
Abstract
Mechanisms governing the activity of neuronal nitric oxide synthase (nNOS), the major source of nitric oxide (NO) in the nervous system, are not completely understood. We report here a protein-protein interaction between nNOS and NOSIP (nitric oxide synthase-interacting protein) in rat brain in vivo. NOSIP and nNOS are concentrated in neuronal synapses and demonstrate significant colocalization in various regions of the central and peripheral nervous systems. NOSIP produces a significant reduction in nNOS activity in a neuroepithelioma cell line stably expressing nNOS. Furthermore, overexpression of NOSIP in cultured primary neurons reduces the availability of nNOS in terminal dendrites. These results thus suggest that the interaction between NOSIP and nNOS is functionally involved in endogenous mechanisms regulating NO synthesis. Furthermore, we found that the subcellular distribution and expression levels of NOSIP are dynamically regulated by neuronal activity in vitro as well as in vivo, suggesting that NOSIP may contribute to a mechanism via which neuronal activity regulates the synaptic availability and activity of nNOS.
- nitric oxide
- synaptic spines
- hippocampal neurons
- protein-protein interactions
- seizures
- inflammatory pain
Introduction
Nitric oxide (NO) and its signaling mediators govern various cellular functions in the nervous system such as apoptosis, differentiation, development, and synaptic plasticity (Dawson and Dawson, 1998; Boehning and Snyder, 2003; Gibbs, 2003). Because of the potency and broad spectrum of the cellular actions of NO and its brief duration of action, mechanisms that regulate NO synthesis with respect to time and space are crucial in determining the biological functions of NO. However, the complex molecular mechanisms that underlie the activation and localization of NO synthases (NOSs) are still incompletely understood (Dawson et al., 1998; Alderton et al., 2001; Boehning and Snyder, 2003). Although all NOS isoforms can be expressed in the nervous system (Alderton et al., 2001), neuronal NOS (nNOS) constitutes the predominant source of NO in neurons and localizes to synaptic spines.
Several protein-protein interactions regulating the localization of nNOS at postsynaptic density have been described (Brenman et al., 1996; Jaffrey and Snyder, 1996; Jaffrey et al., 1998, 2002; Christopherson et al., 1999; Fang et al., 2000; Alderton et al., 2001; Watanabe et al., 2003). For instance, the postsynaptic density protein PSD-95 binds nNOS via its N-terminal PDZ (PSD-95/Discs large/zona occludens-1) domain and holds nNOS in a functional complex with the NMDA subtype of glutamate receptors (Christopherson et al., 1999). Another synaptic protein, CAPON (C-terminal PDZ ligand of nNOS), competes with PSD-95 for interaction with nNOS in the postsynaptic density of synaptic spines (Jaffrey et al., 1998) and is required for formation of a ternary complex with synapsin (Jaffrey et al., 2002). Furthermore, syntrophin binds nNOS at the neuromuscular junction and maintains it at the sarcolemmal membrane (for review, see Govers and Oess, 2004). These interactions do not occur with other NOS isoforms and mainly affect the localization rather than the activity of nNOS (Alderton et al., 2001; Nedvetsky et al., 2002).
Binding of calcium-calmodulin to nNOS is an important trigger for enzymatic activation, which is achieved after increases in intracellular calcium brought about by diverse pathways, for example, after activation of NMDA receptors in neurons (Dawson et al., 1996; Alderton et al., 2001). With respect to endothelial NOS (eNOS), several recent studies have addressed the complex machinery that regulates NO production with respect to time and space, including substrate and cofactor availability, protein-protein interactions, phosphorylation, acylation, and cellular localization (Govers and Oess 2004). However, the mechanisms modulating the activity of nNOS are not completely clear (Dawson et al., 1998; Alderton et al., 2001). We hypothesized that the activity of NOS might be regulated by protein-protein interactions and have previously reported the identification of two novel proteins, NOSIP (nitric oxide synthase-interacting protein) and NOSTRIN (eNOS traffick inducer), interacting with eNOS in yeast two-hybrid screens (Dedio et al., 2001; Zimmermann et al., 2002).
Here we describe how the protein NOSIP interacts and colocalizes with nNOS in synaptic spines and influences its activity as well as subcellular distribution. Interestingly, we found that NOSIP is itself regulated by neuronal activity in terms of both the localization and the levels of expression. Thus, the interaction between nNOS and NOSIP may contribute to a mechanism via which neuronal activity regulates the synaptic availability and activity of nNOS.
Materials and Methods
Constructs. The rat nNOS cDNA was obtained as a kind gift from Solomon Snyder (Johns Hopkins University, Baltimore, MD) and cloned into a eukaryotic expression vector pcDNA3.1 (Invitrogen, Gröningen, The Netherlands). DNA fragments spanning amino acids 1-726 (named ΔRed hereafter) and 727-1464 (named nNOSred hereafter) were amplified by PCR, sequenced, and cloned in frame with an N-terminal flag tag into a modified pcDNA3.1 vector. A plasmid expressing enhanced green fluorescent protein (pEGFP-N1; Invitrogen) served as a transfection control. Human NOSIP cDNA was cloned into a modified pcDNA3.1 expression vector coding for an N-terminal myc tag. Expression of the NOSIP cDNA using the Semliki Forest virus (SFV) system was done according to the previously described protocol (Dedio et al., 2001).
Antibodies. Immunohistochemical analysis was done using a rabbit antiserum against human NOSIP (anti-NOSIP), which recognizes NOSIP protein in human and rat (Dedio et al., 2001, Dreyer et al., 2003), and with a sheep polyclonal antibody to rodent nNOS (Chemicon, Hofheim, Germany). To aid identification of cell types expressing NOSIP, sections were costained with anti-NOSIP and mouse monoclonal anti-NeuN (1:200; Chemicon). A rabbit polyclonal antibody to synapsin (Chemicon) and a monoclonal antibody to MAP-2 (Sigma, Deisenhofen, Germany) were used to identify synaptic spines and neuronal dendrites, respectively. A mouse monoclonal antibody to GABA (Sigma) was used to detect GABAergic interneurons. Immunoprecipitations were performed using anti-NOSIP, antisera to nNOS (sheep polyclonal; Chemicon), or flag tag (M2; Sigma). For Western blot detection, antibodies against NOSIP, nNOS (Transduction Laboratories, Lexington, KY), flag tag, or myc tag (9E10; Santa Cruz Biotechnology, Heidelberg, Germany) were used in combination with horseradish peroxidase (HRP)conjugated antibodies against mouse or rabbit IgG (Amersham Biosciences, Braunschweig, Germany). Anti-GFP (Chemicon) and anti-hemagglutinin (HA) (Hoffmann-La Roche, Basel, Switzerland) antibodies were used to detect expression of GFP and HA-tagged NOSIP, respectively, in transfected neurons. In some experiments, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Molecular Probes, Eugene, OR) and tetramethylrhodamine isothiocyanate (TRITC)conjugated phalloidin (Sigma) were used to counterstain cellular nuclei and the actin cytoskeleton, respectively.
Cell culture. COS-7 cells were maintained in DMEM containing 10% FCS and 1% streptomycin and penicillin and were transfected using the DEAE-dextran method by application of expression plasmids in 5.7 ml of serum-free medium mixed with 300 μl of DEAE-dextran (1 mg/ml) and 12 μl of chloroquine (50 mg/ml) to cells cultured in 10 cm dishes. After incubation for 2.5 hr, cells were treated with 10% DMSO in PBS for 2 min and cultured in DMEM and 10% FCS for 30-48 hr before use. A673 cells were maintained in DMEM containing 10% FCS and 1% streptomycin and penicillin and treated with dibutyryl-cAMP (500 μm; Sigma) to enhance expression of nNOS 48 hr before the experiment (Boissel et al., 2003). A673 cells were transfected using Polyfect (Qiagen, Hilden, Germany).
Primary hippocampal neurons were isolated from rats at embryonic day 17 and cultured on poly-l-lysine-coated (Sigma) glass coverslips in six-well plates in Neurobasal medium with B-27 supplement as described (Swiercz et al., 2002). In some experiments, primary hippocampal neurons at 14-21 d after culture [14-21 d in vitro (DIV)] were treated with either NMDA (1 μm; Sigma) and glycine (10 μm; Sigma) or with tetrodotoxin (TTX, 0.5 μm; Tocris Neuramen) for 30 min and then fixed in 4% paraformaldehyde at 2 hr after treatment. In some experiments, neurons were treated with kainate (20 μm; Sigma) for 12, 24, or 40 hr, fixed, and processed for immunofluorescence as described below.
Coimmunoprecipitation and Western blotting. Cells were lysed 24 hr after transfection in lysis buffer (20 mm Tris-HCl, pH 7.4, 137 mm NaCl, 0.05% Triton X-100, 1 mm PMSF, 1 μg/ml aprotinin, 100 μg/ml leupeptin, and 1 mm orthovanadate) on ice, and rat brain tissue was lysed in 2× lysis buffer consisting of 20 mm Tris-HCl, pH 7.4, 2 mm EDTA, 150 mm NaCl, 2% Triton X-100, 1 μg/ml aprotinin, and 1 mm orthovanadate immediately after removal. Lysates were cleared by centrifugation, and the supernatants normalized for protein content were subjected to preclearing with protein A/G agarose (Santa Cruz Biotechnology) for 30 min. Purified supernatants were incubated for 2 hr with specific antisera or with the corresponding preimmune sera or unrelated sera as indicated, and the immune complexes were precipitated after a 60 min incubation with protein A/G agarose by centrifugation. The precipitates were washed twice with 0.5 m LiCl and 100 mm Tris-HCl, pH 7.4, and once with 10 mm Tris-HCl, pH 7.4. For Western blotting, samples were dissolved in sample buffer (200 mm Tris-HCl, pH 6.8, 6% SDS, 20% glycerol, 10% DTT, and 0.1 mg/ml bromophenol blue), separated by SDS-PAGE, and transferred to polyvinylidene difluoride membranes. The membranes were washed for 30 min in PBS with Tween 20 (PBST) and blocked for 15 min in PBST containing 10% nonfat dry milk. Incubation with the primary antibodies was performed for 2 hr at 37°C, followed by extensive washing with PBST and incubation for 1 hr at 37°C with the secondary antibody. After washing with PBST, blots were developed with the ECL Western blotting detection system (Amersham Biosciences).
Arginine-citrulline assay. A673 cells were transfected with human NOSIP cDNA or pEGFP or mock-transfected with an empty expression vector (Polyfect; Qiagen). Transfection efficiency was judged from pEGFP transfections to be ∼40%. Twenty-four hours after transfection, cells were incubated at 37°C for 30 min in HEPES buffer, pH 7.4 (containing 150 mm NaCl, 5 mm KCl, 1 mm MgSO4, 1 mm CaCl2, 10 μm l-arginine, and 0.2 mg/ml glucose). Subsequently, cells were stimulated with the calcium-ionophore A23187 (3 μm; Sigma) for 15 min in the presence of 3.3 μCi/ml l-[14C]arginine and cofactors, and the conversion from l-[14C]arginine to l-[14C]citrulline was measured (Nuszkowski et al., 2001). Briefly, cells were denatured, and the soluble cellular components were dissolved in 20 mm HEPES-Na, pH 5.5, and applied to 2 ml columns of Dowex AG50WX-8 (Na+ form). The radioactivity corresponding to the l-[14C]citrulline content of the eluate was quantified by liquid scintillation counting. NOS-specific l-[14C]citrulline production was calculated as the difference of the respective radioactivity count to the value of cells treated with the NOS inhibitor N5-[imino(nitroamino)methyl]-l-ornithine (l-NNA; 100 μm) 15 min before and during stimulation.
Kainate model of epilepsy. The local governing committee approved all animal experimental protocols. Adult C57BL/6 mice were intraperitoneally injected with kainic acid (25 mg/kg) dissolved in saline (0.9% sodium chloride). We chose this moderate dose of kainate to avoid extensive cell death. Control animals received intraperitoneal injections of an equal volume of saline. The severity of behavioral seizures was assessed according to the behavioral scale (Hu et al., 1998) for 2 hr after kainate injection. At 24 hr after kainate or saline injections, mice were perfused transcardially with 4% paraformaldehyde (PFA), and the brains were processed for immunohistochemistry using rabbit anti-NOSIP as detailed below.
Complete Freund's adjuvant model of peripheral hindpaw inflammation. Unilateral peripheral hindpaw inflammation was induced by intraplantar injection of 100 μl of complete Freund's adjuvant (CFA; Sigma) into the plantar surface of the right hindpaw of adult male Wistar rats under halothane anesthesia (Merck, Planckstadt, Germany), as described previously (Kolhekar et al., 1997; Walker et al., 1999). Control animals were injected with an equal volume of saline. The uninjected contralateral hindpaw of each animal served as an internal control. At different time points after intraplantar injection of CFA (6, 12, 24, or 48 hr) or saline (12 hr), rats were perfused transcardially with 4% PFA and the dorsal root ganglia (DRGs) of the L4 and L5 nerves were isolated and processed for immunohistochemistry using rabbit anti-NOSIP.
Immunohistochemistry, quantification, and data analysis. Mice were perfused with 4% PFA, and the brains were extracted and postfixed overnight in 4% PFA. Immunohistochemistry was performed on vibratome sections (50 μm) or cryosections (20 μm) with biotinylated secondary antibodies as described in detail previously (Dreyer et al., 2003) using Vectastain Elite HRP kits and 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA) as a substrate according to the manufacturer's instructions. Care was taken to ensure that the development of the HRP-mediated end color reaction was terminated within the linear range of enzymatic activity. Sections from treated animals (kainate or CFA) and mock-treated animals were stained in the same experiment and photographed under identical illumination conditions using a cooled CCD camera (Leica, Bensheim, Germany). In experiments involving CFA injections, stained neurons in DRGs of the L4 and L5 lumbar nerves ipsilateral and contralateral to the side of CFA injection were counted. Data were averaged from at least 8-10 sections from each animal and from at least three animals per group. C- and A-δ-fiber neurons were identified on the basis of size (Stucky and Lewin, 1999). Upregulation in the number of NOSIP-positive neurons was calculated as the percent increase in stained neurons in the ipsilateral DRGs over contralateral DRGs. ANOVA and then a post hoc Fisher's exact test were performed to judge statistically significant differences (p ≤ 0.05).
Immunofluorescence and confocal analysis. Vibratome-cut brain sections (50 μm) or cultured cells grown on coverslips were incubated overnight with primary antibodies in 5% normal serum in PBS, washed, and treated with FITC- or TRITC-conjugated secondary antibodies (Dianova, Hamburg, Germany) as previously reported (Swiercz et al., 2002). Sections or coverslips were washed, mounted in Mowioll, and visualized using a laser-scanning confocal microscope (TCS-AOBS; Leica, Bensheim, Germany). In some experiments, fluorescence intensity per unit area was measured over selected areas (e.g., nucleus and cytoplasm) in stained cells using the Leica confocal software. In experiments involving quantification of staining intensity or number of cells demonstrating a defined pattern of staining, at least 20-25 cells were analyzed per group per experiment, and data were averaged from at least three independent experiments. In experiments involving kainate injections, staining intensity per unit area was measured densitometrically (NIH Image software) over the piriform cortex, dentate gyrus, and CA1 region of the hippocampus, using unstained areas within the same section for background correction. Data were averaged over at least three or four stained sections per mouse and from at least three mice per test group. Induction levels were calculated as mean fold increase in kainate-injected mice over normalized staining intensities in saline-injected mice. Induction levels from at least three such experiments were averaged and statistically evaluated by randomized ANOVA.
Results
Interaction between NOSIP and nNOS
NOSIP was originally identified as an interactor and modulator of eNOS (Dedio et al., 2001). Because of the similarity of sequence, domain structure, and mode of activation between the two NOS isoforms, we asked whether NOSIP would also associate with nNOS. This was supported by the observation that NOSIP and nNOS interacted in a yeast two-hybrid screen, suggesting a direct interaction between these two proteins (Dedio et al., 2001). Indeed, on heterologous expression, NOSIP was coimmunoprecipitated with nNOS from cotransfected COS cells by an anti-nNOS antibody, whereas an unrelated serum failed to do so (Fig. 1A). Conversely, an antiserum directed against NOSIP selectively coimmunoprecipitated nNOS with endogenous and cotransfected NOSIP (Fig. 1B). To determine whether NOSIP and nNOS are capable of interacting in the nervous system in vivo, we performed coimmunoprecipitation experiments on rat brain lysates. Anti-nNOS but not unrelated serum coimmunoprecipitated NOSIP with nNOS from brain lysates (Fig. 1C), demonstrating that NOSIP and nNOS have the potential to interact in vivo.

Coimmunoprecipitation of NOSIP and nNOS from COS cells transfected as indicated. A, Anti-nNOS but not unrelated serum (control) coimmunoprecipitates heterologously transfected NOSIP. B, Conversely, an anti-NOSIP antibody coimmunoprecipitates heterologously transfected nNOS with endogenous or cotransfected NOSIP. In A and B, rat brain lysates are loaded as positive controls. C, Coimmunoprecipitation of NOSIP and nNOS from rat brain lysates by anti-nNOS but not by control serum. IP, Immunoprecipitation; lys, lysates; pre, preimmune serum; WB, Western blot.
To assess which domains of nNOS physically associate with NOSIP in mammalian cells, we constructed epitope-tagged mutants of nNOS in which the reductase domain of nNOS was either deleted (ΔRed) or expressed alone (nNOSred) (for precise domain boundaries, see Materials and Methods). Flag-tagged ΔRed was coimmunoprecipitated by anti-NOSIP in cells cotransfected with NOSIP and also to a weaker extent in COS cells expressing endogenous levels of NOSIP (Fig. 2A). Similarly, the anti-flag antibody coimmunoprecipitated NOSIP from cell lysates containing both NOSIP and flag-tagged ΔRed (Fig. 2B). In contrast, NOSIP was not significantly coimmunoprecipitated with nNOSred (Fig. 2C). These experiments thus show that the interaction with NOSIP does not involve the reductase domain of nNOS but is rather mediated via motifs located in the N-terminal region of nNOS. This finding is consistent with our initial observation from yeast two-hybrid screens also implicating the N terminus of nNOS (amino acids 1-728) in interacting with NOSIP (data not shown). Furthermore, cotransfection experiments of deletion constructs in a yeast interaction assay suggested that the nNOS binding site is located within the first 200 amino acids of the NOSIP sequence (data not shown).

Coimmunoprecipitation of NOSIP and flag-tagged deletion constructs of nNOS from COS cells transfected as indicated. A, The anti-NOSIP antibody but not preimmune serum coimmunoprecipitates flag-tagged nNOS lacking the reductase domain (ΔRed). Immunoprecipitation with anti-flag serves as a positive control. B, Conversely, anti-flag but not unrelated antibodies coimmunoprecipitate NOSIP from cotransfected cells. Immunoprecipitation with anti-NOSIP serves as a positive control. C, Anti-NOSIP fails to substantially coimmunoprecipitate the flag-tagged reductase domain of nNOS (nNOSred). Immunoprecipitation with the anti-flag antibody serves as a positive control. IP, Immunoprecipitation; pre, preimmune serum; WB, Western blot.
Colocalization of NOSIP and nNOS on heterologous coexpression
Next, we addressed whether NOSIP and nNOS colocalize in situ and whether they mutually influence their subcellular distribution in living cells. NOSIP is described to exhibit two distinct subcellular localization patterns and can reside either in the cytoplasm or in the nucleus of various cell types (König et al., 2002). After transient transfection, NOSIP is predominantly targeted to the nucleus, and cotransfection of NOSIP with the control vector (for example and summary, see Fig. 3A,D) or with EGFP (Fig. 3D) did not change the nuclear localization of NOSIP in COS cells. However, coexpression of nNOS with NOSIP led to a striking retention of NOSIP in the cell cytoplasm (Fig. 3B) (p < 0.05 in comparison with vector- or EGFP-transfected cells). Cotransfected NOSIP and nNOS appeared to be distributed in aggregates or clusters at the cell membrane and demonstrated near-complete colocalization (Fig. 3B,D). In contrast, after cotransfection with the reductase domain of nNOS, NOSIP was predominantly found in the cell nucleus, with only a minor fraction evident in the cytoplasm (Fig. 3C,D) (p > 0.05). These results further support a physical association between nNOS and NOSIP in mammalian cells.

Confocal analysis of the localization of NOSIP and nNOS in cotransfected COS cells. In all panels, arrowheads in the differential interference contrast (DIC) images point to the cell nucleus. A, Anti-NOSIP labels the cell nucleus when NOSIP is cotransfected with pcDNA3; for control, an empty vector was used. B, In COS cells transfected with NOSIP and nNOS, NOSIP is retained in the cytoplasm, where it colocalizes with nNOS. C, A construct expressing the flag-tagged reductase domain of nNOS (nNOSred-flag) fails to colocalize with NOSIP. D, Quantitative analysis of cotransfected COS cells for the differential distribution of NOSIP between nucleus and cytosol (expressed as a fraction of the total number of cells analyzed). *Significant differences in cell fractions showing cytoplasmic or nuclear staining, respectively, with respect to other transfection groups (p < 0.001, ANOVA followed by post hoc Fisher's exact test).
Effect of NOSIP on catalytic activity of nNOS
An important question, which arose at this point, was whether protein-protein interactions between nNOS and NOSIP affect the enzymatic activity of nNOS. To address this, we used an immortalized neuroepithelioma cell line, A673, stably expressing moderate levels of nNOS (Boissel et al., 2003) but devoid of eNOS (data not shown). A673 cells were either mock-transfected or transfected with myc-tagged NOSIP, and activity of nNOS after stimulation with the calcium ionophore A23187 was estimated by measuring the conversion of 14C-labeled arginine to citrulline in intact cells. Transfection efficiency of myc-tagged NOSIP was judged to be ∼40%. The fraction of A23187-induced 14C-labeled citrulline production, which could be inhibited by pretreatment with the NOS inhibitor l-NNA, was taken as a measure of specific nNOS activity. Cells transfected with NOSIP demonstrated significantly lower specific nNOS activity than mock-transfected cells (∼30%; p < 0.002 in four independent experiments, each performed in triplicate) (a representative experiment is shown in Fig. 4). Furthermore, because NOSIP was overexpressed in only ∼40% of the total number of A673 cells used for determining nNOS activity, the degree of inhibition of nNOS activity represented in Figure 4 actually represents an underestimation. These differences in citrulline production did not arise because of variations in nNOS levels across treatment groups, as judged by Western blot analysis (Fig. 4, top). Thus, the interaction between NOSIP and nNOS negatively affects the catalytic activity of nNOS.

Influence of NOSIP on nNOS activity. nNOS activity in intact A673 cells was measured as conversion of [14C]arginine to [14C]citrulline. Compared with mock- or GFP-transfected cells (data not shown), expression of NOSIP reduced the A23187-induced nNOS activity by ∼30%. Transfection efficiency was judged to be ∼40% by using pEGFP as a transfection control (data not shown). The expression levels of nNOS protein remain unchanged by transfection of empty vector (mock) or myc-NOSIP, as demonstrated by Western blot (WB) analysis of corresponding lysates (top). **p < 0.05.
Expression of NOSIP in the nervous system
Previous studies have precisely characterized the expression pattern of nNOS (Eliasson et al., 1997; Guirado et al., 2003), whereas little is known about the distribution of NOSIP in the nervous system. When we compared the staining patterns of anti-NOSIP or anti-nNOS on adult rat brain sections, we found that both NOSIP and nNOS were expressed in similar populations of neurons in the somatosensory cortex, piriform cortex, cerebellum, and hippocampus (Fig. 5). In the somatosensory cortex, several pyramidal cells were labeled with anti-NOSIP and anti-nNOS (Fig. 5A,B, arrowheads, arrows). The overall number of neurons expressing NOSIP over the entire somatosensory cortex is higher than the overall number of neurons expressing nNOS. Consistent with previous reports (Eliasson et al., 1997; Lumme et al., 2000; Guirado et al., 2003), nNOS was found to be only very weakly expressed in hippocampal pyramidal neurons but strongly expressed in interneurons that are scattered in between the pyramidal cell layers and enriched in the hilus of the dentate gyrus (Fig. 5C). Interestingly, both NOSIP and nNOS are abundant in hilar interneurons (Fig. 5C). Unlike nNOS, NOSIP is also highly expressed in the pyramidal cells in CA3 and CA4 sectors but only poorly expressed in CA2 and CA1 sectors of the hippocampus (see below). In the cerebellum, Purkinje cells expressed both nNOS and NOSIP, whereas NOSIP but not nNOS was moderately expressed in cerebellar granule cells (Fig. 5D). Furthermore, anti-NOSIP immunoreactivity was observed in a large number of neurons in the brainstem (data not shown).

Immunohistochemical determination of NOSIP and nNOS expression in rat brain. A, Staining of all layers of the somatosensory cortex (arrowheads point to stained neurons in layer V). B, Higher magnification of A (arrows). The arrowhead in B points to a neuron with predominant nuclear localization of NOSIP. C, Staining of interneurons (arrows) scattered in the hilus and around the granule cell layer of the dentate gyrus (DG). D, In the cerebellum, both NOSIP and nNOS are expressed in Purkinje cells (arrowheads), but only NOSIP is clearly evident in granule cell layer (arrows). Scale bars: A, C, 100 μm; B, D, 20 μm.
We then performed confocal coimmunofluorescence analysis of brain sections to determine whether individual neurons in the brain have the ability to coexpress NOSIP and nNOS. Interestingly, we observed that nNOS immunoreactivity was found over a majority of neurons in layer V of the cortex (Fig. 6A). Therefore, particularly, layer V cortical neurons coexpress NOSIP and nNOS, where both nNOS and NOSIP were observed to be concentrated in clusters along the soma as well as the apical dendrites (Fig. 6A). In addition, anti-NOSIP immunoreactivity was evident in the cytoplasm and dendrites of most neurons (Fig. 6A) and was occasionally seen in the cell nucleus (Fig. 5B, arrowhead). A few neurons in the dentate gyrus also demonstrated colocalization of NOSIP and nNOS (data not shown).

Colocalization of NOSIP and nNOS in neurons. A, Magnified view of layer V pyramidal neurons of the rat somatosensory cortex, which are abundant in immunoreactivity for NOSIP and nNOS. Arrowheads indicate costained clusters on neurons; arrows indicate NOSIP expression in apical dendrites; and asterisks indicate somatic labeling. B, C, Coimmunofluorescence analysis of localization of NOSIP and nNOS (B) or synapsin and nNOS (C) in cultured primary hippocampal neurons at 21 d in vitro. Spines labeled on dendritic trees are magnified in the lower panels; arrows point to colabeled spines.
To characterize the subcellular distribution of NOSIP and nNOS in neuronal cells, we performed confocal immunofluorescence analysis on cultured rat hippocampal neurons. Young, developing hippocampal neurons at DIV 3-5 expressed NOSIP and nNOS throughout the developing dendrites and axons and in axonal growth cones (data not shown). In contrast, mature (DIV 20-28) neurons demonstrated a punctate pattern of labeling along the dendritic tree, with anti-nNOS and anti-NOSIP resembling the typical pattern of synaptically localized proteins (Fig. 6B). Most NOSIP-positive puncta were also immunoreactive for nNOS and vice versa (Fig. 6B, overlay). The synaptic nature of the majority of these puncta was evident from colabeling experiments with synaptic marker proteins such as synapsin (Fig. 6C).
Subcellular localization of NOSIP is modulated by synaptic activity
Although NOSIP antigen was evident in the somatic cytoplasm of cultured hippocampal neurons, approximately one-third of these cells showed additional strong labeling of the cell nucleus with anti-NOSIP, whereas the rest of the population showed predominant cytoplasmic staining. Based on our previous observation that NOSIP exhibits two distinct subcellular localization patterns, i.e., cytosolic versus nuclear (König et al., 2002), and that nNOS leads to the cytoplasmic retention of NOSIP on heterologous cotransfection in COS cells (see above), we addressed the cytoplasmic versus nuclear localization of NOSIP in neurons more closely. Because all neurons studied demonstrated only weak somatic expression of nNOS, no correlation was evident between the endogenous expression of nNOS and the nuclear targeting of NOSIP. We then asked whether NOSIP trafficking in the neuronal somata is subject to regulation by mechanisms, which are dependent on the activation state of neurons. When we treated neurons with the NMDA receptor agonist NMDA (1 μm), and a coagonist, glycine (10 μm), we observed a significant increase in the number of neurons demonstrating predominantly cytoplasmic localization and a corresponding decrease in cell number demonstrating predominantly nuclear localization of NOSIP over the basal state (for typical example and summary of relative staining intensities, see Fig. 7A,D). Application of glutamate (100 μm) had similar effects (data not shown). Because cultured hippocampal neurons exhibit a significant amount of spontaneous activity in the basal, unstimulated state in culture (Ju, 2004), we addressed the relative distribution, i.e., nuclear versus cytoplasmic, of NOSIP in neurons silenced by treatment with TTX. At concentrations known to abolish spontaneous activity in cultured hippocampal neurons (0.5 μm), TTX led to an increase in neurons showing a predominantly nuclear localization of NOSIP and a corresponding decrease in cell number showing predominantly cytoplasmic expression (for typical example and summary of relative expression levels, see Fig. 7A,D). In contrast to the neuronal somata, neither NMDA and glycine nor TTX treatment showed any readily obvious effects on the localization of NOSIP at terminal dendrites and synaptic spines (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These observations suggest that inhibition of neuronal activity leads to an accumulation of NOSIP in the cell nucleus, whereas an increase in neuronal activity triggers mechanisms facilitating the targeting of NOSIP to the cytoplasmic compartment.

Immunofluorescence analysis of localization of NOSIP in cultured primary hippocampal neurons at 21 d in vitro. DAPI was used to label nuclei. A, Neuron with NOSIP localization in the cell nucleus and in the cytoplasm. B, Neurons treated with NMDA (1 μm) and glycine (10 μm) demonstrate a primarily cytoplasmic localization of NOSIP. C, Neurons treated with TTX (0.5 μm) demonstrate a primarily nuclear localization of NOSIP. D, Quantitative measurement of NOSIP immunofluorescence over the cell cytoplasm or nucleus in neurons, expressed as the difference between cytoplasmic and nuclear staining intensities, in the basal state and on treatment with NMDA and glycine or with TTX. *Significant difference from the basal state (p < 0.05, ANOVA followed by post hoc Fisher's exact test). AU, Arbitrary units.
Activity-dependent regulation of NOSIP expression in vivo
The expression of nNOS as well as its interacting proteins such as CAPON, PIN (protein inhibitor of NOS), and PSD-95 has been reported to be upregulated by stimuli triggering long-lasting activation of neurons in the central and peripheral nervous systems (Che et al., 2000a,b; Huh et al., 2000; Lumme et al., 2000; Ohnuma et al., 2000; Sasaki et al., 2000; Jiang et al., 2003; Wiggins et al., 2003). We therefore asked whether the expression of NOSIP is also regulated by neuronal activity in vivo and in vitro. We chose kainate-induced seizures as a model to investigate the activity-induced regulation of nNOS and NOSIP expression in the forebrain, especially because nNOS has been reported to be transcriptionally regulated by kainate at time points before seizure-induced cell death is observed and because seizure-related changes have been linked to NOS activation (Huh et al., 2000; Lumme et al., 2000; Chavko et al., 2001; Bagetta et al., 2002). Consistent with previous reports, intraperitoneal injection of 25 mg/kg kainate led to stage 2-3 seizures in mice (Hu et al., 1998). At 24 hr after induction of seizures, the intensity of immunofluorescence for NOSIP was found to be increased in several regions of the cortex such as the piriform (Fig. 8A-C), somatosensory, and cingulate (data not shown) compared with control. Similar results were obtained when we stained sections with a biotinylated secondary antibody and the HRP method, with the added advantage of being able to detect the architecture of NOSIP-expressing pyramidal cells at a higher sensitivity (Fig. 8C). Interestingly, whereas saline-injected mice demonstrated minor, perinuclear immunoreactivity for NOSIP antigen, the cytoplasm in the entire soma and primary dendrites were richly stained with anti-NOSIP in kainate-injected mice (Fig. 8C). Under the same conditions, NOSIP was abundantly found in several small nonpyramidal cells (Fig. 8A,B, arrows), which, based on their abundant localization in layer I, morphology, and failure to costain with anti-GABA (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), likely represent astrocytes, which is interesting in light of reports of an upregulation of nNOS in astrocytes after seizures (Catania et al., 2003).

Upregulation of NOSIP expression by neuronal activity in the brain in vivo. Antigen levels were judged from immunofluorescence (A, B, D-G) or peroxidase immunohistochemistry (C) in brain. NOSIP expression in mice injected intraperitoneally with 25 mg/kg kainate or saline is shown in the piriform cortex (A-C) and the hippocampus (D-G). B, C, Higher-magnification views of pyramidal neurons from the piriform cortex. Arrows in A and B indicate upregulation of NOSIP in cortical neurons. Arrowheads in D and E indicate upregulation of NOSIP in the dentate gyrus, whereas the arrow points to NOSIP upregulation in pyramidal cell layer of the CA1 region. F, G, Higher-magnification views of pyramidal neurons in CA1 and CA3 sectors of the pyramidal cell layer, respectively. DG, Dentate gyrus. Scale bars: A, D, E, 200 μm; B, C, F, G, 40 μm.
The most striking upregulation of NOSIP antigen in the kainate model was seen in the hippocampus (Fig. 8D,E). Whereas the granule cells of the dentate gyrus and pyramidal cells of the CA1 and CA2 sectors only poorly expressed NOSIP in naive or saline-injected mice, NOSIP was abundantly expressed in these regions in kainate-injected mice (Fig. 8D-F). Quantification of the intensity of NOSIP immunofluorescence demonstrated significant upregulation in kainate-injected mice in comparison with saline-injected mice in the piriform cortex (2.10- ± 0.38-fold) as well as in the CA1 sector (5.15- ± 0.8-fold) and the dentate gyrus (5.54- ± 0.57-fold) (p < 0.005 in all three regions, ANOVA followed by post hoc Fisher's exact test). In contrast, nNOS expression did not change significantly in any of the principal cell populations in the hippocampus (data not shown), consistent with previous reports of interneuron-specific upregulation after kainate injection (Lumme et al., 2000). In kainate-injected mice, NOSIP, like nNOS, was also found to be highly expressed in cells that were scattered in the molecular layer of the hippocampus, many of which comprise GABAergic interneurons, as probed by costaining with anti-GABA (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Consistent with activity-induced upregulation of NOSIP in vivo, application of kainate to neurons in vitro also led to an obvious increase in anti-NOSIP immunoreactivity (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material), which was distributed over the entire soma and dendrites. Kainate-induced upregulation of NOSIP in vitro was detectable at 24 and 40 hr but not at 12 hr after treatment. Unlike NOSIP, nNOS immunoreactivity was visibly increased in only a small population of neurons treated with kainate (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material).
Because NOSIP is coexpressed with nNOS in peripheral nociceptive neurons of the DRGs (Dreyer et al., 2003) and nNOS is upregulated in DRG neurons after peripheral inflammation (Luo et al., 1999; Cizkova et al., 2002), we chose peripheral hindpaw inflammation as a model to study the potential regulation of NOSIP expression in peripheral neurons by persistent nociceptive activity (Walker et al., 1999). DRG sections ipsilateral to the inflamed hindpaw demonstrated a significant increase in NOSIP-positive C- and A-δ-fiber neurons [classified based on size, as described by Stucky and Lewin (1999)] compared with the contralateral DRGs, which received input from the untreated hindpaw (Fig. 9A,B). Upregulation of NOSIP was observed as early as 6 hr after the onset of inflammation. Most NOSIP-positive neurons were also colabeled with isolectin-B4 or expressed substance P, both of which serve as markers for the major subpopulations of nociceptive and thermoreceptive neurons (data not shown).
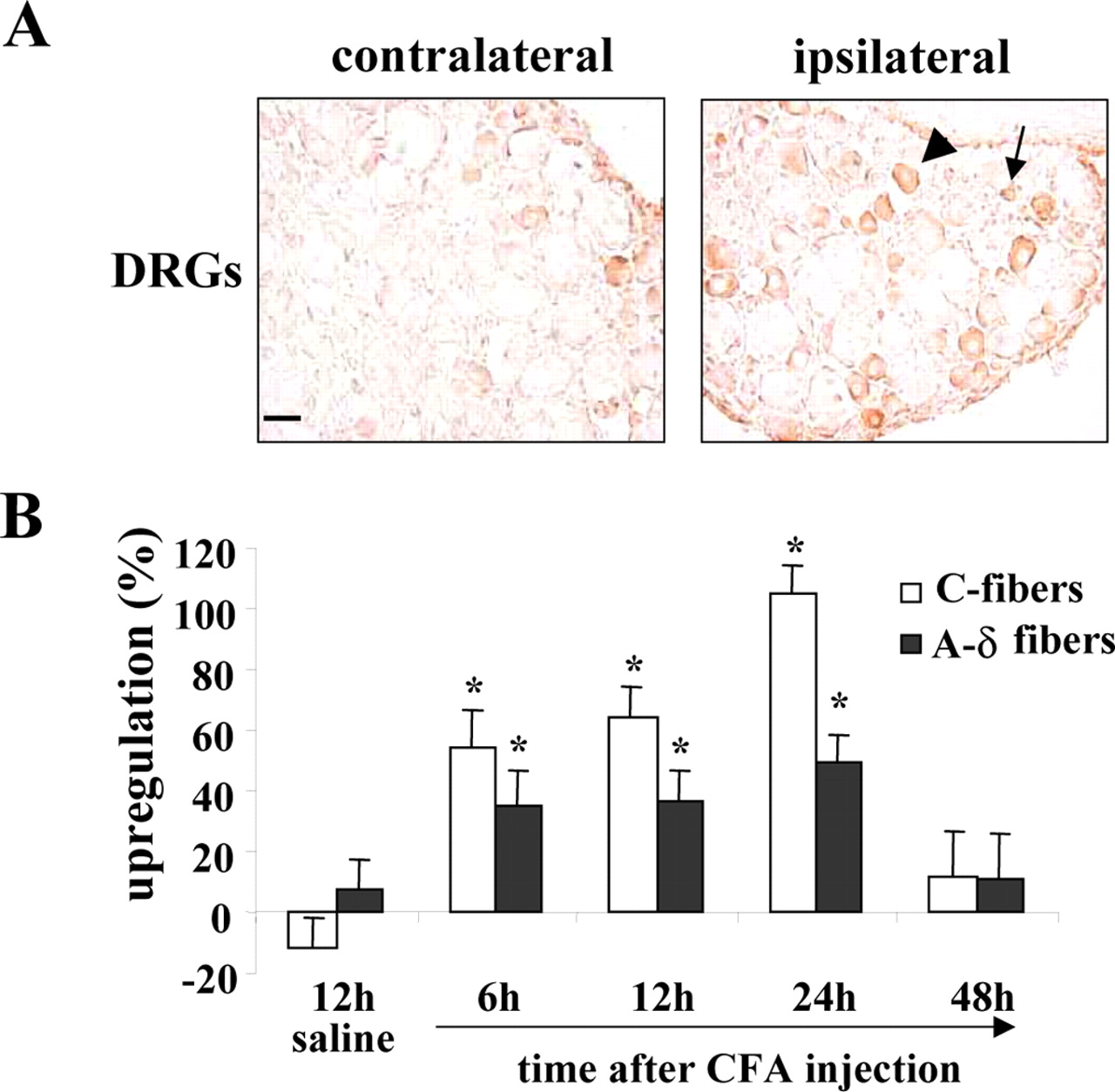
Upregulation of NOSIP expression by neuronal activity in peripheral sensory neurons in vivo. A, NOSIP expression in rats, which received unilateral injections of CFA (100 μl) in the hindpaw. Ipsilateral and contralateral L4 DRGs stained with anti-NOSIP are shown. The arrow indicates small-diameter (C-fiber) neurons, and the arrowhead indicates intermediate-diameter (A-δ-fiber) neurons. B, Quantification of NOSIP-expressing cells in rats injected with CFA or saline (control). The y-axis represents an increase in NOSIP-positive cells in the DRGs ipsilateral to injection over the uninjected contralateral side (in percent). *Significant differences from respective cell types in saline-injected rats (p < 0.05, ANOVA followed by post hoc Fisher's exact test).
Thus, paradigms of increased neuronal activity in the central and peripheral nervous systems, which are reported to produce an upregulation of nNOS, also lead to a concurrent upregulation of NOSIP. Taken together, our results indicate that NOSIP and nNOS are not only coexpressed in several neuronal populations but also coregulated in their expression levels by neuronal activity in vivo.
Modulation of nNOS localization by NOSIP
The observation that neuronal activity consistently increased cytoplasmic (rather than nuclear) localization of NOSIP in neurons in vitro as well as in vivo led to the question of how the localization of nNOS is then influenced by the differential intracellular distribution of its interacting protein. We therefore simulated activity-induced cytoplasmic overexpression of NOSIP in cultured neurons using a Semliki Forest virus expressing HA-tagged NOSIP (SFV-NOSIP). SFV particles expressing GFP (SFV-GFP) served as a control. Although α viruses induce toxicity at 24-48 hr after infection, they produce rapid overexpression of cytoplasmic proteins and have been extensively used as the method of choice for analyzing the functional significance of protein-protein interactions in primary hippocampal neurons (Malinow and Malenka, 2002; Lundstrom et al., 2003). We carefully monitored infected primary neurons for virally induced toxicity by observing the morphology of dendrites, cellular contours, and signs of vacuolization of the soma in phase contrast microscopy as well as by analyzing phalloidin-based fluorescence staining of the neuronal actin cytoskeleton. At 12 hr after infection, a large number of neurons demonstrated GFP immunoreactivity in the soma and dendrites without showing signs of toxicity. No differences were evident in the subcellular distribution of nNOS in GFP-positive neurons versus untransfected neurons (Fig. 10A). In contrast, in a majority of NOSIP-positive neurons, nNOS immunoreactivity was obviously lessened in terminal tertiary dendrites and was restricted to the primary and secondary dendrites (Fig. 10B). This effect of SFV-NOSIP was specific for nNOS and was not observed with other dendritic proteins such as MAP-2 (data not shown). A blinded, semiquantitative analysis of the intensity of nNOS immunofluoresence in dendrites versus cytoplasm revealed that HA-NOSIP but not GFP diminished the localization of nNOS in distal dendrites by at least 50% (Fig. 10B) (p = 0.002 compared with GFP-transfected cells, ANOVA followed by post hoc Fisher's exact test). These results suggest that an increase in the somatic expression of NOSIP leads to a sequestration of nNOS into the soma, thereby limiting its trafficking to distal dendrites.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of expression of Semliki Forest virus constructs encoding GFP (SFV-GFP; A) or HA-tagged NOSIP (SFV-HA-NOSIP; B) on immunofluorescence staining of nNOS in cultured primary hippocampal neurons at 21 d in vitro. Anti-GFP (B) and anti-HA (A) were used to detect expression of the respective proteins. C, Quantification of nNOS immunofluorescence over primary, secondary, or distal tertiary dendrites normalized to the staining intensity in the somatic cytoplasm. *Significant differences in staining ratio in NOSIP-expressing compared with GFP-expressing neurons (p = 0.002, ANOVA followed by post hoc Fisher's exact test).
Discussion
The present study describes a novel functional interaction between nNOS and NOSIP and their coexpression, colocalization, and regulation in the nervous system. NOSIP was originally identified as a protein, which interacts with the oxygenase domain of eNOS and translocates eNOS from its active sites at the cell membrane to the cell interior (Dedio et al., 2001). Although we have not found any evidence for an interaction of NOSIP with inducible NOS (Dedio et al., 2001) (J. Dedio and W. Müller-Esterl, unpublished observations), it is striking that the molecular interaction of NOSIP with eNOS and nNOS is very similar in terms of the locus of interaction as well as its functional consequences.
Perhaps the most interesting finding of this study is that NOSIP modulates the activity as well as the localization of nNOS. Although several protein interactions have been described for nNOS, after PIN, NOSIP is the second protein reported so far to inhibit NO production. Although we only observed a modest inhibition of nNOS activity by NOSIP, two factors deserve consideration. First, because of the technical aspects specific to our assay system (e.g., a low transfection efficiency), the degree of inhibition observed in vitro may represent an underestimation. Second, a 30% decrease in the activity of nNOS could be very critical in the highly localized cellular milieu of synaptic spines on terminal dendrites, where nNOS is expressed.
Two mechanisms for the inhibition of nNOS activity are conceivable. First, NOSIP could inhibit nNOS directly by interfering with the electron flux. In the case of eNOS, however, enzymatic activity was unaffected by NOSIP in a cell-free system, strongly arguing against such a direct mechanism of inhibition (Dedio et al., 2001). Alternatively, the inhibition of nNOS activity could be indirectly caused by a NOSIP-induced relocation of nNOS away from its sites of action at the cell membrane. Indeed, an overexpression of NOSIP leads to sequestration of nNOS in the perikaryon and appears to limit its availability at distal dendrites in primary hippocampal neurons. This change could arise either from sequestration of newly synthetized nNOS and failure to traffic it to distal sites or from a relocation of synaptic nNOS toward the somata away from the distal dendrites. Although further studies are required to elucidate the precise mechanisms underlying our observations, the latter possibility appears more likely in lieu of the suppression of de novo synthesis of cellular proteins by α viruses and the ability of NOSIP to translocate eNOS from the plasma membrane to the cell interior (Dedio et al., 2001). Although previous studies have demonstrated that protein-protein interactions can induce changes in the coupling of nNOS to the postsynaptic density (e.g., CAPON) (Jaffrey et al., 1998) or to the sarcolemmal membrane at the neuromuscular junction (e.g., syntrophin) (for review, see Govers and Oess, 2004), this is the first report, to our knowledge, demonstrating spatial changes in nNOS targeting induced by an interacting protein in neuronal dendrites via confocal microscopy. However, in light of the diverse protein-protein interactions associated with nNOS, further detailed studies addressing dendritic trafficking of nNOS in neurons and its modulation and orchestration via its multiple interacting proteins are mandatory.
Both eNOS and nNOS are very widely expressed proteins and play crucial roles in the regulation of neural, cardiovascular, and vegetative functions. Given the broad expression of NOSIP in non-neural tissues (Dedio et al., 2001; König et al., 2002) as well as its abundance in the central and peripheral nervous systems as described here, it is conceivable that NOSIP is functionally linked to eNOS as well as nNOS in a wide range of tissues and cell types. In the nervous system, NO is primarily derived from nNOS and is functionally implicated in diverse cellular processes ranging from axonal guidance in developing neurons (Hess et al., 1993) to synaptic transmission and plasticity in mature neurons (Dawson and Dawson, 1998; Boehning and Snyder, 2003; Gibbs, 2003). We observed an extensive colocalization of NOSIP and nNOS in immature, developing neurons as well as at synaptic sites in mature neurons in culture, suggesting that NOSIP has the capacity to affect nNOS activity and localization at many critical stages during the lifetime of a neuron.
NO has gained prominence as an atypical neurotransmitter and a regulator of the strength of synaptic transmission, which has been best studied so far in the hippocampus (O'Dell et al., 1994). In the hippocampal CA1 sector, NO has been shown to function as a retrograde messenger released from postsynaptic pyramidal neurons, which facilitates presynaptic function and thereby contributes to long-term potentiation (LTP) of hippocampal synaptic transmission (O'Dell et al., 1991; Schuman and Madison, 1991). Interestingly, eNOS but not nNOS serves as the prime source of NO in hippocampal CA1 and CA2 pyramidal cells (O'Dell et al., 1994; Kantor et al., 1996), whereas nNOS is dominant in hippocampal interneurons (Lumme et al., 2000; Guirado et al., 2003). Because of its ability to attenuate the catalytic activity of both nNOS and eNOS and its expression in pyramidal cells as well as interneurons, NOSIP is thus ideally poised to modulate synaptic strength and LTP in the hippocampus via fine tuning of the local NO production. Apart from its role in neurotransmission, NO is also functionally involved in epilepsy, ischemia, and a wide range of neurodegenerative diseases (Dawson et al., 1998). Both nNOS and eNOS are upregulated in animal models of epilepsy, and NO has been causally linked to neuronal cell death after status epilepticus (Huh et al., 2000; Lumme et al., 2000; Chavko et al., 2001; Bagetta et al., 2002). We were therefore intrigued to observe a massive upregulation of NOSIP after kainate-induced seizures in the CA1 and CA2 regions and in the dentate gyrus, both being regions that are relatively spared of neurotoxicity in comparison with the CA3 and CA4 regions. Similarly, nNOS is upregulated and eNOS is ectopically expressed in peripheral sensory neurons after peripheral injury (Luo et al., 1999; Levy et al., 2000; Cizkova et al., 2002), and the generation of NO has been functionally associated with nociceptive hypersensitivity and chronic pain (Meller and Gebhart, 1993). Interestingly, we observed that NOSIP is also upregulated in nociceptive C- and A-δ-neurons after peripheral inflammation. These results, together with the negative modulation of NOS function by NOSIP, suggest that an activity-dependent upregulation of NOSIP may constitute a protective or compensatory mechanism to counteract excessive production of NO in neurons. Interestingly, this kind of modulation is not unique for NOSIP but is a feature shared by several other proteins interacting with nNOS, including CAPON, PIN, and PSD-95, which are known to be upregulated in models of enhanced neuronal activity and pathology in the central and peripheral nervous systems (Che et al., 2000a,b; Ohnuma et al., 2000; Jiang et al., 2003; Wiggins et al., 2003). Thus, nNOS is likely subject to tight regulatory control via activity-dependent modifications in the expression of several nNOS-interacting proteins.
Another interesting observation of this study was that not only the expression level but also the subcellular localization of NOSIP is regulated by neuronal activity. In cultured primary hippocampal neurons, the localization of NOSIP varied among nuclear, cytoplasmic, and synaptic, consistent with an active shuttling of NOSIP observed in mammalian cell lines (M. Schleicher, S. Oess, and Müller-Esterl, unpublished observations). Although we do not currently understand the functional implications of these findings, it is noteworthy that almost all of the few proteins that have been described to shuttle between synapses and nuclear loci in neurons were revealed to play key roles in synaptic plasticity and activity-dependent long-term changes in neuronal function (Curtis and Finkbeiner, 1999; Thomas and Huganir, 2004). We were intrigued by the observations that a silencing of neuronal activity favors a nuclear localization of NOSIP, whereas a predominantly non-nuclear localization is favored during basal activity or NMDA receptor-evoked activity of neurons. More detailed studies are required to study the significance of NOSIP trafficking in the context of synaptic transmission and plasticity.
In conclusion, we report here a functional interaction between nNOS and NOSIP and describe the expression of NOSIP in the nervous system and its modulation by neuronal activity. Our results suggest that NOSIP regulates NO production in the nervous system by modulating the localization and activity of nNOS, and potentially also of eNOS, and lay a basis for testing these hypotheses in null mouse mutants of NOSIP and NOS enzymes.
Footnotes
- Received June 9, 2004.
- Revision received October 7, 2004.
- Accepted October 7, 2004.
This work was supported by an Emmy Noether Program grant from the Deutsches Forschungsgemeinschaft (R.K.) and by the Sonderforschungsbereich 553 (S.O., W.M.-E.). We thank S. Snyder for providing the nNOS cDNA, S. Jaffrey for advice, U. Förstermann for providing the A673 cell line, and S. Graf for technical assistance.
Correspondence should be addressed to Rohini Kuner, Department of Molecular Pharmacology, Pharmacology Institute, Im Neuenheimer Feld 366, University of Heidelberg, 69120 Heidelberg, Germany. E-mail: rohini.kuner{at}urz.uni-heidelberg.de.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410454-12$15.00/0
* J.D. and M.S. contributed equally to this work.