width was twofold larger in MuSKΔCRD (Fig. 3A,B). Furthermore, AChR clusters were reduced in number by 40% (Fig. 3C), volume by 25% (Fig. 3D), and intensity by 14% (Fig. 3E). All AChR clusters were innervated in WT and MuSKΔCRD; however, MuSKΔCRD embryos showed aberrant extension of motor axons, bypassing AChR clusters and growing excessively toward the periphery of the muscle (Fig. 3F). Although the number of primary and secondary branches was not significantly affected, the length of primary as well as secondary branches was increased by 75% and 46%, respectively, in MuSKΔCRD mutants compared with WT embryos (Fig. 3G–J).

Aberrant NMJ formation in E18.5 MuSKΔCRD embryos. A, Confocal images of whole-mount left hemidiaphragms from E18.5 WT and MuSKΔCRD embryos stained with α-BTX to visualize AChR clusters. Right panels, Enlarged images of boxed regions in left panel. White dashed lines indicate the synaptic endplate band and include most AChR clusters. Right panels, Insets, Higher-magnification views of AChR clusters. B–E, Quantifications of the endplate band width (B), the AChR cluster number (C), volume (D), and intensity (E). Numbers of AChR clusters analyzed: 806 in WT and 604 in MuSKΔCRD. F, Confocal images of whole-mount left hemidiaphragms from E18.5 WT and MuSKΔCRD embryos stained as in Figure 2B. G–J, Quantitative analysis of the length (G, H) and the number (I, J) of primary and secondary nerve branches. Number of primary branches analyzed: 273 in WT and 309 in MuSKΔCRD; secondary branches: 266 in WT and 310 in MuSKΔCRD. Data are mean ± SEM. *p < 0.05 (Mann–Whitney U test). **p < 0.01 (Mann–Whitney U test). ***p < 0.001 (Mann–Whitney U test). ns, Not significant. N = 6 embryos per genotype. Scale bar, A, 300 μm; F (merged image), 50 μm.
Together, these results indicate that MuSK CRD deletion drastically perturbs NMJ formation as exemplified by reduced prepatterned and neural AChR clusters as well as exuberant neurite outgrowth.
MuSKΔCRD adult mice exhibit immature and fragmented NMJs
As previously mentioned, MuSKΔCRD mutant mice are viable at birth. Therefore, we wondered whether NMJ defects observed during development would be also detected in newborn and adult mutant mice. NMJ phenotype analyzed in P5 WT and MuSKΔCRD whole-mount preparations of diaphragm revealed AChR cluster deficit and neurite outgrowth defects in MuSKΔCRD similar to those observed in MuSKΔCRD embryos (Fig. 4A–E). To assess whether the presynaptic and postsynaptic counterparts were still affected in MuSKΔCRD adult mice, we analyzed NMJ morphology on isolated muscle fibers from WT and MuSKΔCRD TA muscles at P20 and P60 (Fig. 5). At birth, the shape of the endplates is ovoid and as NMJs mature, the endplates begin to acquire their branched postnatal topology (Marques et al., 2000; Kummer et al., 2006). In P20 MuSKΔCRD mice, most of the endplates analyzed were ovoid and compact compared with the perforated WT ones, suggesting that NMJs are immature (Fig. 5A, left). Both analyzed AChR endplates of WT and MuSKΔCRD mice were innervated as confirmed by nerve terminal staining, which colocalized with AChR clusters (Fig. 5A, left). However, quantitative analysis revealed that the number of AChR clusters as well as nerve terminal area per NMJ were significantly reduced by 49% and 58%, respectively, in MuSKΔCRD mice compared with WT mice without affecting the final overlap area between presynaptic and postsynaptic elements (Fig. 5B–D).

Diaphragm innervation defects in P5 MuSKΔCRD mice. A, Confocal images of whole-mount P5 WT and MuSKΔCRD left hemidiaphragms stained as Figure 2B. White dashed lines indicate the synaptic endplate band and include most AChR clusters. B–E, Quantitative analysis of the endplate band width (B), the AChR cluster number (C), volume (D), and intensity (E). Numbers of AChR clusters tested: 263 in WT and 120 in MuSKΔCRD. Data are mean ± SEM. *p < 0.05 (Mann–Whitney U test). ***p < 0.001 (Mann–Whitney U test). N = 4 embryos per genotype. Scale bar, 50 μm.

Immature and fragmented NMJs in MuSKΔCRD adult mice. Whole-mount isolated muscle fibers from P20 and P60 WT and MuSKΔCRD TA were stained with NF (red) and Syn (red) antibodies together with α-BTX (AChRs, green). A, Confocal images of synapses from P20 and P60 WT and MuSKΔCRD mice. For P60 NMJs, top views of the reconstructed image are represented on the right side. B–D, Quantification analysis of the AChR cluster area (B), the Syn area (C), and overlap area of presynaptic and postsynaptic elements (D) in P20 WT and MuSKΔCRD mice. E–H, Quantification analyses of the number of fragments per AChR clusters (E), the AChR cluster area (F), the Syn area (G), and the overlap ratio of presynaptic and postsynaptic elements (H) in P60 WT and MuSKΔCRD mice. Data are mean ± SEM of at least 50 NMJs. *p < 0.05 (Mann–Whitney U test). **p < 0.01; ***p < 0.001 (Mann–Whitney U test). ns, Not significant. N = 6 animals per genotype. Scale bar (merged image), 10 μm.
At P60, WT NMJs formed a continuous branched postnatal topology and exhibited a typical “pretzel-like” structure (Fig. 5A, right). At this stage, 10% of the analyzed NMJs in MuSKΔCRD were similar in shape to WT ones (data not shown). However, the structure of most MuSKΔCRD synapses (90% of the analyzed NMJs) was severely altered with the following characteristics: the postsynaptic network was discontinuous and isolated AChR clusters were frequently observed, suggesting fragmentation of the NMJs. Indeed, the number of AChRs fragments per NMJ was increased by fourfold (Fig. 5E). Moreover, the total occupied AChR clusters area per NMJ was significantly reduced by 40%, suggesting a loss of AChR-rich domains (Fig. 5F). Axonal branches appeared discontinuous and fragmented in correlation with the postsynaptic apparatus parceling. The nerve terminal area was reduced by 39% without affecting the overlap area between presynaptic and postsynaptic elements (Fig. 5G,H). Similar NMJ defects were observed in other muscle types including fast-twitch extensor digitorum longus and slow-twitch soleus (data not shown).
Together, our findings indicate that the CRD deletion of MuSK perturbs NMJs maturation that finally leads to a severe dismantlement of the postsynaptic and presynaptic apparatus in adult mice.
MuSKΔCRD mice display altered NMJ ultrastructures
To analyze the morphological alterations of muscle and NMJs at the ultrastructural level in MuSKΔCRD mice, we performed electron microscopic analysis on TA muscle of P120 WT and MuSKΔCRD mice (Fig. 6A–M). Although presynaptic specialization of nerve terminals appeared normal in MuSKΔCRD mice with abundant mitochondria in the axoplasm, quantification analyses revealed a 67% decrease in MuSKΔCRD synaptic vesicle density compared to WT mice without affecting the mean synaptic vesicle diameter (Fig. 6H,I). Moreover, on the postsynaptic side, ∼30% of MuSKΔCRD NMJs exhibited highly disorganized or very few junctional folds (JFs) compared with WT mice (78% decrease of JFs in MuSKΔCRD; Fig. 6A–F,J). In some cases, compared to WT, the distance between JFs and muscle fibers was increased and abundant accumulation of mitochondria beneath the postsynaptic membrane was observed in MuSKΔCRD mice (Fig. 6G). However, no morphological difference between WT and MuSKΔCRD overall muscle structure could be detected (Fig. 6K). Indeed, the sarcomeric organization and the distance between Z-lines were similar between WT and MuSKΔCRD mice (Fig. 6L). In addition, MuSKΔCRD mice did not show any alteration in the myelin sheath diameter (Fig. 6M).

Disorganized NMJ ultrastructures in MuSKΔCRD mice. A–G, Representative electron micrographs of P120 WT and MuSKΔCRD TA NMJs. A–C, Examples of WT NMJs. D–G, Examples of MuSKΔCRD NMJs. B, E, Higher-magnification views of A and D, respectively. H–J, Quantification analyses of the synaptic vesicle density (H), diameter (I), and the number of JFs (J) in MuSKΔCRD compared to WT mice. K, Representative electron micrograph of P120 WT and MuSKΔCRD TA structure. L, Quantification of the distance between Z-lines in MuSKΔCRD and WT mice. M, Representative electron micrograph of myelin sheath in P120 WT and MuSKΔCRD TA. Data are mean ± SEM. *p < 0.05 (Mann–Whitney U test). N = 4 animals per genotype. ns, Not significant; N, nerve; MF, muscle fiber; SVs, synaptic vesicles; m, mitochondria; SBL, synaptic basal lamina. Black arrows indicate presynaptic membrane. Black arrowheads indicate postsynaptic membrane. White arrows indicate Z-line. Stars indicate M-line. Scale bars: A–G, 500 nm; H, J, 1 μm.
Collectively, these results indicate that MuSK CRD deletion alters both presynaptic and postsynaptic ultrastructures, which may result in a motor activity deficit.
MuSKΔCRD mice progressively develop muscle weakness, fatigue, and motor defects
During the first 2 weeks after birth, MuSKΔCRD mice developed normally without any gross physical signs of muscle weakness compared with WT mice. After this period, changes in the trunk region became progressively evident by the abnormal spine curvature caused by the shrinkage of the pelvic and scapular region. The CT scan analysis performed on P90 WT and MuSKΔCRD mice illustrated the kyphosis developed by MuSKΔCRD mice (Fig. 7A). kyphotic index (KI; see Material and Methods) used to appreciate the kyphotic severity degree was significantly reduced in MuSKΔCRD compared to WT, confirming the presence of a severe spine deformation in mutant mice (Fig. 7B) (Laws and Hoey, 2004). To assess whether motor function was altered in MuSKΔCRD mice, we performed a grip test assay on young and adult mice. Whereas the latency to fall from the rail increased with age in WT mice, MuSKΔCRD mice exhibited poor motor performance from P20 to P90 as determined by a reduced latency to fall (latency decrease: P20, 62%; P40, 33%; P60, 64%; P90, 72%; Fig. 7C). To further investigate the origin of motor defect in MuSKΔCRD mice, we measured the grip strength of the forelimbs (Fig. 7D) and the hindlimbs (Fig. 7E). In MuSKΔCRD as in WT mice, the forelimb and the hindlimb grip strength increased with age. However, the grip strength of MuSKΔCRD mice was reduced at all time points compared to WT mice, indicating a muscle weakness (forelimb grip strength decrease: P20, 26%; P40, 23%; P60, 20%; P90, 30%; hindlimb grip strength decrease: P20, 37%; P40, 18%; P60, 23%; P90, 21%; Fig. 7D,E).

MuSKΔCRD mice progressively develop muscle weakness, fatigability, and decreased muscle contraction. A, Micro-CT scans of P90 WT and MuSKΔCRD mice. B, P120 WT and MuSKΔCRD KI. C, Latency to fall quantifications during a rail-grip test at various time points (P20, P40, P60, and P90). D, E, Quantification of forelimb (D) and hindlimb (E) grip strengths in WT and MuSKΔCRD mice. F, Representative examples of twitch and tetanic contractions evoked by stimulation of the motor nerve in WT and MuSKΔCRD P120 isolated mouse hemidiaphragms. The phrenic nerve was stimulated either with single or tetanic stimuli (600 ms duration) at 20, 40, 60, 80, and 100 Hz. G, H, Peak amplitudes of nerve-evoked single twitch and tetanic stimulations in WT and MuSKΔCRD mice. I, Example of repeated tetanic nerve stimulation (60 Hz, 600 ms duration at 1 Hz) that induced a degree of fatigue more pronounced in MuSKΔCRD than in WT. J, Quantification of the fatigability in WT and MuSKΔCRD. K, Example of spontaneous twitch induced by unique stimulation observed in MuSKΔCRD muscles. Bottom line indicates the stimulator. Calibration scales in WT apply to MuSKΔCRD. L, Confocal images of whole-mount P90 WT and MuSKΔCRD left hemidiaphragms stained as in Figure 2B. White arrows indicates loss of postsynapse. White arrowheads indicates denervated postsynapses. White stars indicate fragmented NMJ. Data are mean ± SEM. *p < 0.05 (Mann–Whitney U test). **p < 0.01 (Mann–Whitney U test). N = 3 animals per genotype in A, B, L. N = 6 animals per genotype in C–E. N = 5 animals per genotype in F–K. Scale bar: L, 50 μm.
To confirm the occurrence of muscle weakness in MuSKΔCRD, we analyzed the ability of P120 WT and MuSKΔCRD left hemidiaphragms to evoke twitches and tetanic contractions in response to phrenic nerve stimulation at different frequencies ex vivo. As shown in Figure 7F, WT as MuSKΔCRD muscles developed and maintained tetanic contractions, in a wide range of nerve-stimulation frequencies. However, MuSKΔCRD muscles developed less force than WT ones. Indeed, in MuSKΔCRD mice, the strength of muscle twitch upon nerve stimulation was significantly reduced compared to WT mice, both upon single and tetanic stimulations (strength decrease of muscle twitch: single twitch, 59%; T40 Hz, 54%; T60 Hz, 47%; T80 Hz, 44%; T100 Hz, 42%; Fig. 7G). The developed muscle specific force, defined as the muscle strength (mN) normalized to the muscle weight (g) was significantly reduced in MuSKΔCRD compared to WT muscles, both upon single and tetanic (T100 Hz) stimulations by 52% and 24%, respectively (Fig. 7H). One expected pathophysiological consequence of NMJ structural changes is fatigable muscle weakness as observed in myasthenia (Hantaï et al., 2013). We therefore evaluated the muscle fatigue strength after a train of tetanic nerve stimulations (T60 Hz) and found that MuSKΔCRD muscles exhibited a degree of fatigue more pronounced than WT muscles (fatigability increase: 30%; Fig. 7I,J). Interestingly, we also observed spontaneous twitches after one twitch induced by unique stimulation in MuSKΔCRD mice, suggesting the presence of muscle denervation processes (Fig. 7K) (Heckmann and Ludin, 1982). To further confirm this issue, non-innervated AChR clusters could be detected in P90 MuSKΔCRD whole-mount diaphragm preparations (Fig. 7L).
Our data demonstrate that MuSK CRD deletion compromises motor performances, affects muscle strength, and leads to increased muscle fatigability. This is in agreement with clinical symptoms generally observed in patients suffering from CMS.
Lithium chloride rescues NMJ phenotype of MuSKΔCRD mice
Deletion of MuSK CRD, impairing Wnt/MuSK interaction, is likely to perturb Wnt signaling at the NMJ. Indeed, treatment of WT primary myotubes with Wnt11, a member of the Wnt family known to interact with MuSK CRD and required for AChR clustering (Jing et al., 2009; Zhang et al., 2012), induced a 4.5-fold increase in the number of AChR clusters that was fully abolished (80% decrease) in Wnt11-treated MuSKΔCRD primary myotubes, demonstrating that deletion of MuSK CRD alters Wnt-induced AChR clustering (Fig. 8A). In addition, β-catenin translocation to the nucleus was strongly reduced in Wnt11-treated MuSKΔCRD compared with Wnt11-treated WT primary myotubes, indicating that Wnt activation of the canonical pathway is affected in MuSKΔCRD muscle cells (Fig. 8C). Because previous reports suggest that the Wnt canonical pathway is involved in neuromuscular synapse formation (Li et al., 2008; Liu et al., 2012; Wu et al., 2012a), we then reasoned that forced activation of the Wnt β-catenin signaling pathway during development could thwart impaired NMJ formation and compensate at least partially MuSKΔCRD NMJ phenotype. To test this hypothesis, we set up a pharmacological approach using lithium chloride (LiCl), a well-known reversible inhibitor of Gsk3 kinase and activator of Wnt/β-catenin signaling (Klein and Melton, 1996; Stambolic et al., 1996; Wada, 2009). LiCl treatment of MuSKΔCRD primary myotubes resulted in a sixfold increase in the number of AChR clusters and increased β-catenin translocation to the nucleus compared with Wnt11-treated MuSKΔCRD myotubes, suggesting that LiCl is able to rescue AChR clustering and Wnt canonical signaling in MuSKΔCRD myotubes (Fig. 8B,C). We then tested the effect of LiCl treatment on NMJ formation in vivo in MuSKΔCRD mice. Repeated intraperitoneal injections of LiCl or placebo (NaCl) from E12 to E18.5 in pregnant mice were performed, and the phenotype of E18.5 LiCl-treated MuSKΔCRD NMJs was compared with NaCl-treated MuSKΔCRD and WT NMJs (Fig. 8D). Remarkably, LiCl treatment almost fully rescued the postsynaptic phenotype in E18.5 MuSKΔCRD embryos (Fig. 8D). The endplate band width in LiCl-treated MuSKΔCRD embryos was reduced by 23% compared to NaCl-treated mutant embryos and was similar to the endplate band of WT embryos (Fig. 8E). Moreover, LiCl-treated MuSKΔCRD embryos significantly gained AChR clusters in number (+ 103%), volume (+ 186%), and intensity (by 20%) and were almost indistinguishable from WT embryos (Fig. 8F–H). In addition, presynaptic defects were improved in LiCl-treated MuSKΔCRD. The increased length of primary and secondary branches observed in MuSKΔCRD was reduced by 35% and 28%, respectively, in LiCl-treated MuSKΔCRD embryos being almost similar to WT embryos (Fig. 8I,J). In addition, the number and length of bypassing neurites were reduced by 33% and 138%, respectively, in LiCl-treated MuSKΔCRD embryos compared to NaCl-treated ones (Fig. 8K,L). Together, these results indicate that LiCl treatment almost fully rescued both presynaptic and postsynaptic defects of MuSKΔCRD mutants, with NMJs being phenotypically indistinguishable from WT NMJs.

LiCl treatment rescues NMJ defects in MuSKΔCRD embryos. A, B, Quantitative analysis of the number of AChR clusters in myotubes isolated from WT or MuSKΔCRD primary cultures and treated or not with Wnt11 (A) or LiCl (B). C, Examples of Wnt11-treated WT, Wntt11-treated MuSKΔCRD, and LiCl-treated MuSKΔCRD primary myotubes stained with β-catenin together with DAPI to visualize β-catenin translocation to nuclei (white arrowheads). D, Confocal images of whole-mount E18.5 WT, NaCl-treated MuSKΔCRD, and LiCl-treated MuSKΔCRD left hemidiaphragms stained as in Figure 2B. White dashed lines indicate the synaptic endplate band and include most AChR clusters. E–L, Quantification of the endplate band width (E), AChR cluster number (F), volume (G), and intensity (H). Number of AChR clusters tested: 2235 in WT, 846 in NaCl-treated MuSKΔCRD embryos, and 1714 in LiCl-treated MuSKΔCRD embryos. F–I, Quantitative analyses of the length of primary and secondary nerve branches (I, J), the number and the length of bypassing neurites (K, L). At least 300–400 primary and secondary nerve branches were analyzed per condition. Data are mean ± SEM. *p < 0.05 (two-way ANOVA or Mann–Whitney U test). **p < 0.01 (two-way ANOVA or Mann–Whitney U test). ***p < 0.001 (two-way ANOVA or Mann–Whitney U test). ns, Not significant. N = 6 embryos per genotype. Scale bar: C (merged image), 20 μm; D, 50 μm.
To further investigate the beneficial effect of LiCl treatment on NMJ maintenance in adulthood, MuSKΔCRD mice were injected with LiCl or placebo (NaCl) from P10 to P60 and NMJ morphology and motor functions as well as β-catenin translocation to subsynaptic nuclei were analyzed (Fig. 9). LiCl treatment resulted in a strong increase of β-catenin translocation to subsynaptic nuclei in LiCl-treated MuSKΔCRD compared with NaCl-treated MuSKΔCRD isolated P40 TA muscle fibers, as shown in the intensity plot profiles measuring the fluorescence intensity of β-catenin and DAPI along the segmented lines (Fig. 9A). In addition, we found that NMJ structures of P40 TA-isolated muscle fibers from LiCl-treated MuSKΔCRD were increased in size compared with NaCl-treated MuSKΔCRD NMJ (Fig. 9B). The AChR cluster and Syn area in LiCl-treated mutants were increased by 40% and 105%, respectively, compared to NaCl-treated mice (Fig. 9C,D). Importantly, the number of AChRs fragments per NMJ in LiCl-treated mutants was decreased by 44% compared to NaCl-treated mice (Fig. 9E). Moreover, LiCl treatment significantly improved MuSKΔCRD mice latencies to fall from the rail grip as well as fore and hindlimb strength compared to NaCl-treated MuSKΔCRD mice (Fig. 9F–H). Together, these data demonstrate that LiCl treatment to postnatal MuSKΔCRD mice improves the NMJ morphological defects and muscle strength and restores β-catenin translocation to synaptic nuclei, suggesting that MuSK CRD plays a role during NMJ maintenance in adulthood likely in part via activation of the Wnt β-catenin signaling pathway.

LiCl treatment restores NMJ morphological defects and motor function in adult MuSKΔCRD mice. A, Confocal images of synapses from P40 NaCl-treated or LiCl-treated MuSKΔCRD whole-mount isolated TA muscle fibers stained with β-catenin (red) antibody together with α-BTX (AChRs, green) and DAPI (blue). Examples of intensity plot profiles measuring the fluorescence intensity of β-catenin and DAPI along the segmented lines corresponding to a subsynaptic nucleus are represented on the right side: (a), NaCl-treated MuSKΔCRD; (b), LiCl-treated MuSKΔCRD. B, Confocal images of synapses from P40 WT, NaCl-treated, or LiCl-treated MuSKΔCRD whole-mount isolated TA muscle fibers stained with NF (red) and Syn (red) antibodies together with α-BTX (AChRs, green). C–E, Quantification analyses of the AChR cluster area (C), the Syn area (D), and the number of fragments per AChR clusters (E). F, Latency to fall quantifications during a rail-grip test at various time points (P20, P40, P60, and P90). G, H, Quantification of fore (G) and hindlimb (H) grip strength in WT, NaCl-treated, or LiCl-treated MuSKΔCRD mice. Data are mean ± SEM of at least 50 NMJs. *p < 0.05 (two-way ANOVA). **p < 0.01 (two-way ANOVA). ***p < 0.001 (two-way ANOVA). ns, Not significant. N = 6 animals per genotype. Scale bar: A, B (merged image), 10 μm.
Discussion
Here, we have investigated the functional role of the MuSK-Wnt binding domain (CRD) during NMJ formation and maintenance in vivo. To this end, we generated mutant mice deficient for MuSK CRD. Deletion of MuSK CRD leads to severe alteration of both presynaptic and postsynaptic elements during early muscle prepatterning (E14) and NMJ differentiation (E18.5) mainly characterized by the following: (1) a drastic deficit in AChR clusters and (2) exuberant axonal growth bypassing AChR clusters. Moreover, MuSK CRD deletion is pathogenic in adult mice, inducing CMS-like symptoms, including kyphosis, NMJ dismantlement, muscle weakness, and fatigability as previously observed in other mice models of CMS (Gomez et al., 1997; Chevessier et al., 2008, 2012; Bogdanik and Burgess, 2011; Webster et al., 2013). We also report that NMJ innervation defects in MuSKΔCRD mice can be rescued in vivo by LiCl treatment. Together, our data uncover a critical role for MuSK CRD in NMJ formation and function in adulthood.
Wnts proteins are known to be involved in muscle prepatterning early during NMJ formation (Wu et al., 2010). Moreover, in zebrafish, Wnt11r induced aneural AChRs clustering requires the CRD of Unpplugged/MuSK (Jing et al., 2009). However, zebrafish lacking muscle prepatterning are able to form NMJ and are fully motile, leaving open the question of the exact role of the prepatterning in NMJ functioning (Jing et al., 2009; Gordon et al., 2012). Here, we demonstrate that deletion of MuSK CRD in mammals severely impairs muscle prepatterning because the number of AChR clusters is drastically reduced (63%) and non-innervated AChR clusters are strongly increased (30%) in E14 MuSKΔCRD embryos. Why deletion of MuSK CRD does not fully abolish muscle prepatterning remains unclear. Three hypotheses could explain this observation: (1) It has been shown in vitro that deletion of MuSK CRD reduces but does not fully inhibit the binding activity of Wnt proteins to MuSK (Zhang et al., 2012; Barik et al., 2014). Thus, we cannot exclude that Wnts elicited muscle prepatterning requires other domains in MuSK. (2) Frizzled (Fzd) receptors are expressed in skeletal muscle cells and could mediate Wnt signaling to contribute to muscle prepatterning (Strochlic et al., 2012; Avilés et al., 2014). (3) It has been suggested that MuSK and Lrp4 expression in early fused myofibers is sufficient to auto-activate MuSK and initiate muscle prepatterning (Kim and Burden, 2008; Burden et al., 2013). Wnts binding to MuSK CRD could therefore maintain or reinforce MuSK activation to amplify muscle prepatterning.
Our results demonstrate that MuSKΔCRD expression at the NMJ is increased by 70% in mutant compared with WT mice. In zebrafish, Wnt proteins have been shown to regulate the level of MuSK expression at the plasma membrane via activation of Wnt-induced MuSK endocytosis (Gordon et al., 2012). Therefore, deletion of MuSK CRD could disturb MuSK translocation from the plasma membrane to intracellular compartment, leading to MuSK membrane accumulation and reduction of Wnt/MuSK downstream signaling. Accordingly, we show that Wnt11-induced AChR clustering as well as β-catenin translocation to the nucleus are impaired in MuSKΔCRD primary myotubes, suggesting that deletion of MuSK CRD alters downstream Wnt canonical signaling during NMJ formation.
With innervation, nerve terminals release agrin that binds to Lrp4 and subsequently increases MuSK phosphorylation further enhancing AChR clustering (Kim et al., 2008; Zhang et al., 2008, 2011). Among the key molecules involved in NMJ formation, MuSK, Lrp4, Rapsyn, and Dok7 knock-out mice lack both aneural and agrin-induced AChR clusters (DeChiara et al., 1996; Gautam et al., 1999; Okada et al., 2006; Weatherbee et al., 2006). In contrast, although muscle prepatterning is severely affected in MuSKΔCRD embryos, our results show that, at E18.5, upon innervation, NMJs are able to form but are abnormal with reduced AChR cluster number (30%), volume, and density, suggesting that (1) prepatterning is not indispensable but necessary for normal progression of NMJ differentiation, and (2) innervation only partially compensates early AChR clusters deficit. Because we show that MuSK CRD deletion does not affect agrin-induced AChR clustering in primary myotubes, AChR clusters defects observed are likely to result from early developmental defects. However, Wnt proteins may bind to other receptors at the postsynaptic membrane, including Fzd, thus activating signaling mechanisms leading to inhibition of agrin-induced AChR clustering. Consistent with this, it has been recently shown that Fzd9 is highly expressed in skeletal muscle when NMJs form and its overexpression in culture myotubes impairs agrin-induced AChR clustering (Avilés et al., 2014).
In MuSK and LRP4 null mutant mice, motor axons grow extensively throughout the muscle (DeChiara et al., 1996, Weatherbee et al., 2006). Similarly, both during muscle prepatterning and later during NMJ formation, MuSKΔCRD motor axons overshoot AChR clusters and grow aberrantly all over the muscle. However, in contrast to MuSKΔCRD embyros, MuSK and LRP4 null mutant exhibited increased nerve branching/arborization. These results underline the importance of the MuSK CRD in regulating a muscle retrograde stop signal for motor axons although not required for presynaptic nerve arborization. In support to this hypothesis, studies of conditional invalidation or overexpression of muscle key components of Wnt canonical signaling, including Lrp4 and β-catenin in vivo in mice, suggest a role for Wnt canonical signaling to direct a retrograde signaling required for presynaptic differentiation (Li et al., 2008; Liu et al., 2012; Wu et al., 2012a, 2012b).
Intriguingly, despite severe NMJ formation defects, MuSKΔCRD mice are viable at birth and reach adulthood without any obvious abnormal phenotype during the first 2 weeks. In contrast, mice deficient for MuSK fail to form NMJs and die at birth due to respiratory failure (DeChiara et al., 1996). These data suggest that the remaining activity of MuSK deleted from its CRD is sufficient to prevent mutant mice from lethality. However, 2 weeks after birth, MuSKΔCRD mice start to develop CMS-like symptoms. Morphological analysis of adult MuSKΔCRD NMJs reveals abnormal endplates architecture, with an immature phenotype at P20 followed by a severe dismantlement at P60. This has been observed in all muscles analyzed, including diaphragm, TA, soleus, and extensor digitorum longus, suggesting that MuSK CRD is necessary for all striated muscles to guarantee NMJ integrity. This abnormal NMJ phenotype is likely to be the consequence of early NMJ defects during development because NMJ defects similar to those observed in mutant embryos are detected in P5 MuSKΔCRD diaphragms. In this hypothesis, in addition to its role for synapse positioning early during NMJ formation, muscle prepatterning would also have an unsuspected role in NMJ functioning in adult mice. However, we cannot exclude a specific MuSK CRD-dependent role of Wnts during NMJ maintenance in adulthood. Further investigations are required to discriminate the role of MuSK CRD during NMJ formation in embryos and maintenance in adulthood.
NMJ fragmentation is often associated with muscle weakness as it has been described in CMS patients (Slater et al., 2006). Indeed, MuSKΔCRD adult mice develop fatigable muscle weakness highlighted by abnormal performance in the grip test assay, ex vivo isometric diaphragm contraction, and fatigability measurement in response to nerve stimulation. Interestingly, one mutant mouse over five mice tested for isometric diaphragm contraction exhibits spontaneous twitches after a unique stimulation, a phenomenon often caused by muscle denervation (Heckmann and Ludin, 1982). In line with this observation, analysis of the NMJ innervation pattern in P90 MuSKΔCRD mice reveals the presence of non-innervated AChR clusters suggesting a denervation-like process.
Electron microscopy analyses of adult MuSKΔCRD NMJs reveal defects in presynaptic vesicle density and postsynaptic folds structure associated with either few or highly disorganized JFs. AChR clusters and JFs are required for the genesis of an efficient endplate potential leading to the activation of the voltage-gated sodium channels concentrated in the depth of the JFs (Engel and Fumagalli, 1982; Marques et al., 2000). Thus, reduced presynaptic vesicle density, fragmented endplates, and disorganized or fewer JFs are most probably responsible for the fatigable muscle weakness observed in adult MuSKΔCRD mice. Our investigation of the muscle morphology does not reveal major changes, except mild muscle atrophy with reduced fiber size (data not shown). This muscle atrophy may participate to the muscle weakness, but it may also result from defective NMJ maintenance because muscle activity is required for muscle trophicity (Schiaffino et al., 2007).
LiCl is currently used as a pharmacological reagent to treat bipolar, Parkinson's, and Hungtinton's diseases (Klein and Melton, 1996; Schou, 2001; Chiu et al., 2011; Yong et al., 2011). Remarkably, LiCl treatment rescues β-catenin signaling and improves the impaired NMJ defects in both MuSKΔCRD embryos and adult mice, suggesting that the defects observed during NMJ formation and maintenance in MuSKΔCRD mice are in part due to inhibition of the Wnt canonical signaling pathway. However, given that Gsk3 is a pivotal kinase interacting with multiple signaling pathways, including PI3K-PTEN-Akt-mTOR or Ras-Raf-MEK-ERK, we cannot rule out the possibility that LiCl regulates other signaling pathways involved in NMJ development (McCubrey et al., 2014). Interestingly, LiCl was shown to be efficient in the treatment of occulopharyngeal muscular dystrophy through activation of the Wnt canonical pathway and to improve skeletal muscle strength in mouse models of myotonic dystrophy (Jones et al., 2012; Abu-Baker et al., 2013). Our results provide the first evidence that LiCl or other Gsk3 inhibitors might also be used as therapeutic reagents or in complement to the treatment currently available of neuromuscular disorders associated with Wnt-MuSK deficiency.
Footnotes
This work was supported by Association Française contre les Myopathies Grant 14960 to L. Strochlic and Grant 18046 to C.L., Centre National de la Recherche Scientifique, and Paris Descartes University. We thank Markus Rüegg for MuSK atibodies and technical advice; Sophie Nicole and Suzie Lefebvre for critical reading of the manuscript; Eric Krejci and Jean and Annie Cartaud for experimental advice; Anne-Sophie Armand and Capucine Trollet for help with muscle primary cultures; and Jean-Maurice Petit for technical advices and image processing. The mouse mutant line was established at the Mouse Clinical Institute (Institut Clinique de la Souris) in the Targeted Mutagenesis and Transgenesis Department with funds from the Institut National de la Santé et de la Recherche Médicale and the Association Française contre les Myopathies (Association Française contre les Myopathies Grant 14960 to L. Strochlic).
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Laure Strochlic, Centre National de la Recherche Scientifique Unité Mixte de Recherche 8119, Université Paris Descartes, PRES Sorbonne Paris Cité, Paris, France. laure.strochlic{at}inserm.fr





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}