
Abstract
Neurexins are presynaptic cell-adhesion molecules that bind to postsynaptic cell-adhesion molecules such as neuroligins and leucine-rich repeat transmembrane proteins (LRRTMs). When neuroligins or LRRTMs are expressed in a nonneuronal cell, cocultured neurons avidly form heterologous synapses onto that cell. Here we show that knockdown of all neurexins in cultured hippocampal mouse neurons did not impair synapse formation between neurons, but blocked heterologous synapse formation induced by neuroligin-1 or LRRTM2. Rescue experiments demonstrated that all neurexins tested restored heterologous synapse formation in neurexin-deficient neurons. Neurexin-deficient neurons exhibited a decrease in the levels of the PDZ-domain protein CASK (a calcium/calmodulin-activated serine/threonine kinase), which binds to neurexins, and mutation of the PDZ-domain binding sequence of neurexin-3β blocked its transport to the neuronal surface and impaired heterologous synapse formation. However, replacement of the C-terminal neurexin sequence with an unrelated PDZ-domain binding sequence that does not bind to CASK fully restored surface transport and heterologous synapse formation in neurexin-deficient neurons, suggesting that no particular PDZ-domain protein is essential for neurexin surface transport or heterologous synapse formation. Further mutagenesis revealed, moreover, that the entire neurexin cytoplasmic tail was dispensable for heterologous synapse formation in neurexin-deficient neurons, as long as the neurexin protein was transported to the neuronal cell surface. Furthermore, the single LNS-domain (for laminin/neurexin/sex hormone-binding globulin-domain) of neurexin-1β or neurexin-3β, when tethered to the presynaptic plasma membrane by a glycosylinositolphosphate anchor, was sufficient for rescuing heterologous synapse formation in neurexin-deficient neurons. Our data suggest that neurexins mediate heterologous synapse formation via an extracellular interaction with presynaptic and postsynaptic ligands without the need for signal transduction by the neurexin cytoplasmic tail.
Introduction
Neurexins are conserved presynaptic cell-adhesion molecules that are essential for synaptic transmission (Ushkaryov et al., 1992; Missler et al., 2003). Vertebrates contain three neurexin genes (Nrx1, Nrx2, and Nrx3) that produce α- and β-neurexins from independent promoters (Rowen et al., 2002; Tabuchi and Südhof, 2002). Neurexins bind to multiple postsynaptic ligands, including neuroligins (Ichtchenko et al., 1995,1996), dystroglycan (Sugita et al., 2001), leucine-rich repeat transmembranes (LRRTMs; Ko et al., 2009a; de Wit et al., 2009; Siddiqui et al., 2010), and cerebellins that in turn bind to postsynaptic GluRδ2 receptors (Uemura et al., 2010). Expression of these ligands in a nonneuronal cell induces synapse formation by cocultured neurons (Scheiffele et al., 2000; Chubykin et al., 2005; Ko et al., 2009a,b; Linhoff et al., 2009; Uemura et al., 2010), which we refer to as heterologous synapse formation. It is likely that heterologous synapse formation involves engagement of presynaptic neurexins, but the only direct currently available evidence for this hypothesis is the observation that a neuroligin-1 (NL1) mutation that blocks neurexin binding also blocks heterologous synapse formation by NL1 (Ko et al., 2009b). Neurexins are thought to act in synapse formation as transducers of an extracellular signal (Shipman and Nicoll, 2012; Owald et al., 2012), but no direct evidence for this hypothesis is available.
Neurexins intracellularly interact with the PDZ-domain protein CASK (Hata et al., 1996), an atypical kinase that phosphorylates neurexins in vitro (Mukherjee et al., 2008). CASK also binds to protein 4.1 (Cohen et al., 1998; Hsueh et al., 1998) and forms a trimeric CASK-protein 4.1-neurexin complex that nucleates actin (Biederer and Südhof, 2001). Other PDZ-domain proteins likely also bind to neurexins, such as Syd-1, whose binding to neurexins regulates synapse formation in Drosophila (Owald et al., 2012), consistent with the notion that neurexins transduce extracellular signals via interactions with intracellular PDZ-domain proteins. However, the precise function of neurexins and their mechanism of action remain unknown.
Here, we have developed an shRNA-mediated knockdown (KD) approach of the mRNAs of all neurexins [neurexin triple knockdown (Nrx TKD)] in cultured neurons to further characterize the fundamental properties of neurexins. Nrx TKD reduced the levels of CASK, which binds to the cytoplasmic sequence of neurexins, but did not decrease the density of synapses formed between the cultured neurons. However, Nrx TKD blocked formation of heterologous synapses between presynaptic neurons and postsynaptic nonneuronal cells expressing NL1 or LRRTM2, but not postsynaptic nonneuronal cells expressing SlitTrk6. Rescue experiments revealed that all neurexin forms tested restored heterologous synapse formation in Nrx TKD neurons, and that mutations in the cytoplasmic tail of a neurexin did not impair heterologous synapse formation, as long as the mutation did not impede surface transport of the neurexin. Surprisingly, even surface-exposed lipid-anchored neurexins were capable of supporting synapse formation, suggesting that the cytoplasmic sequences of neurexins are dispensable for heterologous synapse formation.
Materials and Methods
Generation of KD and expression lentiviruses.
Nrx TKD was effected with an L315 lentiviral vector that can express four shRNAs (Pang et al., 2010a,b; see Fig. 1A). Briefly, previously published Nrx1, Nrx2, and Nrx3 shRNAs were cloned downstream of two human H1 and U6 RNA-polymerase III promoters (shRNA sequences, Nrx1, GTGCCTTCCTCTATGACAACT; Nrx2, GAACAAAGACAAAGAGTAT; Nrx3, ATGCTACACTTCAGGTGGACA; Zhang et al., 2010). Because a single Nrx2 shRNA downstream of the first U6 promoter did not achieve sufficient KD efficiency, we increased the Nrx2 KD efficiency by cloning the same shRNA also downstream of the second U6 promoter (see Fig. 1A). Empty L315 lentivirus was used as a control. Lentiviral Nrx1β, which is amplified from mouse cDNA library, and Nrx3β (NM_138970.4) rescue constructs were mutated to be shRNA resistant without changing the protein sequence. A double human influenza hemagglutinin (2×HA) epitope tag was inserted into the extracellular sequence of vectors encoding Nrx3beta contain an insert in splice site #4 (Nrx3βSS4+), Nrx3beta lacking an insert in splice site #4 (Nrx3βSS4−), and Nrx1beta containing in insert in splice site #4 (Nrx1βSS4+), but not into Nrx1beta lacking an insert in splice site #4 (Nrx1βSS4−; see Fig. 2C). The cytoplasmic alanine substitutions and the PDZ-domain binding sequence deletions in Nrx3β were generated by PCR using the shRNA-resistant Nrx3βSS4− 2×HA-tagged construct as a template. To generate the Nrx3β-NL1 tail construct, we separately amplified the NL1 transmembrane and cytoplasmic sequences (698-843 aa; NCBI reference sequence, NP_446320.1) and the extracellular regions of Nrx3β-SS4 2×HA with overlapping forward and reverse primers, and joined the two fragments. To generate Nrx3β-PDZ/NL1, Nrx3βSS4− 2×HA was amplified by PCR with reverse primers in which the last 10 Nrx3 residues were exchanged for those of NL1. Similarly, Nrx3β-Δ55 + 10 was generated by PCR with reverse primers containing the Nrx3β transmembrane region and the last 10 Nrx3 amino acids followed by a stop codon. Neurexin rescue constructs were cloned into the standard lentiviral vector downstream of a synapsin promote (nucleotides 1527–1998 of the human synapsin gene promoter; Südhof, 1990). In preliminary tests, we identified the NCAM120 glycosylphosphatidylinositol (GPI) anchor sequence as most effective for surface transport of Nrx3 extracellular sequences. We generated the NCAM120 GPI attachment constructs by cloning synthetic DNA fragments (Integrated DNA Technologies) into the standard lentiviral backbone vector downstream of the synapsin promoter, such that the resulting “GPI-exhibit construct” included the NCAM120 signal peptide (residues 1–19), followed by multiple cloning sites (MCSs), a 2×HA tag, and the NCAM120 GPI-anchor attachment sequence (residues 685–725*; NCBI reference sequence, NP_035005.2). The nucleotide sequence of the construct from the ATG to the stop codon is as follows (NCAM120 sequences are underlined, cloning sites are in italics, and the HA epitope sequence is in bold): ATGCTGCGAACTAAGGATCTCATCTGGACTTTGTTTTTCCTGGGAACTGCAGTTTCCACCGGTTTCGAATCTAGATGTACAGCTAGCTATCCATATGATGTTCCAGATTATGCTTATCCATATGATGTTCCAGATTATGCTGCAGCTCACTTTGTGTTCAGGACCTCAGCCCAGCCCACGGCCATCCCAGCCACCCTGGGCGGAAGCTCCACCTCCTACACCTTGGTCTCATTGCTTTTCTCTGCGGTGACTCTTCTTCTGCTCTGA. shRNA-resistant Nrx3βSS4− (residues 35–340) and Nrx1βSS4− (residues 49–344) were PCR amplified and cloned in frame into the MCSs of the GPI-exhibit construct to generate Nrx3β-GPI and Nrx1β-GPI (see Fig. 5A). To create stalk modifications, the stalk region of the Nrx3β-GPI was cut out with hpa1-nhe1 restriction enzymes, and then synthetic DNA fragments for each three stalk constructs were cloned by Gibson assembly (New England Biolabs) into the resulting vector. For actin imaging, N-terminally GFP-tagged β-actin was cloned into the standard lentiviral backbone vector downstream of the ubiquitin promoter. Vectors used in COS cell transfections (pCMV5-mVenus, pCMV5-NL1-mVenus, pCMV5-LRRTM2-mVenus, pDis-Slitrk6) were described previously (Ko et al., 2009b). The endoplasmic reticulum (ER) marker pEYFP-ER was obtained from Clontech. Lentiviral particles were produced by calcium phosphate transfection of HEK293T cells (ATCC) as described previously (Pang et al., 2010a,b).
Quantification of mRNA levels by quantitative RT-PCR.
Neurexin KD efficiencies and effect of RNAi on selected mRNAs were determined using quantitative RT-PCR. Gene expression profiling was performed using the Fluidigm Biomark dynamic array according to the manufacturer's protocol. Briefly, RNA was collected from cultures infected with control or Nrx TKD or Nrx TKD with rescue Nrx3β at DIV14 using the RNAqueous kit (Ambion). One nanogram of the isolated RNAs were subjected to target- specific reverse transcription and 18 cycles of PCR preamplification with a mix of primers specific to the target genes. Preamplified cDNAs were then processed for real-time PCR analysis on BioMark 96.96 Dynamic Array integrated fluidic circuits (Fluidigm). To quantify total Nrx mRNA levels, probes were selected to detect all target protein variants. All assays for quantitative RT-PCR analyses were purchased from IDT: Nrx1 (forward, ACTACATCAG TAACTCAGCACAG; reverse, ACAAGTGTCCGTTTCAAATCTTG; probe, CTTCTCCTTGA CCACAGCCCCAT), Nrx2 (forward, ATCATCACTTGGACACTCAGC; reverse, ACAATGAG GGACAGCACC; probe, CAGGAGGTCATCTGTGTTCTGGGTG), Nrx3 (forward, CCTTTG TCCTTTCCTCCGATG; reverse, CACTGATAATGAACGCCTCCA; probe, CCTTTTTCCT GCAGCCACTCCTCT), NL1 (Mm.PT.45.9950210), NL2 (Mm.PT.45.10816796), NL2 (Mm.PT.45.10816796), NL3 (Mm.PT.51.11910785), SlitTrks1 (Mm.PT.45.17156849.g), SlitTrks2 (Mm.PT.45.8986803), SlitTrks3 (Mm.PT.45.17200598), SlitTrks4 (Mm.PT.45.13878337), SlitTrks5 (Mm.PT.45.12246115), LRRTM1 (Mm.PT.45.14015856), LRRTM2 (Mm.PT.45.12386236), LRRTM3 (Mm.PT.45.10830812), LRRTM4 (Mm.PT.45.11146838), CL1/Lphn1 (Mm.PT.47.6572453), CL2/Lphn2 (Mm.PT.47.12868555), Rbfox3 (Mm.PT.45.6200011), Neuroflilament (Mm.PT.45.8814045), Syt 1 (Mm.PT.45.7011015), Glt-1 (Slc1a2) (Mm.PT.45.12612086), Gpr6 (Mm.PT.45.17141248.g), DARPP-32 (Mm.PT.51.15997313), Eya1 (Mm.PT.45.6448569), and Actin-B (Mm.PT.51.14022423). To ensure the specificity of the amplification, all assays were tested with dilutions of mouse hippocampal cDNA dilutions. We analyzed only the RT-PCR assays that showed high efficiency (90–110%) and linear amplification (r2 > 98). Expression values were calculated relative to Actin-B, then each gene was normalized to its average expression in the control cultures. Heat map was generated via HeatMapViewer (www.broadinstitute.org).
Immunoblotting analyses.
For immunoblotting of proteins from cultured neurons, neurons were washed three times with PBS, lysed with Laemmli sample buffer containing DTT, and boiled for 5 min. Lysates were subjected to SDS-PAGE and immunoblotting using fluorescently labeled secondary antibodies (donkey anti-rabbit IRDye 800CW, 1:15,000; donkey anti-mouse IRDye 680, 1:15,000; LI-COR Bioscience) and signal detection with an Odyssey Infrared Imager and Odyssey software (LI-COR Biosciences), Signals were normalized for valosin-containing protein (VCP) and β-actin (for primary mouse monoclonal and rabbit polyclonal antibodies, respectively) probed on the same blots as loading controls. Antibodies used were as follows: monoclonal mouse antibodies to β-actin (Sigma; 1:2000), neuroligins (4C12; 1:1000), synaptobrevin-2 (Cl69-1; 1:2000), synaptotagmin-1 (Synaptic Systems; 1:2000), and CASK (Millipore; 1;1000), and rabbit polyclonal antibodies to snap25 (p215; 1:1000), Mint1-2 (U1693; 1:1000), Veli (T813; 1:1000), liprin-α3 (4396; 1:5000), VCP (K330; 1:1000), and neurexins (A473; 1:500).
Neuronal cultures, heterologous synapse formation assays, immunocytochemistry, and image acquisition and analyses.
Hippocampal neurons were cultured from newborn mice of either sex and neuronal transfections done by calcium phosphate transfection as described previously (Pang et al., 2010a,b; Ko et al., 2009b), and heterologous synapse formation assays were performed with COS-7 cells (Chubykin et al., 2005; Ko et al., 2009b). For quantifying the amount of heterologous synapse formation and of the surface transport of HA-tagged neurexins, COS cells were transfected using FuGene-6 (Roche) with plasmids expressing mVenus, the NL1- or LRRTM2-mVenus fusion proteins, Flag-NL1, or pDisplay-SlitTrk6, trypsinized after 24 h and plated onto DIV14–DIV16 hippocampal neurons. After 24 h coculture, cocultured neurons and COS cells were incubated for 10 min in PBS containing 0.5 mm CaCl2 and 1 mm MgCl2 without fixation with anti-HA monoclonal antibody (Covance; 1:250), washed, fixed with 100% methanol at −20°C, and labeled with rabbit anti-synapsin antibodies (E028; 1:1000). Samples were analyzed by confocal microscopy. Heterologous synapse formation was measured by quantifying with MetaMorph image analysis software (Molecular Devices) the ratio of synapsin fluorescence intensity over the transfected COS cells to the ratio of the fluorescence of the protein transfected into the COS cells (i.e., NL1-mVenus, LRRTM2-mVenus, mVenus, or myc-tagged pDisplay-SlitTrk6). Surface transport of HA-tagged neurexins was measured by quantifying the ratio of fluorescence signals derived from surface HA tags and intracellular synapsin labeling in regions of cultured neurons away from COS cells. For immunocytochemistry, cultured neurons were fixed for 10 min in 100% methanol at −20°C or 4% PFA, 4% sucrose in PBS at room temperature; blocked with 0.1% Triton X-100, 3% BSA, and 5% goat serum in PBS for 30 min; incubated with primary and secondary antibodies in blocking buffer for 60 min; and stained with Alexa-405-, Alexa-488-, Alexa-546-, or Alexa-633-labeled goat anti-mouse, goat anti-rabbit, or goat anti-guinea pig secondary antibodies (Invitrogen). Primary antibodies used were rabbit antibodies to synapsin (E028; 1:1000), VGAT (1:500; Millipore), guinea pig antibodies to vGLUT1 (1:1000; Millipore), and monoclonal mouse antibodies to the HA tag (1:250; Converge), MAP2 (1:2000; Sigma), GAD65 (1:500; Developmental Studies Hybridoma Bank), the Myc epitope (9E10 1:500; Developmental Studies Hybridoma Bank), and NeuN (Millipore 1:500). For determining synapse densities per 50 μm dendrite, Z-stacked images obtained from confocal microscope were converted to maximal projection and analyzed using the count nuclei tool of MetaMorph software (Molecular Devices). Images were acquired with a Leica TCS2 confocal microscope using constant image settings. For β-actin-GFP localization experiments, single-plane four channel confocal images were acquired for Actin-GFP, Nrx3β-HA, NL1-flag, and vGLUT1 using a Carl Zeiss LSM510 confocal microscope. We calculated the Manders' colocalization coefficients for each fluorescent signal pair that are localized on COS cell (Manders et al., 1993) using the ImageJ program (US National Institutes of Health) and the plug-in JACOP (Bolte and Cordelières, 2006). This program calculates the sum of colocalized pixels for each fluorescent channel divided by the total number of labeled pixels (maximal colocalization coefficient, 1).
Miscellaneous.
For all experiments, samples and data were “blinded” to the experimenter. Statistical significance was determined by Student's t test, and all data are expressed as means ± SEM.
Results
Characterization of Nrx TKD neurons
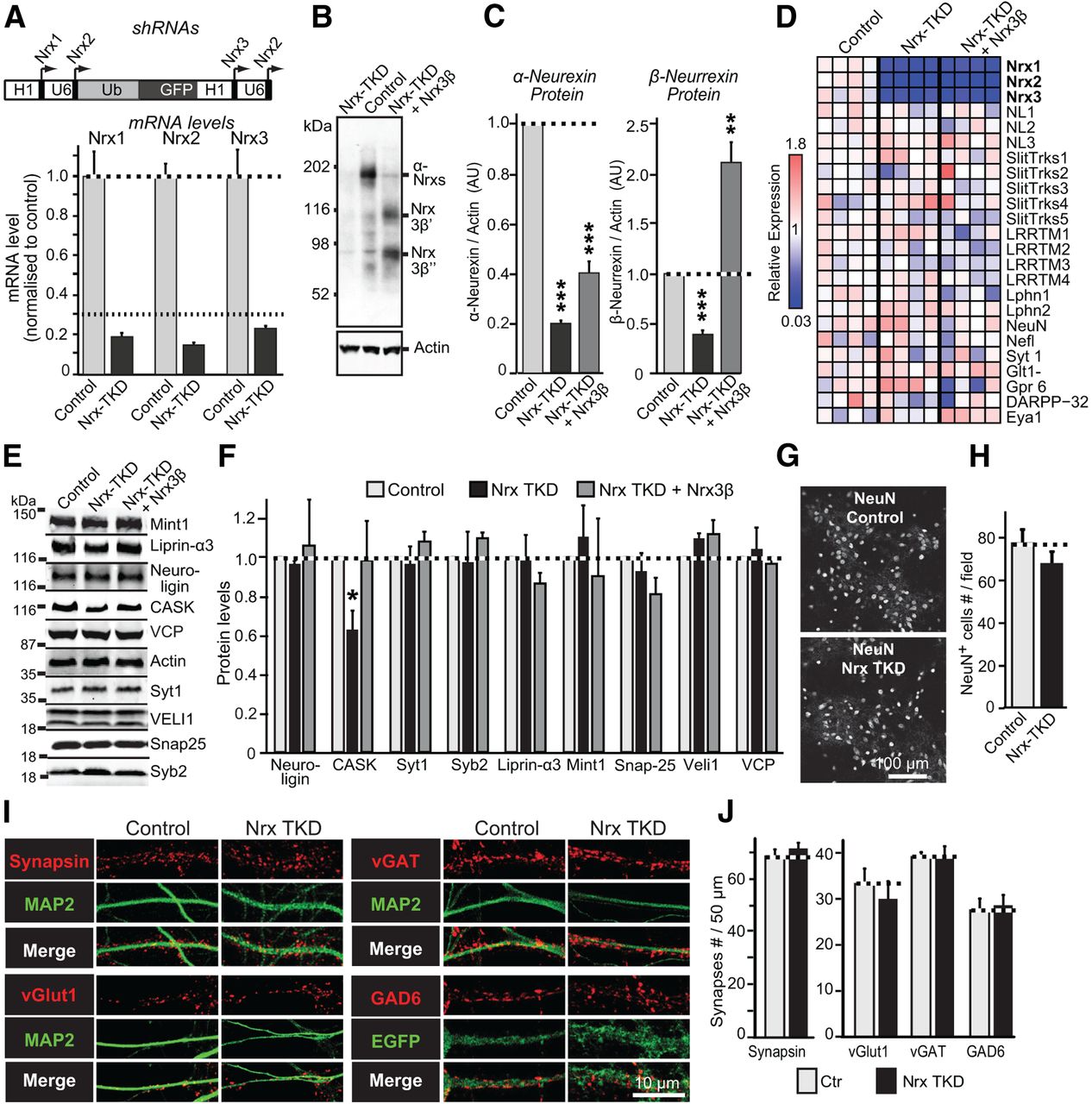
We cultured hippocampal neurons from newborn mice and used a single lentivirus encoding four shRNAs to knock down expression of all endogenous neurexins mRNAs to <25% of control levels, resulting in the neurexin triple knockdown (Fig. 1A). Direct protein measurements by quantitative immunoblotting with a pan-neurexin antibody confirmed that the Nrx TKD decreased the levels of α-neurexins by ∼80%, and of β-neurexins by ∼60% (Fig. 1B,C). Since at the protein level α-neurexins are expressed at much higher levels than β-neurexins (Fig. 1B), the overall decrease in total neurexin protein also corresponded to almost 80%, validating the Nrx TKD approach. When we reexpressed a β-neurexin (Nrx3β) as a rescue protein in Nrx TKD neurons, we found that, compared to control neurons, total β-neurrexin levels (now composed largely of the expressed Nrx3β) were increased ∼200% (Fig. 1B,C).

Generation and characterization of Nrx TKD neurons. A, Design of the lentiviral Nrx TKD vector (top) and quantifications of Nrx1, Nrx2, and Nrx3 mRNAs in Nrx TKD neurons (bottom). H1, Human H1 promoter; U6, human U6 promoter; Ub, ubiquitin promoter. For mRNA quantifications, β-actin was used as endogenous control in quantitative RT-PCR measurements of neurons infected with control or Nrx TKD lentiviruses; levels are normalized to those of β-actin and the control. B, C, Nrx TKD decreases neurexin protein levels; this decrease is reversed by reexpression of Nrx3β. B depicts a representative immunoblot stained with a pan-neurexin antibody (A473), with β-actin immunoblot as a loading control (bottom). C depicts quantifications of α- and β-neurexins using fluorescently labeled secondary antibodies (normalized for β-actin). In control neurons, α-neurexins are more abundant than β-neurexins; Nrx3β rescue protein is present in two forms (Nrx3β′ and Nrx3β″) that may correspond to different glycosylation states. D, Quantification of mRNA levels in control infected hippocampal neurons or hippocampal neurons infected with the Nrx TKD lentivirus without or with expression of Nrx3β. mRNAs were measured by quantitative RT-PCR using Fluidigm dynamic arrays. Hippocampal neurons were infected at DIV3, and mRNA levels were quantified at DIV14. Rows represent the evaluated genes and columns individual cultures of neurons. The heat map (blue to red) represents the relative expression of a gene normalized to its average expression in the control cultures. mRNA levels of endogenous neurexins are shown on the top three rows. The Nrx3 RT-PCR assay is specific to mouse and does not detect the human Nrx3β rescue mRNA, validating the continued effectiveness of the Nrx TKD even after rescue overexpression. NL, Neuroligin; Slitrks, Slit- and Trk-like; CL/Lphn1–2, G-protein-coupled receptor CIRL/latrophilin; NeuN/Rbfox3, neuronal nuclei; Gpr6, G-protein-coupled receptor 6; Glt1, glial high-affinity glutamate transporter; Eya1, eyes absent 1 homolog. E, F, Effect of the Nrx TKD on the levels of selected synaptic proteins. Syt1, Synaptotagmin-1; Syb2, synaptobrevin-2. Protein levels were determined by quantitative immunoblotting with fluorescently labeled secondary antibodies (E, representative immunoblots; F, summary graphs of protein levels determined with fluorescent secondary antibodies). Actin and VCP were used as loading controls. VCP levels relative to actin showed no significant difference between three groups. G, H, Nrx TKD does not impair neuronal viability, as determined by measurements of the density of NeuN-positive neurons (G, representative images; H, summary graph). I, J, Nrx TKD does not decrease synapse density, as determined by synapsin staining (I, representative images; J, summary graph). Data are means ± SEM (A, n = 3; C, n = 5; D, n = 4; F, n = 3–4; H, n = 3; J, n = 4 independent cultures). Statistical analyses were performed by Student's t test comparing test samples to the control. *p < 0.05; **p < 0.01; ***p < 0.001.
We asked whether the Nrx TKD alters the expression of specific genes or proteins that may be functionally related to neurexins. Using quantitative RT-PCR on a Fluidigm Dynamic Array (Pang et al., 2011), we measured the expression of 14 candidate synaptic cell-adhesion molecules as well as selected synaptic proteins in control neurons, Nrx TKD neurons, and Nrx TKD neurons with Nrx3β rescue (Fig. 1D). We observed no significant change in gene expression as a function of the Nrx TKD in any mRNA we measured; in particular, we noted no change in the expression of the neurexin ligands neuroligins, LRRTMs, and latrophilins. We also performed quantitative immunoblotting analyses of eight selected synaptic proteins to test whether their levels were changed. We did not detect changes in seven of these synaptic proteins (neuroligin, synaptotagmin-1, synaptobrevin-2, SNAP-25, Mint1, Velis, and liprin-α3), but observed a significant decrease in CASK in Nrx TKD neurons (Fig. 1E,F). The decrease in CASK levels in Nrx TKD neurons was reversed by reexpression of Nrx3β, suggesting that it is specific. These data indicate that neurexins stabilize CASK in neurons, and validate the physiological importance of CASK-binding to neurexins (Hata et al., 1996).
In a further set of experiments to characterize the Nrx TKD neurons, we examined the effect of the Nrx TKD on neuronal viability (Fig. 1G,H) and on the density of synapses formed between neurons in culture (I,J). We found that the Nrx TKD had no significant effect on neuronal survival as assessed by the number of viable neurons present in control versus Nrx TKD conditions and did not alter synapse density. These results are consistent with previous data from α-neurexin triple KO mice, which also failed to uncover a significant change in excitatory synapse density in neurons that lacked α-neurexins but still expressed β-neurexins (Missler et al. 2003).
Effect of the Nrx TKD on heterologous synapse formation
We next tested the effect of the Nrx TKD on heterologous synapse formation by neurons, using COS-7 cells expressing NL1 as a postsynaptic target. We found that KD of individual neurexins in neurons did not impair heterologous synapse formation, whereas Nrx TKD severely decreased synapse formation (Fig. 2A,B). Thus, individual neurexins act redundantly during heterologous synapse formation. The decrease in heterologous synapse formation by Nrx TKD was rescued by Nrx1β and Nrx3β, suggesting that the decrease was not due to an off-target KD effect. Both variants of splice site 4 (SS4) of both Nrx1β and Nrx3β were able to rescue heterologous synapse formation in Nrx TKD neurons (Fig. 2C,D). An HA epitope that we had introduced into most neurexin rescue constructs enabled us measure the surface transport of these neurexins, and to examine their localization in heterologous synapses. All wild-type neurexins tested were efficiently transported to the surface and highly enriched in presynaptic terminals over the NL1-expressing COS cells (Fig. 2C,D), consistent with their rescue activity.

Effect of Nrx TKD on heterologous synapse formation. A, B, Nrx TKD, but not single neurexin KDs, impair heterologous synapse formation. COS-7 cells expressing a NL1-mVenus fusion protein were cocultured with hippocampal neurons infected with lentiviruses mediating KDs of individual neurexins or the Nrx TKD; the latter was analyzed without or with expression of Nrx3β rescue protein. After cell fixation and permeabilization, transfected COS cells were visualized by mVenus contained in NL1, while synapses were stained for synapsin (A, representative images; B, summary graphs of synapse formation quantified as the ratio of presynaptic synapsin to postsynaptic mVenus). C, D, Specificity of the Nrx TKD effect on heterologous synapse formation. COS cells expressing mVenus, NL1-mVenus fusion protein, or myc-tagged SlitTrk6 were cocultured with neurons that were infected with control lentiviruses, or lentiviruses expressing the Nrx TKD, without or with coexpression of various Nrx1 and Nrx3 splice variants as indicated. For analyses, coverslips containing live neurons were first incubated with HA antibody (to label surface-exposed Nrx1β and Nrx3β containing HA epitopes) and then fixed, permeabilized, and labeled for synapsin. Heterologous synapse formation was quantified as the ratio of presynaptic synapsin in neurons to postsynaptic mVenus in COS cells, and surface transport of neurexins was assessed as the ratio of HA to synapsin in regions devoid of COS cells. Note that Nrx1βSS4− was not HA-tagged, and is thus not surface stainable (C, representative images; D, summary graphs). Data in B and D are means ± SEM (B, n = 4; D, n = 4 independent cultures). Statistical analyses were performed by Student's t test comparing test samples to the positive control. ***p < 0.001.
To examine whether the Nrx TKD impairs heterologous synapse formation induced by proteins that do not bind to neurexins, we examined heterologous synapse formation induced by SlitTrk6, which acts by binding to presynaptic PTPδ (Takahashi et al., 2012). We found that heterologous synapse formation induced by SlitTrk6 was unchanged by the Nrx TKD, indicating that SlitTrk6 acts independently of neurexins, and validating the specificity of the Nrx TKD effect on NL1-induced heterologous synapse formation (Fig. 2C,D).
Role of the neurexin cytoplasmic sequences in heterologous synapse formation
The Nrx TKD-mediated impairment in heterologous synapse formation and its rescue by reexpression of Nrx3β provided us with an assay to dissect the mechanisms of neurexin-mediated synapse formation. Neurexins contain a highly conserved cytoplasmic sequence that binds to FERM (F for 4.1 protein, E for ezrin, R for radixin and M for moesin) domain proteins such as protein 4.1 (Biederer and Südhof, 2001), and to PDZ-domain proteins such as CASK (Hata et al., 1996). Thus, a plausible hypothesis is that the neurexin cytoplasmic tail may transduce an extracellular binding signal into an intracellular synapse-assembly signal. Consistent with this hypothesis, binding of the PDZ domain containing RhoGAP protein Syd-1 to the cytoplasmic tail of neurexins is essential for synapse formation in Drosophila (Owald et al., 2012).
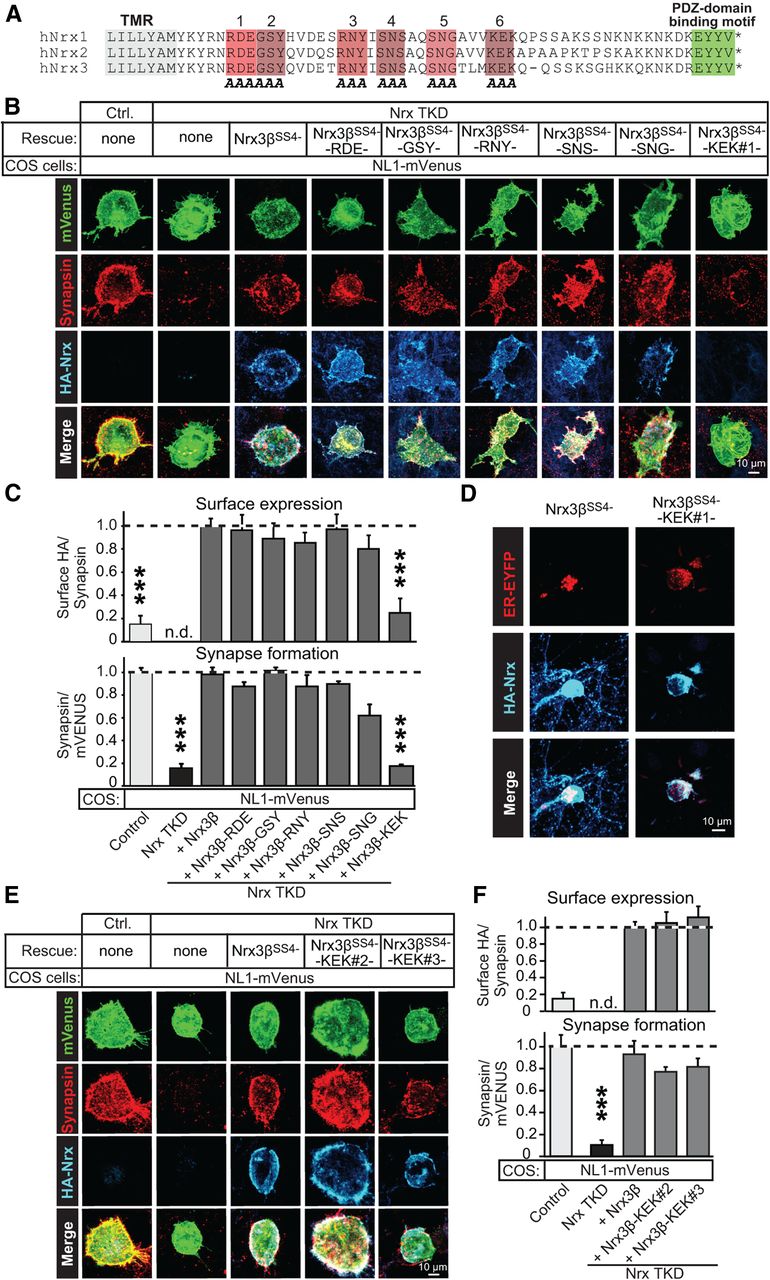
We selected six conserved three-residue sequence blocks in the cytoplasmic tail of Nrx3β for alanine substitution mutagenesis (Fig. 3A). Five of the six alanine substitution mutants were efficiently transported to the neuronal cell-surface and fully supported heterologous synapse formation in Nrx TKD neurons (Fig. 3B,C). The sixth mutant (which targeted the “KEK” sequence in the cytoplasmic neurexin tail) impaired surface transport of Nrx3β and blocked rescue of heterologous synapse formation in Nrx TKD neurons.

Analysis of point mutations in the Nrx3β cytoplasmic tail. A, Alignment of the human Nrx1, Nrx2, and Nrx3 intracellular sequences shows a high level of conservation. The six blocks of conserved three-residue sequences targeted for alanine (A) substitution mutagenesis are highlighted in different shades of red and numbered on top. The C-terminal PDZ-domain binding sequences are highlighted in green. TMR, Transmembrane region. B, C, Analysis of six alanine substitution mutants in the C-terminal cytoplasmic sequence of Nrx3β (A). COS cells expressing NL1-mVenus were cocultured with control or Nrx TKD hippocampal neurons; the latter were also infected with control lentiviruses or lentiviruses expressing wild-type or mutant HA-tagged Nrx3β (B, representative images of heterologous synapses; C, summary graphs of synapse formation and surface expression of various Nrx3β proteins). For analysis approaches, see legend to Figures 2, C and D. D, Fluorescence images of hippocampal neurons cotransfected with the endoplasmic reticulum marker pEYFP-ER and with wild-type Nrx3β or mutant Nrx3β-KEK containing alanine substitutions in the KEK sequence (A). E, F, Same as B and C, except that two additional mutations of the cytoplasmic KEK sequence of Nrx3β were analyzed: a mutation changing KEK to CQC (Nrx3β-KEK2), and a deletion of the entire TLMKEK sequence (Nrx3β-KEK3). Data shown in C and F are means ± SEM (n = 3 independent cultures). Statistical analyses were performed by Student's t test comparing test samples to the positive control. ***p < 0.001. n.d., Not determined.
The lack of surface transport of the KEK mutant Nrx3β suggests that this mutant Nrx3β may become trapped in the ER. To test this hypothesis, we investigated the localization of Nrx3β and Nrx3β-KEK in transfected neurons. Whereas Nrx3β was localized to both the cell body and neuronal dendrites, Nrx3β-KEK was completely retained in the cell body of transfected neurons. In the cell body, mutant Nrx3β-KEK but not wild-type Nrx3β was confined to a perinuclear structure that also accumulated the ER marker pEYFP-ER (Fig. 3D). These results suggest that the hydrophobic alanine residues substituting the KEK sequence may have impeded export of mutant Nrx3β-KEK protein out of the ER, and that incomplete trafficking of Nrx3β-KEK explains its functional impairment.
To further test this hypothesis, we generated other mutations in the KEK sequence of Nrx3β and analyzed them functionally. Indeed, we found that mutating the KEK sequence to cysteine-glutamine-cysteine (Nrx3β-KEK 2), or deleting the entire KEK sequence and the three preceding residues (TLMKEK; Nrx3β-KEK 3), did not impair surface transport of Nrx3β, demonstrating that the KEK sequence as such is not essential for surface transport. Moreover, the new KEK sequence mutants were fully active in heterologous synapse formation (Fig. 3E,F). Thus, the KEK sequence as such is also not required for heterologous synapse formation. Viewed together, these results suggest that most of the cytoplasmic tail of neurexins is not essential for the ability of Nrx3β to restore heterologous synapse formation in Nrx TKD neurons.
We next generated Nrx3β mutants with substitutions in the C-terminal PDZ-domain binding sequence of Nrx3β. When we exchanged the last three amino acids of the PDZ-domain binding sequence in Nrx3β for alanines or simply deleted these three residues, the resulting mutant Nrx3β proteins again were not efficiently transported to the neuronal cell surface (Fig. 4). This observation is consistent with previous results suggesting that the neurexin PDZ-domain binding sequence is important for cell-surface transport (Fairless et al., 2008). However, when we exchanged the last 10 residues of Nrx3β with those of NL1 (which contains a PDZ-domain binding sequence with a different specificity than neurexins, and does not bind to CASK; Irie et al., 1997), the resulting hybrid protein (Nrx3β-PDZ/NL1) was efficiently transported to the neuronal cell surface and was fully able to rescue heterologous synapse formation in Nrx TKD neurons (Fig. 4). Since the NL1 PDZ-domain binding sequence does not interact with CASK or related class II PDZ-domain containing proteins, this result suggests that the inability of Nrx3β with a deletion or with alanine substitutions of the C-terminal PDZ-domain binding sequence to rescue the loss of heterologous synapse formation in Nrx TKD neurons is caused by the effect of these mutations on the surface transport of Nrx3β, and not by a loss of CASK binding as such. Moreover, this result demonstrates that the surface transport of Nrx3β does not require a specific PDZ-domain binding sequence, but can be promoted by any such sequence, thereby extending the conclusions of Fairless et al. (2008).

The specific PDZ-domain binding sequence of Nrx3β is not essential for heterologous synapse formation. A, B, COS cells expressing NL1-mVenus were cocultured with control (Ctrl.) or Nrx TKD hippocampal neurons; the latter were additionally infected with control lentiviruses or lentiviruses expressing wild-type or mutant Nrx3β with either (1) an alanine substitution of the C-terminal three residues (Nrx3β-PDZ/A), (2) a deletion of the C-terminal three residues (Nrx3β-PDZΔ3), or (3) an exchange of the 10 C-terminal residues of Nrx3β for those of NL1 (Nrx3β-PDZ/NL1), which includes a different type of PDZ-domain binding sequence [A, representative images; B, quantifications of synapse formation mediated by the various Nrx3β proteins (top) and their surface expression (bottom)]. Data shown in B are means ± SEM (n = 3 independent cultures). Statistical analyses were performed by Student's t test comparing test samples to the positive control. ***p < 0.001. n.d., Not determined.
Lipid-anchored extracellular neurexin domains rescue heterologous synapse formation in Nrx TKD neurons
Our data raise the question of whether the cytoplasmic tail of neurexins is at all essential for heterologous synapse formation. Initial attempts to test this question included deletion of the entire neurexin cytoplasmic tail and, alternatively, using the pDisplay (Invitrogen) system to deliver Nrx1β and Nrx3β extracellular domains on neuronal membranes. However, both approaches failed due to poor surface transport of the resulting proteins. Thus, we pursued three alternative strategies (Fig. 5A). We (1) deleted most of the cytoplasmic tail of Nrx3β except for the last ten amino acids to retain the PDZ-domain binding sequence (Nrx3β-Δ55 + 10; Fairless et al., 2008), (2) fused the extracellular Nrx3β sequences to the transmembrane region and cytoplasmic tail of NL1 (Nrx3β-NL1tail), a normally postsynaptic protein, and (3) attached a GPI anchor sequence to the Nrx1β or the Nrx3β extracellular sequences, thus producing Nrx1β and Nrx3β proteins that are bound to the cell surface by a lipid anchor (Nrx1β-GPI, Nrx3β-GPI).

Heterologous synapse formation does not require cytoplasmic neurexin signaling. A, Domain structures of wild-type and mutant Nrx3β that either lacks the entire cytoplasmic tail except for the last 10 residues (Nrx3β-Δ55 + 10), contains the complete NL1 cytoplasmic tail instead of the Nrx3 cytoplasmic tail (Nrx3β-NL1tail), or is anchored on the membrane surface by a GPI moiety (Nrx3β-GPI). B, C, Nrx3β cytoplasmic tail mutants fully sustain heterologous synapse formation (B, representative images; C, synapse formation quantifications). COS cells expressing NL1-mVenus fusion protein were cocultured with control neurons or with Nrx TKD neurons that express the indicated rescue proteins and analyzed as described for Figure 2C. D, E, Heterologous synapse formation induced by LRRTM2 is blocked by the Nrx TKD, but rescued by GPI-anchored Nrx3β. Experiments were performed as in B and C, except that the COS cells expressed LRRTM2 instead of NL1. Data shown in C and E are means ± SEM (C, n = 4; E, n = 3 independent cultures). Statistical analyses were performed by Student's t test comparing test samples to the positive control. **p < 0.01; ***p < 0.001.
All four β-neurexin proteins lacking the neurexin cytoplasmic sequence were efficiently transported to the neuronal cell surface, and fully rescued heterologous synapse formation in Nrx TKD neurons (Fig. 5B,C). Moreover, heterologous synapse formation triggered by postsynaptic LRRTM2, which is a high-affinity neurexin ligand similar to neuroligins (Ko et al., 2009b; de Wit et al., 2009; Siddiqui et al., 2010), was also blocked by the Nrx TKD, but could again be rescued by GPI-anchored Nrx3β (Fig. 5D,F), demonstrating the general applicability of the mechanism we describe here. Thus, the surface-exposed extracellular β-neurexin domains are sufficient to restore heterologous synapse formation in Nrx TKD neurons.
Extracellular neurexin sequences required for heterologous synapse formation
The extracellular sequences of β-neurexins comprise a short β-specific sequence, a single LNS domain that corresponds to the sixth LNS domain of α-neurexins, and a “stalk” sequence that is threonine rich and O-glycosylated (Ushkaryov et al., 1994). In addition, we identified a previously overlooked acidic cysteine-loop sequence that is conserved in all neurexins (Fig. 6A). Since the GPI-anchored extracellular sequence of Nrx1β and Nrx3β suffices to restore heterologous synapse formation in Nrx TKD neurons (Fig. 5C), we asked whether the glycosylated stalk and/or the cysteine-loop sequences contribute to this activity. We deleted either the stalk or the cysteine-loop sequence of Nrx3β in the context of the GPI-anchored protein, or substituted glycine for most of the glycosylated serine and threonine residues in the stalk sequence (Fig. 6A).

The Nrx3β LNS domain mediates heterologous synapse formation. A, Domain structure of Nrx3β (top) and sequence alignment of human Nrx1, Nrx2, and Nrx3 and of the Nrx3 stalk and cys-loop mutants in the region between the LNS domain and the transmembrane region (bottom; mutated residues or deletions are underlined). B, C, Analysis of the effect of mutations in the O-linked sugar stalk region and the cys-loop of Nrx3β on heterologous synapse formation. NL1-mVenus expressing COS cells were cocultured with hippocampal neurons infected with control or the Nrx TKD lentiviruses without or with rescue with wild-type or mutant Nrx3β [B, representative images; C, summary graphs of synapse formation (top) and Nrx3β surface expression (bottom); for analysis methods, see Figure 3]. Data are means ± SEM (n = 3 independent cultures). Statistical analyses were performed by Student's t test comparing test samples to the positive control. *p < 0.05; **p < 0.01; ***p < 0.001.
The lack of a glycosylated stalk significantly diminished, but did not block, the activity of Nrx3β in heterologous synapse formation (Fig. 6B,C). In contrast, the cysteine loop deletion had no effect. It is possible that the O-glycosylated stalk of Nrx3β enhances heterologous synapse formation by enabling a better presentation of the Nrx3β LNS domain on the neuronal surface, while the cysteine loop is probably not involved in heterologous synapse formation.
Neurexins and actin do not colocalize in presynaptic specializations of heterologous synapses
In a final set of experiments, we probed the localization of neurexins in heterologous synapses induced by NL1 and compared it to that of presynaptic actin and synaptic vesicles and postsynaptic NL1. The neurexin cytoplasmic sequence forms a trimeric complex with CASK and protein 4.1 that nucleates actin assembly in vitro (Biederer and Südhof, 2001). To test whether actin is recruited to the cytoplasmic sequence of neurexins in a living neuron, a process that might contribute to synapse formation and be compensated for in neurons expressing Nrx3β lacking a normal cytoplasmic tail, we imaged actin in presynaptic terminals (Fig. 7A). We rescued the loss of heterologous synapse formation in Nrx TKD neurons with wild-type Nrx3β and coexpressed actin-GFP in the same neurons. We observed excellent colocalization of presynaptic Nrx3β with postsynaptic NL1 in heterologous synapses as described above, and additionally detected a similar colocalization of Nrx3β with the synaptic vesicle marker vGluT1. However, we found that actin was relatively excluded from the location of Nrx3β and vGluT1, suggesting that Nrx3β does not recruit actin to the synapse, a finding that is consistent with the relative lack of actin from presynaptic specializations (Fernández-Busnadiego et al., 2010).

Actin localization during heterologous synapse formation. A, Comparative localization of β-actin-GFP coexpressed with Nrx3β (top) or with Nrx3β-NL1tail (bottom) in Nrx TKD neurons that were cocultured with COS cells expressing Flag-tagged NL1. Panels show representative images of a heterologous synapse stained for actin-GFP, vGluT1, NL1, and Nrx3β (via its HA tag) at two magnifications. B, Summary graph of the Manders' coefficients as the degree of colocalization for each fluorescent signal pairs. Data shown are means ± SEM (n = 4 independent cultures, A, B). Statistical analyses were performed by Student's t test comparing test samples to Manders' coefficients of NL1 + Nrx3β interaction. *p < 0.05; **p < 0.01.
We also analyzed the relative localization of actin in presynaptic terminals formed by heterologous synapses of Nrx TKD neurons expressing the Nrx3β-NL1 fusion protein containing the cytoplasmic tail of NL1. Strikingly, now we observed strong presynaptic colocalization of actin with both Nrx3β-NL1 and vGluT1 (Fig. 7A). This result validates the absence of actin recruitment by wild-type Nrx3β described above by demonstrating that under other conditions, actin can in principle be recruited into presynaptic terminals. Quantifications in multiple independent experiments confirmed that actin is selectively excluded from synaptic junctions in heterologous synapses formed by wild-type Nrx3β, but is recruited to such synapses when the Nrx3β-NL1tail hybrid protein was present (Fig. 7B). Together, these results suggest that neurexins do not physiologically recruit actin to membranes, but that the cytoplasmic tail of NL1 does and may contribute to the high enrichment of actin in postsynaptic spines.
Discussion
Here, we use a triple KD targeting all neurexin mRNAs (the Nrx TKD) in cultured hippocampal neurons, and employ heterologous synapse formation of these TKD neurons on to nonneuronal cells expressing postsynaptic cell-adhesion molecules as a tool to investigate the role and mechanism of action of neurexins in synaptogenesis. Our experiments resulted in six principal observations that have significant implications for the properties of neurexins, as follows.
The Nrx TKD significantly decreased the levels of CASK protein in hippocampal neurons, providing direct evidence that the in vitro interaction of neurexins with CASK (Hata et al., 1996) is physiologically significant (Fig. 1). At the same time, the Nrx TKD did did not impair formation of synapses between cultured neurons, either because neurexins are not essential for synapse formation, or because the Nrx TKD, as all shRNA-mediated KDs, was only partial.
The Nrx TKD blocked the ability of hippocampal neurons to form heterologous synapses on nonneuronal COS cells expressing either NL1 or LRRTM2, but had no effect on heterologous synapse formation on COS cells expressing SlitTrk6 (Figs. 2, 5D). This observation demonstrates that neurexins are not essential for heterologous synapse formation as such (which agrees well with the conclusion that they are not generally essential for synapse formation), but that, consistent with NL1 mutagenesis experiments (Ko et al., 2009a), they are required for heterologous synapse formation induced by NL1 or by LRRTM2, suggesting that such synapse formation involves binding of these neurexin ligands to neurexins.
The loss of heterologous synapse formation in Nrx TKD neurons was rescued by all tested mutant neurexins that contain changes in the cytoplasmic sequences, as long as the mutant neurexins reached the cell surface (Figs. 3⇑⇑⇑–7). Moreover, the extracellular domains of Nrx1β or of Nrx3β, when expressed on the neuronal cell surface via a lipid GPI anchor, were sufficient for rescue of heterologous synapse formation in hippocampal Nrx TKD neurons (Fig. 5). These results not only confirm that it is an extracellular interaction of neurexins that mediates heterologous synapse formation, but also suggest that neurexins act by cis interactions on the neuronal cell surface, and not by a direct intracellular signal transduction event (see the discussion below).
Of the extracellular Nrx3β sequences, only the LNS domain and the O-linked sugar region (the stalk) were required for full rescue of heterologous synapse formation in Nrx TKD neurons (Fig. 6). This result suggests that a properly surface-displayed neurexin LNS domain is sufficient for induction of heterologous synapse formation.
Extending previous studies (Fairless et al., 2008), we found that mutating the C-terminal PDZ-domain binding sequence of Nrx3β blocks Nrx3β transport to the neuronal cell surface. Importantly, simply substituting the NL1 PDZ-domain binding sequence for the Nrx3β PDZ-domain binding sequence restored surface transport (Fig. 4), despite the completely different PDZ-domain binding specificities of these sequences (Hata et al., 1996; Irie et al., 1997). These data demonstrate that it is not the binding of a specific PDZ-domain protein such as CASK that promotes surface transport and targeting of neurexins, and that the C-terminal PDZ-domain binding sequence mutations used indirectly impair neurexin targeting and neurexin-mediated heterologous synapse formation by inhibiting surface transport.
Actin is not enriched presynaptically in the heterologous synaptic junctions formed by binding of presynaptic neurexins to postsynaptic NL1, suggesting that neurexins do not recruit actin to these junctions (Fig. 7). Strikingly, actin became enriched in the junctions when heterologous synapse formation was mediated by mutant Nrx3β containing the NL1 cytoplasmic tail, suggesting that the cytoplasmic NL1 sequences recruit actin, which may be important for the role of NL1 in dendritic spines.
Arguably, our most important observation is that the expression on the neuronal surface of a single neurexin LNS domain together with the stalk region is sufficient for rescuing heterologous synapse formation in Nrx TKD neurons (Fig. 5). The most plausible hypothesis to account for this observation is that neurexins act via binding to a coreceptor on the presynaptic surface, with the coreceptor subsequently mediating the signal transduction events necessary for synapse formation (Fig. 8). This coreceptor could either directly induce presynaptic specializations, or could be bivalent for neurexins and bind to the few remaining neurexins in the Nrx TKD neurons, which then might in turn activate presynaptic differentiation via their intact cytoplasmic tails. The latter possibility seems less probable because it is difficult to understand why the remaining neurexins could not simply by themselves mediate heterologous synapse formation if this possibility was correct.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model of neurexin action in heterologous synapse formation. Comparison of the signal transduction model of neurexins mediated by CASK bound to the neurexin cytoplasmic tail (left) versus the signal transduction model emerging from the current study whereby the LNS domain of neurexins serves as an adaptor that transduces a neuroligin-binding signal to an unidentified neurexin coreceptor that then mediates the signal transduction event required for heterologous synapse formation (right).
Several alternative hypotheses to account for our data cannot be completely ruled out, although they appear to be unlikely. For example, it is possible that the rescue neurexin LNS domain or stalk region dimerizes directly with the remaining neurexins in the Nrx TKD neurons, and the neurexin LNS domain then rescues heterologous synapse formation by enhancing the number of neurexin-neuroligin contacts in Nrx TKD neurons. We feel that this hypothesis is implausible because biochemical studies failed to detect homodimerization of neurexins, and numerous biophysical studies, including crystal structures, did not observe any dimerization of neurexin LNS domains even at extremely high concentrations of these domains (Araç et al., 2007; Fabrichny et al., 2007; Chen et al., 2011; Miller et al., 2011; Tanaka et al., 2011). Moreover, the stalk region is O-glycosylated with negatively charged carbohydrates, which are unlikely to bind to each other based on fundamental biochemical principles, and are not known to bind to LNS domains. Finally, as mentioned above for the notion of a bivalent neurexin cis ligand, it is difficult to envision why in this scenario the remaining neurexins in the Nrx TKD neurons would not be sufficient to simply mediate heterologous synapse formation by themselves. In other words, why would an increase of extracellular but not of intracellular neurexin sequences be needed to rescue heterologous synapse formation in these neurons? Viewed together, these considerations render this alternative hypothesis highly improbable.
A second alternative hypothesis to account for the rescue effect of extracellular neurexin sequences in Nrx TKD neurons is that any cell adhesion event may initiate heterologous synapse formation, i.e., that the process of synapse formation is nonspecific. This hypothesis is seemingly supported the observation that several postsynaptic proteins induce heterologous synapse formation [e.g., SynCAMs (Biederer et al., 2002), LRRTMs (Linhoff et al., 2009), SlitTrks (Takahashi et al., 2012)]. However, two sets of previous observations argue against this hypothesis. First, postsynaptic proteins that induce synaptogenesis appear to bind to highly specific presynaptic cell-adhesion molecules [e.g., SynCAMs to themselves (Biederer et al., 2002; Fogel et al., 2011), LRRTMs to neurexins (Ko et al., 2009a; de Wit et al., 2009; Siddiqui et al., 2010), and SlitTrks to PTPRs (Takahashi et al., 2012)]. Second, a large number of neuronal cell-adhesion molecules are unable to induce heterologous synapse formation even though they have neuronal binding partners (Biederer et al., 2002; Linhoff et al., 2009).
In summary, using Nrx TKD neurons and the heterologous synapse formation assay as tools, our study dissects the mechanism by which neurexin binding to postsynaptic NL1 and LRRTM2 triggers heterologous synapse formation. Apart from revealing unexpected facets about neurexin trafficking and function (see summary of observations above), our data suggest the possibility that neurexins do not directly transduce a signal from their extracellular ligand binding sequences to the intracellular compartment, but act via a coreceptor, a possibility that differs significantly from current models of neurexin function (Owald et al., 2012). Although our results demonstrate that the neurexin cytoplasmic sequence is unnecessary for the rescuing heterologous synapse formation in Nrx TKD neurons, we do not mean to imply that the neurexin cytoplasmic sequences are not important. It seems likely that a major function of neurexins is to organize synapses (Missler et al., 2003). Our data show that the binding of neurexins to CASK (Hata et al., 1996) is probably functionally important in this process because the Nrx TKD selectively decreased the levels of CASK in neurons (Fig. 1D), and this binding may well contribute to organizing synapses. Thus, the cytoplasmic neurexin sequence may be involved in other intracellular activities that do not directly participate in synapse formation.
Footnotes
- Received March 21, 2013.
- Revision received July 10, 2013.
- Accepted August 3, 2013.
This work was supported by NIMH Grant R37 MH052804 and NINDS Grant NS077906 to T.C.S., and by Swiss National Science Foundation and Stanford University Dean's Postdoctoral Fellowships to O.G. We thank Jacqueline Burre, Antony A. Boucard, Marc V. Fuccillo, and Lu Chen for advice, and the Stanford Neuroscience Microscopy Service (supported by NIH Grant NS069375) for equipment use.
The authors declare no competing financial interests.
- Correspondence should be addressed to Thomas C. Südhof, Stanford University Medical School, 265 Campus Drive, Stanford, CA 94305-5453. tcs1{at}stanford.edu
- Copyright © 2013 the authors 0270-6474/13/3314617-12$15.00/0