
Abstract
The KCNQ family of K+ channels has been implicated in several cardiac and neurological disease pathologies. KCNQ2 (Q2) is a brain-derived gene, which in association with KCNQ3 (Q3) has been shown to provide a molecular basis for the neuronal M current. We have cloned a long (Q2L) and a short (Q2S) splice variant of the human KCNQ2 gene; these variants differ in their C-terminal tail. Northern blot analysis reveals that Q2L is preferentially expressed in differentiated neurons, whereas the Q2S transcript is prominent in fetal brain, undifferentiated neuroblastoma cells, and brain tumors. Q2L, transfected into mammalian cells, produces a slowly activating, noninactivating voltage-gated K+ current that is blocked potently by tetraethylammonium (TEA; IC50, 0.14 mm). Q2S on the other hand produces no measurable potassium currents. Cotransfection of Q2S with either Q2L, Q3, or Q2L/Q3 heteromultimers results in attenuation of K+ current, the suppression being most profound for Q3. Inclusion of Q2S in the heteromultimer also positively shifts the voltage dependence of current activation and alters affinity for the TEA block, suggesting that under these conditions, some Q2S subunits incorporate into functional channels on the plasma membrane. In view of the crucial role of M currents in modulating neuronal excitability, our findings provide important insight into the functional consequences of differential expression of KCNQ2 splice variants: dampened potassium conductances in the developing brain could shape firing repertoires to provide cues for proliferation rather than differentiation.
- K+ channel
- M current
- KCNQ2
- KCNQ3
- cloning
- splice variants
- patch clamp
- neuronal development
- ER retention motif
- RXR(R)
Potassium channels set the resting membrane potential and control electrical excitability in diverse cell types. In the nervous system, these channels are key regulators of signaling because of their role in governing action potential shape and pattern that are ultimately critical to perception, learning, and behavior (Jan and Jan, 1997). Molecular cloning and functional expression studies have revealed that the genes underlying this functional diversity can be classified into several subfamilies. The KCNQ class of voltage-gated potassium channels is evolutionarily distinct from the Kv family of K+ channels (Barhanin et al., 1996). The most notable differences are the absence of a tetramerization domain in the N terminal that mediates subfamily-specific association and a unique pattern of conserved residues in the pore and sixth transmembrane regions (Wei et al., 1996).
To date, five genes that belong to the KCNQ family of potassium channels have been identified, and all are associated with inherited disorders. The first member of this family, KCNQ1, is expressed predominantly in cardiac tissue (Wang et al., 1996), where it associates with the single-transmembrane protein KCNE1 to form the slowly activating potassium currentIKS that is responsible for cardiac action potential repolarization (Barhanin et al., 1996; Sanguinetti et al., 1996). Inherited mutations of the KCNQ1 and KCNE1 genes cause the long QT syndrome, manifested by cardiac arrhythmias and sudden death. The four remaining members of the KCNQ family have been characterized only recently. KCNQ2 and KCNQ3 were identified by positional cloning of epilepsy loci on chromosomes 20q and 8q, respectively (Charlier et al., 1998; Singh et al., 1998). Both of these genes are highly expressed in the brain and share 40% homology to the KCNQ1 gene. Point mutations of these genes have been identified from patients with inherited forms of neonatal epilepsy. Several of these mutations have been shown to cause suboptimal function in heterologous systems (Biervert et al., 1998; Schroeder et al., 1998; Lerche et al., 1999); these subtle changes are thought to heighten excitability and promote epileptiform synchronization. A heteromultimeric association of KCNQ2 and KCNQ3 channels has been proposed as a molecular correlate of the neuronal M current (Wang et al., 1998; Shapiro et al., 2000). KCNQ2 is expressed in presynaptic terminals of the human hippocampus in the absence of KCNQ3 (Cooper et al., 2000), suggesting that it may form functional M currents as a homomultimer or in coassembly with some yet-to-be-described molecule(s). KCNQ4, expressed in sensory outer hair cells, is mutated in dominant deafness (Coucke et al., 1999; Kubisch et al., 1999). KCNQ5, the most recently discovered gene in this subfamily, is expressed in brain and skeletal muscle (Kananura et al., 2000) and is implicated in retinal disorders. Expression of a heteromultimeric combination of KCNQ5 and KCNQ3 also reconstitutes properties of the neuronal M current (Lerche et al., 2000; Schroeder et al., 2000), raising the possibility that the M-current phenotype may be represented by considerable molecular diversity.
The KCNQ2 gene has several alternatively spliced variants that differ in their cytoplasmic C-terminal tail (Nakamura et al., 1998; Tinel et al., 1998). Although there has been very exciting data linking the KCNQ2 gene to epilepsy and M currents, there has been no systematic study of the expression pattern of KCNQ2 splice variants in neurons and their role in neuronal function. In the present study, we addressed this question via a combination of molecular biology and electrophysiological approaches. Our results provide compelling evidence of differential expression of two splice variants during neuronal development and also reveal important implications for the regulation of M-current function dependent on this expression pattern in developing versus adult neurons.
Parts of this paper have been published previously (Iannotti et al., 1998).
MATERIALS AND METHODS
Cloning of KCNQ2 and KCNQ3 and generation of expression constructs. A tblastn search of a proprietary database using human KCNQ1 (GenBank accession number U40990) as the search motif identified a novel expressed sequence tag (EST) that was then used to search the public domain database to identify a partial Merck-WashU clone yn72g11. Full-length information of the long splice variant of KCNQ2 (Q2L) was obtained using 5′ and 3′ RACE-PCR techniques (Bertioli, 1997) on a human brain cDNA (Clontech, Palo Alto, CA); the sequence was confirmed on both strands. The information has been deposited in GenBank (accession number AF074247). The short splice variant of KCNQ2 (Q2S) and KCNQ3 (Q3) were cloned by direct PCR of a human brain library using primers designed to the full-length published sequence (GenBank accession numbers D82346 and AF071478, respectively). All genes were subcloned into the pCDNA3 vector (Invitrogen, Carlsbad, CA) using engineered restriction sites. For coexpression with Q2S, an N-terminal fusion construct of Q2L with enhanced green fluorescent protein (EGFP) was generated by subcloning into pEGFPC1 vector (Clontech). All constructs were confirmed to be error-free by double-stranded DNA sequencing.
Tissue culture and RNA isolation. Human fetal brain tissue from 16- to 22-week-old fetuses was obtained from the Anatomic Gift Foundation (Woodbine, GA). The tissue was processed for isolation of astrocytes as follows (Lee et al., 1992): Cerebral hemispheres were placed in chilled, sterile, calcium-free, and magnesium-free HBSS (Life Technologies, Gaithersburg, MD). Meninges were removed with sterile forceps, and tissue was dissociated initially by repeated trituration through sterile pipettes. Tissue was incubated with 0.05% trypsin and 0.53 mm EDTA (Life Technologies) and 0.15 mg/ml DNase (Sigma, St. Louis, MO) at 37°C for 45 min with gentle shaking. Fetal bovine serum (FBS; 10%; Life Technologies) was added to the suspension to stop trypsinization. The cells were then passed sequentially through 210 and 149 μm polypropylene meshes (Fisher Scientific, Pittsburgh, PA). The filtrate was washed twice and resuspended in complete media (DMEM with high glucose,l-glutamine, and HEPES; 100 U-μg/ml penicillin/streptomycin; 10% FBS; Life Technologies). Cells were plated at a density of 80 million/75 cm2flask. Cultures were incubated at 37°C in 5% CO2 for 2 weeks. Mature cultures consisted of astrocytes, neurons, microglia, and oligodendrocytes. Pure astrocyte cultures were isolated by removing complete media (containing floating microglia) and trypsinizing the cultures three times to remove effectively neurons, oligodendrocytes, and attached microglia. Astrocytes were lysed in Buffer RLT (Qiagen, Valencia, CA) with 0.01% β-mercaptoethanol (β-ME; Sigma) within 24 hr and frozen at −80°C until use. This RNA was used in the Northern blot analysis (see Fig. 2B, Fetal Astrocytes lane).
IMR-32 human neuroblastoma cells (American Type Culture Collection, Rockville, MD) were cultured in MEM supplemented with 10% heat-inactivated FBS and 2 mml-glutamine at 37°C in a 5% CO2 humidified atmosphere. The medium was changed twice a week. Cells were differentiated with 1 mm dibutyryl cAMP and 2.5 μm5-bromodeoxyuridine (Sigma). Cells were lysed in Buffer RLT (Qiagen) with 0.01% β-ME (Sigma) and frozen at −80°C until use. Lysed cells were thawed on ice and homogenized with a QIAshredder (Qiagen). Total RNA was then prepared with the RNeasy Mini Kit (Qiagen) according to the manufacturer's directions. Concentration was determined by absorbance at 260 nm.
Northern blots. For determining tissue distribution, premade mRNA blots from Clontech were used that contained ∼2 μg of polyA RNA per lane. The human brain tumor blot was purchased from Invitrogen. An additional blot was made as follows: Twenty micrograms each of RNA from human adult brain (Clontech), human fetal brain (Clontech), undifferentiated and differentiated IMR-32 lines (see above), and human fetal astrocytes (see above) along with 5 μg of RNA Ladder (Life Technologies) were run on a 1% agarose, 2.2 mformaldehyde, and 1× 3-[N-morpholino]propanesulfonic acid denaturing gel at 50 V for 4.5 hr. RNA was transferred overnight to positively charged nylon membrane (Hybond n +; Amersham, Piscataway, NJ), according to the manufacturer's protocol. The RNA was UV cross-linked to the membrane for 40 sec.
32P-labeled single-stranded antisense DNA probes were generated as follows: Probe 1, designed to detect Q2L only, was generated by primer extension of a 201 bp PCR product corresponding to nucleotides 1257–1461 of the C-terminal tail of Q2L. Probe 2 was designed similarly, but to nucleotides 989–1109 of the coding region of KCNQ2, corresponding to the S5-P linker. This probe was designed to detect all C-terminal splice variants of KCNQ2. Labeled products were purified from unincorporated radioactive nucleotide using ProbeQuant/G-50 Micro Columns (Amersham Pharmacia) according to the manufacturer's recommendations. A 103 bp antisense cyclophilin probe was generated by primer extension using pTRI-cyclophilin (Ambion, Austin, TX). The blots were prehybridized in NorthernMAX Prehybridization Solution (Ambion) at 42°C for 1 hr followed by overnight hybridization at 42°C using the same solution containing 2.0 × 106 cpm/ml labeled probe. The blots were then washed twice at 37°C as follows: 2× SSC and 0.5% SDS, 5 min; 2× SSC and 0.1% SDS, 5 min; 0.1× SSC and 0.5% SDS, 30 min; and briefly with 0.1× SSC. Blots were then exposed to BIOMAX MS-1 imaging film (Kodak, Rochester, NY) at −80°C with two intensifying screens.
Transfection protocols. Twenty-four to 48 hr before transfection, human embryonic kidney 293 (HEK293) cells (American Type Culture Collection) were plated on 60 mm BIOCOAT (Becton Dickinson, Bedford, MA) collagen-treated plastic Petri dishes containing antibiotic-free DMEM plus 10% heat-inactivated FBS, 4.5 gm/l glucose, 4 mm glutamine, and 1 mm pyruvate. COS-7 cells (American Type Culture Collection) were plated on 60 mm uncoated FALCON (Becton Dickinson) plastic Petri dishes containing antibiotic-free DMEM plus 10% FBS. For biophysical characterization, cells were transfected for 3 hr at 37°C in serum- and antibiotic-free DMEM with a mixture containing 18 μl of lipofectamine, 12 μl of PLUS reagent (Life Technologies), and 2 μg of pcDNA3 vector containing Q2L, Q2S, or Q3 cDNAs. One microgram of EGFP cDNA was added in these mixtures as a tracer for patch clamping. For coexpression experiments, EGFP-Q2L cDNA was combined with untagged Q2S or Q3 cDNA at 1:1 or 1:5 molar ratios. After transfection, cells were washed with PBS and fed with DMEM plus 10% FBS supplemented with Gln, pyruvate, and penicillin/streptomycin. COS-7 or HEK293 cells were replated onto uncoated or poly-l-Lys-coated Nunclon (Nunc, Naperville, IL) 35 mm dishes, respectively, 6–24 hr before patch clamping.
Electrophysiology. All recordings were made starting 48 hr after transfection at room temperature (20–22°C) using the conventional whole-cell configuration (Hamill et al., 1981). The extracellular solution contained (in mm): 160 NaCl, 4.5 KCl, 1 MgCl2, 2 CaCl2, 5 HEPES, and 5 glucose, at pH 7.4 by NaOH (∼325 mOsm). The pipette solution contained (in mm): 160 K-aspartate, 5 HEPES, and 4 EGTA or 4 EDTA, at pH 7.2 by KOH (∼305 mOsm). Electrodes were pulled from thin-walled (1.5 mm outer diameter and 1.12 mm inner diameter) borosilicate glass capillaries (World Precision Instruments, Sarasota, FL) on a Brown Flaming P-87 micropipette puller (Sutter Instruments, Novato CA). DC electrode resistance was 3–5 MΩ when measured with the experimental extracellular and pipette solutions. An Axopatch 200A amplifier (Axon Instruments, Foster City, CA) connected to a personal computer by either a TL-1 (Scientific Solutions, Solon, OH) or Digidata 1200 (Axon Instruments) interface was used to obtain membrane currents. The current signal was balanced to zero with the pipette immersed in the bath just before forming a seal on the cell. Liquid junction potentials were measured for the experimental solutions and corrected off-line. Seal resistances ranged from 1 to >10 GΩ. Series resistance was usually <10 MΩ and was not compensated electronically. Digitized data acquisition and voltage-step protocols were implemented with pClamp 6.0 software (Axon Instruments). Data were low-pass filtered at less than one-half the digital sampling rate before digitizing. Solution changes were accomplished by focal application from six linearly arranged glass-lined tubes (Hewlett Packard, Wilmington, DE) positioned ∼100 μm from the recorded cell. Each solution was released from the desired tube by an electronically controlled solenoid valve (BME Systems, Baltimore, MD). This system achieved rapid (<100 msec) equilibration of drug solution in the extracellular phase without perturbing recording characteristics. Current amplitudes were measured using the Clampfit module of pClamp. Origin 5.0 software (Microcal, Northampton, MA) was used to iteratively fit current–voltage relationships to a Boltzmann equation and concentration–response data to a Hill equation (see figure legends for details). All results are expressed as the mean ± SEM. ANOVA was used to compare effects of the different transfections on current density and activation voltage dependence. Newman–Keuls multiple comparisons tests were used to discern significant differences between pairs of treatments where ANOVA indicated a significant main effect. A level of p < 0.05 was considered significant for statistical tests (for relevant p values, see Table 1).
During preliminary experiments, we observed significant “rundown” of the KCNQ currents. We found that removal of Mg2+ ions from the internal pipette solution significantly slowed the rate and extent of current rundown (Kozlowski and Ashford, 1990). Formation of gigaseals was also found to be facilitated by the Mg2+-free internal solution. We therefore used this Mg2+-free pipette solution in all subsequent experiments, thereby preventing rundown from being a confounding factor in any of the data described in this paper.
RESULTS
We identified a unique EST in the database with homology to KCNQ1 and used this information to clone the full-length cDNA of Q2L using RACE-PCR techniques. Our sequence is highly similar to the published sequence of KCNQ2 as revealed by positional cloning of the epilepsy locus (Singh et al., 1998), as well as other reports (Biervert and Steinlein, 1999). We also identified another EST that corresponds to the splice variant Q2S that was published previously by Yokoyama et al. (1996); we subsequently PCR-amplified the gene from a human fetal brain library. The two splice variants differ exclusively in the C-terminal tail (Fig. 1).

Amino acid sequence of the long (Q2L; C terminal in roman type) and short (Q2S; C terminal initalic type) splice forms of KCNQ2.Bold denotes sequence common to Q2L and Q2S. Transmembrane segments are underlined (labeled S1–S6,Pore). The splice junction is indicated by anarrow.The ER retention motif RYRR isboxed. The KCNQ2L sequence has been deposited in GenBank (accession number AF074247).
Expression of KCNQ2 splice variants is regulated during neuronal differentiation
We investigated the relative expression patterns of the two splice variants by comparing Northern blots hybridized to a probe that was unique to the long splice variant (probe 1) and another probe that picked up any splice variant of KCNQ2 (probe 2). We were unable to design a probe unique to the short splice version because the region was too small and too AT rich to make a good probe. Thus same-size transcripts that were positive by both probes 1 and 2 were considered Q2L specific, whereas those that were negative by probe 1 but positive by probe 2 were assumed to represent other splice variants, including Q2S. According to this criterion, the long form of KCNQ2 was a 9.5 kb transcript, whereas the short form was a 1.5 kb transcript. Minor bands of intermediate size may have represented other splice variants.
Relative expression of the splice variants was found to be highly dependent on the developmental stage of the neuronal tissue. As shown in Figure 2A, multiple-tissue Northern blots revealed that a 9.5 kb band corresponding to the long splice variant was the predominant species in adult human brain; this band was not detected in any other tissue. The short variant, on the other hand, was very weak in adult brain but was expressed in testis as a 1.5 kb band (Fig. 2A). Northern blot analysis of regional expression in the adult human brain (Fig. 2A) also showed high expression of Q2L in all regions but the spinal cord; Q2S was by contrast expressed at nearly undetectable levels in these regions. On the other hand, human fetal brain expressed both Q2L and Q2S in high abundance (Fig.2B). Because brain tissue is comprised of 90% glial cells and 10% neuronal cells (Bacci et al., 1999), we evaluated the specificity of this expression pattern to neurons. KCNQ2 was not expressed in fetal astrocytes, the predominant glial cell type (Fig.2B). Equal loading and integrity of the RNAs were ensured by reprobing the blots with the cyclophilin probe (data not shown).
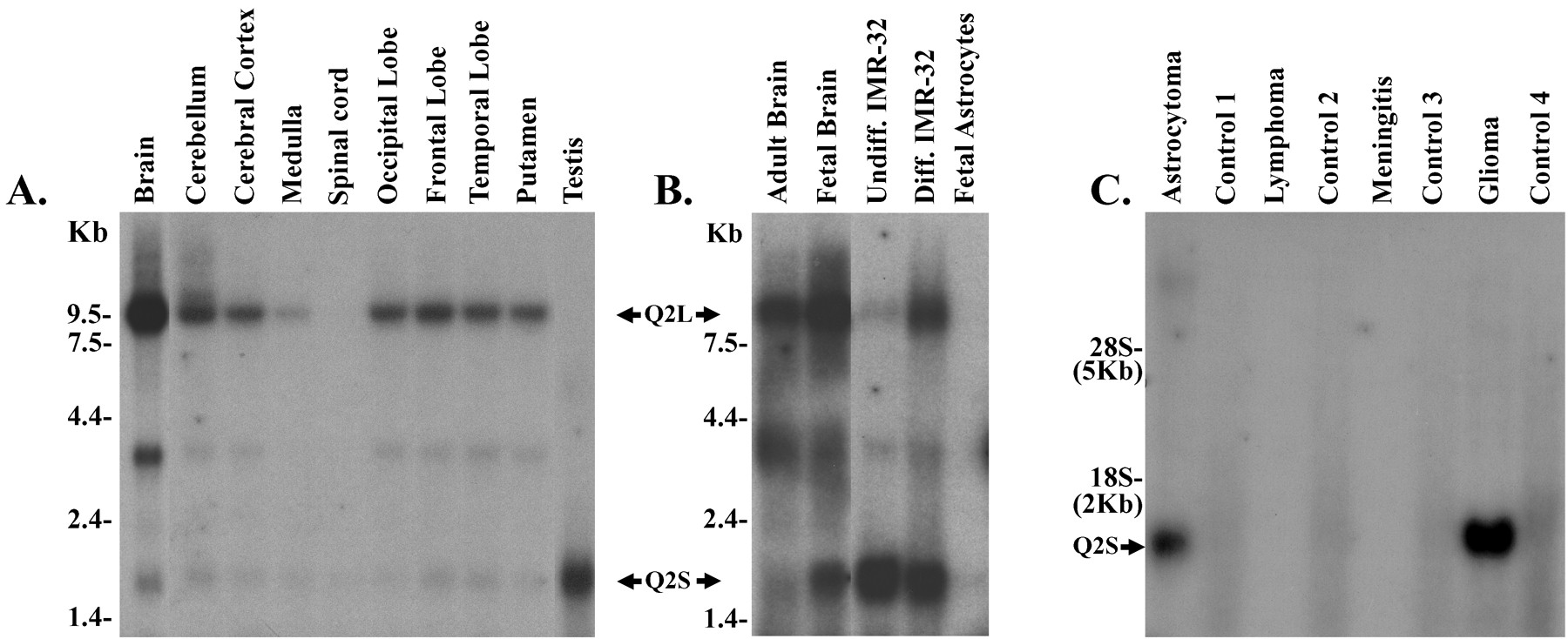
Northern blot analysis of KCNQ2. A, Multiple-tissue Northern blots show expression of the 9.5 kb Q2L transcript in brain and of the 1.5 kb Q2S transcript in the testis. No bands were detected in spleen, heart, kidney, lung, liver, skeletal muscle, colon, placenta, thymus, leukocytes, pancreas, ovary, and small intestine (data not shown). B, Differential expression of Q2L and Q2S transcripts in fetal versus adult brain and in undifferentiated (Undiff.) versus differentiated (Diff.) IMR-32 neuroblastoma cells is shown. No signal was detected in fetal astrocytes. C, Q2S is expressed in tumors that originated in the brain but not in tumors that metastasized to the brain from other regions. Control lanes represent RNA from normal tissue that was excised from the same operational site from the same patient with the tumor. Twenty micrograms of total RNA were loaded on each lane. Cyclophilin probe was used to assess the quality and quantity of RNA in the lanes(data not shown).
We further investigated the expression patterns of these two splice variants in the neuroblastoma line IMR-32 that is a widely used model for neuronal differentiation (Reynolds and Perez-Polo, 1981). Rapidly proliferating, undifferentiated IMR-32 cells exclusively expressed the short form of Q2S (Fig. 2B). After chemically induced differentiation to a neuronal phenotype, however, as revealed by changes in morphology and slowing of proliferation, there was a dramatic upregulation of the Q2L splice variant (Fig.2B), comparable with the pattern seen in adult brain. Thus, the expression of the long splice variant of KCNQ2 was associated with the phenotype of a mature neuron, whereas that of Q2S was associated with proliferating neuronal precursor cells. In agreement with this idea, we found upregulation of the short transcript in brain tumors that originated in the brain but not in tumors that metastasized to the brain from other tissues (Fig. 2C).
Q2L, but not Q2S, expresses a voltage-gated K+ current
In accordance with the previous report by Yu and Kerchner (1998), mock-transfected or nontransfected HEK293 cells expressed a small rapidly activating delayed rectifier K+current that activated at positive voltages (V1/2= approximately −2.5 mV; Fig.3C). The current partially inactivated at high positive potentials and displayed an intermediate tetraethylammonium (TEA) sensitivity (IC50 at +40 mV = 1.6 mm; Table1). Mean current density (+40 mV step) for this endogenous HEK293 K+ current was 6 ± 2 pA/pF (n = 5; see Fig.4D, Table 1).

Transient transfection with Q2L, but not Q2S, produces a delayed rectifier K+ current in HEK293 cells. Currents were recorded during 1 sec depolarizations in 10 mV increments from −70 to 40 mV (holding potential, −70 mV).A, Q2L-transfected cell. B, Q2S-transfected cell. C, Mock-transfected cell (HEK293 endogenous current). Calibration: the same for A–C.D, Conductance–voltage relations constructed from whole-cell current measurements by use of the relation:G/Gmax = (I/(Emax −EK))/(Imax/(E− EK)). Data were fit according to Boltzmann, where G/Gmax= 1/(1 + exp((V − V1/2)/dx)) was used to obtain the values for V1/2 and dx (slope) in millivolts: −25.5 ± 5 and 12 ± 1, −1.8 ± 2 and 9.5 ± 1, and −3 ± 0.4 and 10 ± 0.2 for Q2L (square), Q2S (triangle), and mock transfected (circle), respectively.
Electrophysiological parameters

Q2S inhibits Q2L or Q3 currents but has little effect on Q2L/Q3 heteromultimer expression. Ratios indicate molar equivalents. Currents were recorded as described in Figure 3.A, Q2L (left); Q2L/Q2S, 1:1 (middle); Q2L/Q2S, 1:5 (right).B, Q3 (left); Q3/Q2S, 1:1 (middle); Q3/Q2S, 1:5 (right).C, Q2L/Q3, 1:1 (left); Q2L/Q3/Q2S, 1:1:1 (middle); Q2L/Q3/Q2S, 1:1:5 (right). Calibration: 1 nA (A, B), 2 nA (C); 400 msec (A–C).D, Current density histogram of KCNQ subunit-transfected cells. Current density measurements are expressed in picoamperes per pico-Faradays and were made from whole-cell current recordings taken during a 1 sec depolarizing step from −70 to 40 mV (holding potential, −70 mV). 2L, Q2L; 2S, Q2S.
Transient expression of Q2L in HEK293 cells produced a slowly activating, noninactivating K+ current (Fig. 3A), similar to that reported by others (Wang et al., 1998; Shapiro et al., 2000). After a +40 mV depolarization, tail currents reversed at −78 mV in the experimental solutions, and the reversal potential shifted +55 mV per 10-fold increase in external K+, indicating high-potassium selectivity of these channels (data not shown). Current activation was sigmoidal, and full activation was not reached during 1 sec depolarizations to +40 mV. Half-maximal current activation was determined from a Boltzmann fit to the normalized conductance data, yielding a V1/2 of −25.5 mV and a slope factor of 12 mV (Fig. 3D, Table 1). Q2L currents were highly sensitive to TEA with IC50 = 140 μm at +40 mV (Table 1). Whole-cell currents were typically several nanoamperes in size, and mean current densities were 50 ± 14 pA/pF (+40 mV step; n = 9 cells; Figs. 3A,4D). In contrast, Q2S-transfected cells expressed very small outward currents, and current density and activation voltage did not differ significantly (p > 0.05) from that of currents recorded in mock-transfected HEK293 cells (Figs.3B,D, 4D).
To confirm further the differential ability of KCNQ2 splice variants to express functional currents, we repeated the transient transfections in COS-7 cells, which have no endogenous K+currents. The amplitude and biophysical properties of currents expressed after Q2L transfection in COS-7 cells were nearly identical to those measured in HEK293 cells (data not shown). Q2S expression failed to yield currents significantly larger than background when transfected in the COS-7 cells, in close agreement with the HEK293 cell results.
Q2S suppresses Q2L and Q3 channel function
To determine the physiological significance of coexpression of the two KCNQ2 splice variants in developing neurons, we cotransfected them in varying ratios. An N-terminal EGFP fusion protein of Q2L was generated for this purpose and shown to express channels indistinguishable functionally from those expressed by wild-type Q2L (e.g., Fig. 4A, left). Our rationale for using this construct was that transfected cells would fluoresce green when expressing the Q2L channel and could be patch clamped to evaluate currents when cotransfected with either control vector or a vector containing the short transcript. This strategy ensured that recorded cells expressed the Q2L protein, and thus any difference in current levels observed with the coexpression constructs could be attributed to the influence of Q2S rather than to an artifact stemming from an inadvertent failure to transfect the particular cell with Q2L. Figure 4D shows mean current densities obtained from all experiments in which Q2L was cotransfected with varying molar ratios of Q2S. Currents were suppressed compared with those when Q2L was cotransfected with empty vector. The suppressive effect of Q2S did not reach significance for the 1:1 transfection ratio but became significant (p < 0.05) for the higher ratio (1:5) of the long-to-short splice variant. In addition, the voltage dependence for activation of the current resulting from inclusion of a high molar ratio of the short variant was also shifted in the positive direction (V1/2 = −18 mV), relative to the current expressed by Q2L alone (−26 mV; Fig.5A; Table 1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Q2S affects the voltage dependence of Q2L, Q3, and Q2L/Q3 heteromultimeric channels. A, Q2L (square); Q2L/Q2S, 1:1 (circle); Q2L/Q2S, 1:5 (triangle). B, Q3 (square); Q3/Q2S, 1:1 (circle); Q3/Q2S, 1:5 (triangle). C, Q2L/Q3, 1:1 (square); Q2L/Q3/Q2S, 1:1:1 (circle); Q2L/Q3/Q2S, 1:1:5 (triangle). Data were fit according to Boltzmann as described in Figure 3. Fitted values for V1/2and slope are found in Table 1.
Recent studies (Wang et al., 1998) have established that, in addition to KCNQ2, the KCNQ3 gene is highly expressed in similar regions of the nervous system and that a heteromultimer of KCNQ2 and KCNQ3 forms a molecular basis for the M current in central and peripheral neurons. We therefore evaluated the effects of the short KCNQ2 transcript on currents expressed by KCNQ3 and by Q2L/Q3 cotransfection. KCNQ3 alone expressed K+ currents that showed several biophysical and pharmacological distinctions from Q2L current. Activation kinetics was somewhat more rapid with a less sigmoidal activation threshold. Voltage dependence of activation was more negative with half-maximal activation occurring at −42 mV (Fig.5B). The current was insensitive to TEA blockade with an estimated IC50 of ∼456 mm(Table 1). In contrast to some other recent reports (Schroeder et al., 1998, 2000; Wang et al., 1998; Lerche et al., 2000), Q3 alone was expressed at a current density that did not differ significantly in magnitude from expressed Q2L currents (p > 0.05; Fig. 4B,D; Table 1). As reported previously (Wang et al., 1998; Yang et al., 1998; Shapiro et al., 2000), coexpression of Q2L and Q3 led to a dramatic enhancement of K+ current in both HEK293 (Fig.4C,D; Table 1) and COS-7 (data not shown) cells. The Q2L/Q3 current was intermediate in terms of voltage dependence (V1/2 = −35 mV; Fig. 5C) and TEA sensitivity (IC50 = 34 mm; Table 1), suggesting the formation of heteromultimeric Q2L/Q3 channels; our results are similar to those observed by Shapiro et al. (2000).
Coexpression of Q3 with Q2S at either 1:1 or 1:5 molar ratios led to potent suppression of K+ current (p < 0.001; Fig. 4B,middle, right). The mean currents from Q3/2S-transfected cells were small and not significantly larger than HEK293 cell background currents (Fig. 4D; Table 1), suggesting that Q2S had a potent suppressive effect on surface membrane expression of Q3. However, these small Q2S/Q3 currents were notably more sensitive to TEA blockade than were KQT3 currents (Table 1) and activated at significantly more depolarized potentials than did Q3 (p < 0.001; Fig. 5C).
Somewhat surprisingly, coexpression of Q2L/Q3/Q2S at either 1:1:1 or 1:1:5 molar ratios in HEK293 cells did not have a significant suppressive effect on current densities relative to currents expressed by the Q2L/Q3 heteromultimer (p > 0.05; Fig.4C, middle, right, D). However, in COS-7 cells, current density was reduced by 35% (data not shown). In the HEK293 cells, Q2L/Q3/Q2S (1:1:5) currents showed a +20 mV depolarizing shift in the voltage dependence of activation, yielding a V1/2 of −14 mV (Fig. 5C). Additionally, the sensitivity to TEA blockade was increased from 34 mm for the Q2/Q3 current to 8 and 1.2 mm for the Q2L/Q3/Q2S currents at molar ratios of 1:1:1 and 1:1:5, respectively (Table 1), suggesting participation of Q2S in functional heteromultimeric channel formation on the plasma membrane.
DISCUSSION
The neuronal M current is a voltage-dependent potassium current that opens in the subthreshold range of action potentials, is suppressed by muscarinic agonists (hence the name), and is a critical player in modulating firing patterns and in the release of neurotransmitters (Brown and Adams, 1980; Marrion, 1997). A heteromultimer of KCNQ2 and KCNQ3 has been shown to reconstitute most of the biophysical and pharmacological properties of the M current in peripheral and central neurons (Wang et al., 1998; Shapiro et al., 2000). Numerous studies have also focused on the cloning, expression, and functional anomalies of KCNQ2/Q3 mutations responsible for inherited forms of neonatal epilepsies (Biervert et al., 1998; Charlier et al., 1998; Schroeder et al., 1998; Singh et al., 1998; Yang et al., 1998). Rodent KCNQ2 has been reported to be encoded by a multiexon gene with a variety of splice variants, predominantly in the C terminal (Nakamura et al., 1998; Tinel et al., 1998). Nonetheless, there has been no systematic study of the functional consequences of coexpression of the KCNQ2 splice variants.
We have cloned a long and short splice variant of human KCNQ2, determined their RNA expression in brain cells, and analyzed the physiology of the two variants when transfected alone or together. Our cDNA sequence closely matches those of previous reports (Biervert et al., 1998; Singh et al., 1998; Wang et al., 1998) as do the electrophysiological parameters for Q2L, Q3, and the Q2L/Q3 heteromultimer expressed in mammalian cells, which reconstitutes the M current (Yang et al., 1998; Shapiro et al., 2000). We demonstrate here that although the widely studied long Q2 variant is preferentially expressed in differentiated neurons, Q2S message is prominent in immature proliferating preneurons such as those found in fetal brain, undifferentiated neuroblastoma lines, and brain tumors. Our results are compatible with findings in murine brain of expression of short splice forms in embryonic stages, which disappeared in the adult mouse with the emergence of the longer splice forms (Nakamura et al., 1998).
The neuronal Kv3.1 channel has been reported to have C-terminal splice variants that are also developmentally regulated in response to different signaling pathways (Luneau et al., 1991; Si-qiong and Kaczmarek, 1998). However, no functional differences were observed between the two splice variants; they both expressed K+ currents with identical biophysical and pharmacological properties (Yokoyama et al., 1989; Luneau et al., 1991), leaving open the question of functional implications of the splicing events. The present study imparts physiological relevance to the splice switching of Q2L and Q2S variants during development. In this study, Q2S tranfection alone does not yield functional channels, as reported previously in oocytes (Nakamura et al., 1998). However, cotransfection of the short variant with Q2L, Q3, or the Q2L/Q3 multimer profoundly affects functional expression by shifting the voltage dependence of activation to more depolarized potentials and/or producing overall suppression of potassium conductance.
What is the mechanism by which Q2S causes an inhibition or alteration of channel function? Interestingly, the short C-terminal tail of Q2S contains the endoplasmic reticulum (ER) retention motif RYRR that is not found in other splice variants (Fig. 1). The presence of such motifs in certain membrane proteins has been shown to prevent the molecule from exiting the endoplasmic reticulum. After association with other subunits, however, the retention signal is masked, facilitating trafficking of the complex to the plasma membrane (Zeranue et al., 1999; Margeta-Mitrovic et al., 2000). We hypothesize a somewhat similar mechanism to explain the effects we report here. Q2S is potentially functional but does not produce measurable K+ currents on its own, primarily because a sufficient number of channels fail to reach the plasma membrane [as has been reported for some truncated KCNQ2 proteins found in epilepsy patients (Schwake et al., 2000)]. However, with the inclusion of Q2L and/or Q3 subunits, there could arise a “molecular tug of war”: Q2S subunits in the heteromultimers act to retain the oligomer in the ER, whereas the Q2L/Q3 subunits in the heteromultimers compete with the retention mechanism by pulling the Q2S-containing complexes to the plasmalemma. Such a process is compatible with our observations of suppressed current density in the presence of Q2S (i.e., lower channel number) and also the alteration of properties (i.e., positive shift in voltage-dependent activation; increased TEA sensitivity) because of inclusion of Q2S in functional heteromultimers. Additionally, the observation that this suppressive effect of Q2S on Q2L or Q3 currents is “dose-dependent” (Fig. 4, Table 1) is consistent with a mechanism involving competing affinities between subunits. Interestingly, KCNQ3 (but not KCNQ2L) contains a RQRR domain in its C-terminal tail. This could account for its enhanced expression with Q2L after coassembly as well as suppression of currents with Q2S.
It is possible that the functional alterations seen in the Q2S-containing heteromultimers were caused by contribution of endogenous HEK293 cell channels to the ensemble current, rather than caused by Q2S, particularly in cases in which the total current was reduced substantially [2L/2S (1:5) or 3/2S (1:1 and 1:5) transfections; see Table 1]. Such an argument fails to account, however, for the increased TEA sensitivity and most notably the substantial rightward shift of voltage dependence observed after 2L/3/2S (1:1:5) cotransfections (p < 0.01), where the endogenous current would be expected to comprise <5% of the large total currents observed. Even if an extreme case were envisioned to explain these data in which Q2S assembled with Q3 and prevented it from reaching the plasma membrane, then the remaining predominantly Q2L channels could contribute to the greater TEA sensitivity butwould not account for an activation voltage that is significantly more positive than that measured for the Q2L homomer (i.e., V1/2 = −25.5 mV; p < 0.05). These specific data therefore argue strongly that Q2S must contribute to functional heteromultimeric channels through coassembly with other KCNQ subunits and thus must impart conformational constraints that lead to significant alteration of voltage dependence (Horn, 2000).
Recently, Tinel et al. (1998) have shown that KCNQ3 message is absent at birth in rodent brain and gradually increases thereafter. This observation, in conjunction with our results on high expression of the short variant in immature neurons, suggests that the overall K+ currents through KCNQ2/Q3 channels would be expected to be significantly lower in the developing brain compared with that observed in adult brain. What are the consequences of suppressed potassium conductance early in neuronal development? One possibility is that developing neurons have compensatory low sodium currents and in general have low electrical excitability compared with mature neurons (Barish, 1995). A second possibility is that in immature neurons where the short transcript predominates, reduction in M-current amplitude and/or the shift in voltage dependence could sufficiently alter firing repertoires and consequent calcium influx to provide cues for proliferation rather than differentiation (Ribera, 1999). In agreement with this hypothesis is our finding of increased Q2S transcript in brain tumors. In contrast, with Q2L being the major transcript expressed in mature differentiated neurons, a significantly greater M-current contribution may limit repetitive action potential firing under baseline conditions but also impart a mechanism to impose fine modulatory control on firing pattern via the well established coupling of the M channel to various neurotransmitter-coupled receptors. Intriguingly, in Xenopus laevis spinal neurons, overexpression of potassium currents led to decreased morphological differentiation in vitro (Jones and Ribera, 1994), whereasin vivo (Jones et al., 1995), there were compensatory mechanisms that rescued the neurons from this process. Further biochemical and physiological studies could shed light on the subunit assembly and trafficking of these complexes as well as the mechanisms by which these ion channels participate in the switch from growth to differentiation.
Footnotes
- Received September 5, 2000.
- Revision received November 16, 2000.
- Accepted November 21, 2000.
We are grateful to Lee Hirata and Patricia Sherman for DNA sequencing and primer synthesis, Alan Robbins for advice on KCNQ3 cloning, Eileen Martin for isolation of RNA from human fetal astrocytes, David Gurley for statistical analysis, and Bill Fieles, James Huffmaster, and James Morelli for graphics work.
J.S.S. and C.A.I. contributed equally to this work.
Correspondence should be addressed to Dr. Jeffrey Smith, AW1, AstraZeneca Pharmaceuticals, 1800 Concord Pike, Wilmington, DE 19803. E-mail: jeff.smith{at}astrazeneca.com.
- Copyright © 2001 Society for Neuroscience