

Abstract
Ca2+-triggered exocytosis is a hallmark of neurosecretory granules, but the cellular pathway leading to the assembly of these regulated exocytotic carriers is poorly understood. Here we used the pituitary AtT-20 cell line to study the biogenesis of regulated exocytotic carriers involved in peptide hormone secretion. We show that immature secretory granules (ISGs) freshly budded from thetrans-Golgi network (TGN) exhibit characteristics of unregulated exocytotic carriers. During a subsequent maturation period they undergo an important switch to become regulated exocytotic carriers. We have identified a novel sorting pathway responsible for this transition. The SNARE proteins, VAMP4 and synaptotagmin IV (Syt IV), enter ISGs initially but are sorted away during maturation. Sorting is achieved by vesicle budding from the ISGs, because it can be inhibited by brefeldin A (BFA). Inhibition of this sorting pathway with BFA arrested the maturing granules in a state that responded poorly to stimuli, suggesting that the transition to regulated exocytotic carriers requires the removal of a putative inhibitor. In support of this, we found that overexpression of Syt IV reduced the stimulus-responsiveness of maturing granules. We conclude that secretory granules undergo a switch from unregulated to regulated secretory carriers during biogenesis. The existence of such a switch may provide a mechanism for cells to modulate their secretory activities under different physiological conditions.
- regulated exocytosis
- secretory granules
- membrane remodeling
- organelle biogenesis
- SNARE proteins
- protein sorting
Peptide hormones destined for regulated secretion are packaged into secretory granules that bud from the trans-Golgi network (TGN). These immature secretory granules then undergo a series of ill-defined “maturation” steps that ultimately transform them into mature secretory granules (MSGs). MSGs are regulated exocytotic carriers with distinctive morphology and peptide contents. Recent studies have focused on those maturation processes that impart the characteristic contents and morphology to MSGs. Several constitutively secreted proteins are incorporated into immature secretory granules (ISGs) but become removed during the maturation process (Grimes and Kelly, 1992; De Lisle and Bansal, 1996;Castle et al., 1997). This pathway, named “the constitutive-like” secretory pathway, also mediates the unregulated release of a fraction of newly synthesized peptides from hormone-secreting cells (Kuliawat and Arvan, 1992; Fernandez et al., 1997). In addition, a fraction of lysosomal hydrolases and endosomal endopeptidases escapes sorting at the TGN and subsequently is sorted away from the ISGs (Dittie et al., 1997; Kuliawat et al., 1997; Klumperman et al., 1998). ISGs also differ from MSGs in their size and density. Homotypic fusion between ISGs has been demonstrated and is likely to contribute to the increase in size as ISGs mature (Tooze et al., 1991; Urbe et al., 1998). It is thought that, after granule–granule fusion, soluble contents condense, and excess membranes are removed by vesicle budding (Burgess and Kelly, 1987). In this way the MSGs acquire their higher density and larger size after maturation.
Little is known about how MSGs acquire their function as regulated exocytotic carriers and how granule maturation influences this key property. We are interested in this question particularly because the secretory pattern of a cell can undergo dynamic changes in response to physiological conditions. For instance, tumors often secrete autocrine growth factors in an unregulated manner to enhance their growth. In the case of neuroendocrine tumors, this hypersecretion has been shown to result from dysregulation at the level of newly formed secretory granules (Fernandez et al., 1997). To begin to address this question, we have examined the exocytotic behavior of granules during maturation. Our data indicate that a key step in maturation involves the transition of the granule membrane from an unregulated exocytotic state to a regulated exocytotic state. In addition, we have uncovered an ADP ribosylation factor (ARF)-mediated sorting pathway that is responsible for functional remodeling of the granule membrane. Several trafficking proteins, including vesicle-associated membrane protein-4 (VAMP4) and Synaptotagmin IV (Syt IV), enter the regulated pathway but are removed from the ISGs along this pathway. We provide experimental evidence for the importance of Syt IV as a switch between regulated and unregulated secretion during granule maturation. Modulation of this pathway may provide a mechanism for cells to change their secretory patterns under changing physiological conditions.
MATERIALS AND METHODS
Antisera and reagents
Rabbit anti-porcine adrenocorticotropic hormone (ACTH) antibody was generated as described (Moore et al., 1983) except that antibodies were eluted from the affinity column with 0.1 m glycine, pH 2.0, and neutralized with
 volume of 1 m Tris, pH 9.5. Anti-rat Syt IV polyclonal antiserum was a gift of Dr. Harvey Herschman (University of California Los Angeles). Anti-Syt I monoclonal antibody 48 was from Dr. L. Reichardt (University of California San Francisco). Anti-VAMP2 monoclonal antibody 69.1 was obtained from Synaptic Systems (Gottingen, Germany). Anti-FLAG monoclonal antibody (M2), M2-coupled beads, 8-Br-cAMP, sodium nitroferricyanide (SNF), thrombin, phenylmethylsulfonyl fluoride (PMSF), and iodoacetamide were obtained from Sigma (St. Louis, MO) and anti-FLAG polyclonal antisera from Santa Cruz Biotechnology (Santa Cruz, CA). Brefeldin A (BFA) was purchased from Alexis (San Diego, CA), Na2[35S]SO4from ICN Biochemicals (Costa Mesa, CA), and “Immunoprecipitin” (Staphylococcus aureus) and protein A-agarose beads from Life Technologies (Gaithersburg, MD). Glutathione Sepharose 4B beads and pGEX–KG vector were from Amersham Pharmacia (Piscataway, NJ), and secondary antibodies were from Kirkegaard and Perry Laboratories (Gaithersburg, MD).
volume of 1 m Tris, pH 9.5. Anti-rat Syt IV polyclonal antiserum was a gift of Dr. Harvey Herschman (University of California Los Angeles). Anti-Syt I monoclonal antibody 48 was from Dr. L. Reichardt (University of California San Francisco). Anti-VAMP2 monoclonal antibody 69.1 was obtained from Synaptic Systems (Gottingen, Germany). Anti-FLAG monoclonal antibody (M2), M2-coupled beads, 8-Br-cAMP, sodium nitroferricyanide (SNF), thrombin, phenylmethylsulfonyl fluoride (PMSF), and iodoacetamide were obtained from Sigma (St. Louis, MO) and anti-FLAG polyclonal antisera from Santa Cruz Biotechnology (Santa Cruz, CA). Brefeldin A (BFA) was purchased from Alexis (San Diego, CA), Na2[35S]SO4from ICN Biochemicals (Costa Mesa, CA), and “Immunoprecipitin” (Staphylococcus aureus) and protein A-agarose beads from Life Technologies (Gaithersburg, MD). Glutathione Sepharose 4B beads and pGEX–KG vector were from Amersham Pharmacia (Piscataway, NJ), and secondary antibodies were from Kirkegaard and Perry Laboratories (Gaithersburg, MD).
Cell culture and secretion assays
AtT-20 cells were maintained and secretion assays were performed, as described (Fernandez et al., 1997). Where indicated, BFA was added to the chase medium at 5 μg/ml. Regulated secretion was induced by the addition of 8-Br-cAMP (5 mm) in DMEM for 2–3 hr. To trigger secretion in 15 min, we used a combination of high KCl (50 mm) and sodium nitroferricyanide (1 mm) in DMEM (abbreviated as SNF treatment). Samples were immunoprecipitated with anti-ACTH antibodies. Beads containing the immunoprecipitates were washed sequentially with NDET buffer [containing 1% Nonidet P-40, 0.4% deoxycholate, 1 mg/ml of pepstatin, and (in mm) 66 EDTA, 10 Tris, pH 7.4, and 0.1 PMSF], Urea Buffer (5 m urea in 10 mm HEPES, pH 7.4), and H2O.
Quantitation. Medium and cell extract samples were analyzed by 15% SDS-PAGE, followed by exposure to a PhosphorImager cassette (Molecular Dynamics, Sunnyvale, CA) for 1–3 d. The intensity of sulfate-labeled prohormone proopiomelanocortin (POMC) peptides was quantified with the ImageQuant program (Molecular Dynamics). Unless otherwise indicated, the total radioactivity in POMC and POMC-derived peptides recovered in all media plus cell extract samples was set as 100%. Because the anti-ACTH antibodies used in the immunoprecipitation experiments did not recognize N-terminal fragments, the total label was corrected for the loss of these fragments. By acetone precipitation we have determined the amount of radioactivity incorporated into N-terminal fragments to be 1.8 times that incorporated into mature ACTH (data not shown). This ratio was used to calculate the amount of radioactivity that was lost in immunoprecipitation and was added to the total. Then the amount of secreted or stored peptides at a specific chase time was calculated as the percentage of corrected total label.
Generation of cell lines and production of anti-VAMP4
The GenBank database was searched, using BLAST with sequences conserved between Saccharomyces cerevisiae SNC1p (amino acids 31–70) and rodent VAMP2. One positive expressed sequence tag (EST) from the database (accession number D86817) was used to design an antisense primer (number 1; 5′-ccgccgctcgagtcaagtacggaatttcacaactataagaatgataatc-3′) for a RACE reaction from rat brain cDNA. The full-length product predicted a 16.4 kDa protein homologous to the recently published VAMP4 (Advani et al., 1998). To facilitate epitope-tagging, we generated a vector pcDNAf, which replaced the HindIII site of pcDNA3 (Invitrogen, Carlsbad, CA) with a FLAG epitope, followed by uniqueHindIII and BamHI cloning sites. The full-length VAMP4 sequence was transferred to pcDNAf by PCR amplification by using Primer 1 and a sense primer (number 2; 5′-cgcgggggatccatgcctcccaagtttaag-3′). The resulting clone contained a FLAG epitope (MDYKDDDDKL) at the N terminus of VAMP4 (pFLAG–VAMP4). Stably transfected AtT-20 cells were generated (Chavez et al., 1994), and colonies were screened by immunoblot and immunofluorescence with M2 antibody. The selected clone (AtT-20/FV4) exhibited moderate levels of expression with cisternal staining patterns of the Golgi region. We have found that, on isolation of the stable clone, a fraction of cells lost expression of the transfected protein. Therefore, we routinely assayed the expression of FLAG–VAMP4 by indirect immunofluorescence.
Syt IV primers were designed from the published mouse sequence (accession number U10355) and used in RT-PCR reactions with an AtT-20 cDNA library as the template. The PCR primers (sense primer, 5′-cgcgggaagcttatggctcctatcaccaccagc-3′; antisense primer, 5′-cgcgggctcgagaccatcacag-agcatgtgcca-3′) containedHindIII and XhoI sites to facilitate cloning into pCDM8–FLAG, which allowed insertion of the FLAG epitope at the C terminus. Cell lines were generated as above and screened by immunoblot in the presence or absence of 6 mm sodium butyrate. Clones that had inducible Syt IV expression were selected for secretion assays (see Fig. 9), and those with constitutive Syt IV expression (at fivefold lower level) were selected for localization studies (see Fig. 8).
For production of antibodies a sequence encoding a cytosolic soluble VAMP4 fragment was isolated by using the antisense primer (5′-ccgccgctcga-gttttattttgcatccacgccac-3′) and was cloned into a glutathione S-transferase fusion vector, pGEX–KG. Bacterially produced protein was purified by binding to glutathione beads, followed by cleavage with thrombin, and was used for injection into rabbits (Covance, Berkeley, CA). Tertiary bleeds from immunized animals were used.
RT-PCR
A cDNA library from AtT-20 cells was constructed with the Uni-ZAP cDNA Synthesis Kit from Stratagene (La Jolla, CA). To prepare the library for PCR, we heated 500 μl of packaged library to 70°C for 10 min, followed by phenol/chloroform extraction. The extracted library was ethanol-precipitated and resuspended in water. For VAMP4, one-tenth of the materials was used in a standard 100 μl PCR with Primer 1 and Primer 2 (see above). One-half of the final reaction product was run on a 1.5% agarose gel and stained with ethidium bromide. As controls, these reactions were performed with the cDNA library omitted or substituted with 20 ng of plasmid harboring either a VAMP4 cDNA or a VAMP2 cDNA. For Syt IV, isoform primers were designed from the published mouse sequence.
Immunofluorescence
Cells were fixed, permeabilized, and stained according to the published procedure (Chavez et al., 1994). M2 antibody was used at 1:50 and anti-ACTH at 1:50. Staining was analyzed with a Zeiss Axiophot microscope or a Sarastro 1000 confocal microscope (Molecular Dynamics, San Jose, CA). Images from the Axiophot were generated by scanning 35 mm negatives via a SprintScan slide scanner (Eastman Kodak, New Haven, CT) and colored and merged by using Adobe Photoshop 4.0 (Adobe Systems, Mountainview, CA). Confocal images were merged by using ImageSpace (Molecular Dynamics).
Immunoisolation of FLAG–VAMP4-containing organelles
Cells were pulse-labeled with [35S]sulfate and chased for the indicated times. For experiments including BFA, labeled material was chased for 7 min to allow for the exit of labeled POMC from thetrans-Golgi before the addition of BFA. Chase was continued for 38 min in the presence of BFA before immunoabsorption. Cells were lifted with EDTA and homogenized by eight passes through an EMBL cell homogenizer (8.02 chamber, ball size 5) in Homogenization Buffer [composed of (in mm) 250 sucrose, 10 HEPES, pH 6.9, 1 EDTA, and 1 EGTA] containing 0.1 mm PMSF and 300 μg/ml of iodoacetamide. The homogenate was subjected to centrifugation at 1300 × g for 5 min; the resulting postnuclear supernatants (PNS; 0.8–1.2 mg/ml of protein) were incubated at 4°C with anti-FLAG-coupled (M2) Sepharose beads (see below for preparation) for 2 hr with gentle rotation. After immunoabsorption, bead-bound materials were pelleted at 500 ×g and washed three times in Homogenization Buffer, followed by lysis in 1% Triton X-100/PBS for 30 min at 4°C. Supernatants were diluted twofold with H2O and then precipitated by the addition of 4 volumes of ice-cold acetone. After 1 hr of incubation at −20°C the precipitates were collected by centrifugation in a Beckman GPR centrifuge at 3500 × g for 1 hr at 4°C. The samples were subject to 15% SDS-PAGE and PhosphorImager analysis.
To reduce nonspecific binding, we first preincubated the beads for immunoabsorption experiments for 30 min at 4°C with a high concentration of PNS (∼3 mg/ml protein) prepared from untransfected cells supplemented with 0.5% BSA. In some experiments these incubations also contained a competing FLAG peptide (1 μg of peptides per 100 μl of stock M2 beads); the latter also was included in the immunoadsorption reaction. Organelle lysis during immunoabsorption procedures was monitored by immunoprecipitation of an aliquot of PNS, using anti-ACTH either in the presence or absence of Triton X-100. Values for immunoabsorption experiments were corrected for the number of cells positive for FLAG–VAMP4 in the stable line (152 of 278 or 55%), the percentage of lysis in PNS (10–30%), and the efficiency of immunoabsorption (58%). The efficiency of immunoabsorption was determined by immunoblot analysis of the percentage of total FLAG–VAMP4 recovered on M2 beads (see Fig.3A).
Immunoblot
PNS prepared from unlabeled cells was subject to 15% SDS-PAGE and transferred to nitrocellulose (Micron Separations, Westborough, MA). The blots were probed with anti-VAMP4 polyclonal antibodies (1:1000 dilution) or M2 antibody (1:200 dilution). Binding of antibodies was detected by using HRP-conjugated secondary antibodies, followed by the addition of HRP substrate (ECL, Amersham Pharmacia, Piscataway, NJ) and exposure to x-ray film.
Velocity and equilibrium gradient centrifugation
Golgi membranes were isolated from a 15 cm dish of wild-type cells by velocity centrifugation according to Fernandez et al. (1997). Materials from the peak fraction were precipitated by trichloroacetic acid and immunoblotted with a rabbit anti-Syt IV antiserum (see Fig.8A). For Figure 8C, ISGs and MSGs were isolated by using a modified protocol of Tooze et al. (1991). Velocity centrifugation was performed by using a linear sucrose gradient prepared from 0.3 m (5.3 ml) to 1.2m (5.3 ml) sucrose in 10 mmHEPES-KOH, pH 7.2. PNS from one 15 cm dish of radiolabeled Syt IV-expressing cells was pooled with unlabeled PNS from one 15 cm dish of FLAG–VAMP4-expressing cells. One-half of the pooled PNS (1 ml) was loaded onto the gradient and centrifuged at 25,000 rpm in a Beckman SW41 rotor, with the brake applied at the end of 15 min. Fractions (1.3 ml) were collected from the top after the initial load fraction was discarded. Fractions enriched in ISGs (1, 2) or MSGs (5, 6) were pooled, diluted to 6 ml with cold water, and loaded onto a second gradient. Equilibrium centrifugation was performed by using a linear sucrose gradient prepared from 0.5 m (16 ml) to 1.8 m (16 ml) sucrose in 10 mm HEPES-KOH, pH 7.2. The gradient was centrifuged for 12–16 hr at 25,000 rpm in a Beckman SW28 rotor. Fractions (1.1 ml) were collected from the top after the initial load fraction was discarded. Aliquots from fractions (
 for Syt IV and VAMP4 blots,
for Syt IV and VAMP4 blots,
 for Syt I and VAMP2 blots) were subjected to acetone precipitation, followed by SDS-PAGE/PhosphorImager or immunoblot analysis.
for Syt I and VAMP2 blots) were subjected to acetone precipitation, followed by SDS-PAGE/PhosphorImager or immunoblot analysis.
RESULTS
Newly formed secretory granules are poorly responsive to secretagogues but acquire stimulus-responsiveness during maturation
We have shown previously that the POMC is sulfated in the TGN of AtT-20 cells. Its synchronous labeling and rapid transport to post-Golgi compartments provide powerful tools for studying secretory granule biogenesis (Dumermuth and Moore, 1998). Kinetic analysis has established that the half-time for transport of [35S]sulfate pulse-labeled POMC from the TGN to nascent granules is ∼3 min (Fernandez et al., 1997) (data not shown). On packaging in ISGs, POMC is converted proteolytically to mature ACTH by the prohormone convertase PC1 (Benjannet et al., 1991;Thomas et al., 1991; Zhou et al., 1993; Schmidt and Moore, 1995). ACTH is the final product stored in MSGs for regulated exocytosis.
We first examined the time course with which granules acquire full stimulus-responsiveness. Cells were pulse-labeled with [35S]sulfate for 5 min and chased for 15 min (five half-times) to allow for transport of labeled POMC from the TGN to ISGs. Then the cells were chased further to allow granule maturation to proceed. At different chase times the secretory response of granules was determined by applying a test stimulus and analyzing the secreted materials by immunoprecipitation. To resolve the time course better, we needed a fast-acting secretagogue. We found that AtT-20 cells responded quickly to a combination of sodium nitroferricyanide (a NO donor) plus high [K+] (depolarization)—consistent with a role for NO (Brunetti et al., 1993) and depolarization (Gumbiner and Kelly, 1982) in regulated secretion of ACTH from pituitary cells. This combination of secretagogues, herein referred to as SNF stimulation, thus was used in this kinetic experiment. Figure1A shows quantitation of the “stimulation index,” which is the fold of stimulated secretion in response to SNF. For mature ACTH a steady increase was observed during maturation: the stimulation index was 1.6 after a 15 min chase and increased to 2.7 after a 2 hr chase (Fig.1A, filled bars). Thus, freshly budded granules are only marginally responsive to secretagogues. They gain the ability to undergo regulated exocytosis during an ∼45 min maturation period.

Development of responsiveness of granules to secretory stimuli during maturation. AtT-20 cells were pulse-labeled for 5 min with [35S]sulfate and chased for 15 min to 2 hr (Chase 1). Then the secretory response of granules was tested by a 15 min exposure (Chase 2) to SNF. Parallel cultures were kept unstimulated as controls. Samples were immunoprecipitated with anti-ACTH and analyzed by SDS-PAGE and PhosphorImager. A, Plot of the stimulation index as a function of chase time. The ratios of secretion during Chase 2 from SNF-stimulated to unstimulated cells were calculated and plotted as a function of Chase 1 length. Open bars, Fold of stimulation of labeled POMC; filled bars, fold of stimulation of labeled ACTH. Increasing the chase time led to an increase in the fold of stimulation for ACTH, but not for POMC.B, Plot of the rate of unstimulated secretion as a function of chase time. The sum of labeled POMC, intermediate forms, and ACTH secreted from unstimulated cells at each time point was calculated. Then this amount was normalized to the total label that was recovered from the medium and cell extracts, and it was expressed as the percentage of label secreted per minute. In unstimulated cells the secretory rate peaked between 10 and 30 min after the pulse and then declined to a low level during subsequent chases. The data represent the average of two experiments.
We also quantitated the stimulation index of unprocessed POMC as a function of time. Because conversion of POMC to ACTH in the ISG is time-dependent, POMC provides a marker of young granules, whereas ACTH provides a marker for older granules. The secretion of POMC was not affected significantly by SNF (Fig. 1A, open bars); the stimulation index was 1.4 at 15 min of chase and stayed between 1.2 and 1.4 during subsequent chases. These data further confirmed that older granules are more responsive to stimuli than young granules.
During the critical period in which granules became responsive to SNF, there was also a dramatic change in the rate of unstimulated secretion (Fig. 1B). This unregulated secretion peaked at ∼20–30 min after the pulse label and declined to a low level during subsequent chases. Because this secretory phase occurs after POMC has been packaged into ISGs (t1/2 ∼3 min), it could represent secretion from both the constitutive and the constitutive-like pathways. Supporting the latter argument, some of the materials secreted during this period had undergone the proteolytic event characteristic of ISGs (data not shown). These data show that granules gain responsiveness to secretory stimuli during a critical period of maturation that coincides with a phase of unstimulated secretion from the ISGs.
VAMP4 marks a novel vesicular sorting pathway during granule membrane remodeling
The acquisition of competence for regulated exocytosis suggests that functional remodeling of the granule membrane must occur during maturation. Because this change takes place during a period of high trafficking activity (e.g., unstimulated secretion from the ISGs), we hypothesize that it may involve vesicle-mediated sorting of membrane components away from the ISGs. To investigate this possibility, we sought to identify membrane proteins that may serve as specific markers of this putative sorting pathway. We focused on the VAMP family of trafficking proteins because its members constitute a major group of vesicle proteins controlling membrane fusion. Members of the VAMP family were screened for ones that are expressed in AtT-20 cells and that show the expected pattern of localization, i.e., present in proximal parts of the regulated secretory pathway but absent from mature granules. In the initial screen we found that only VAMP4 met these criteria.
VAMP4 is expressed in AtT-20 cells, as shown by specific RT-PCR amplification with isoform-specific primers (Fig.2A), and also by immunoblot using a rabbit antiserum generated against purified recombinant cytosolic fragment of rat VAMP4 (Fig. 2B,lane 1). Preimmune sera did not generate a signal (data not shown). In preliminary studies we found that the anti-VAMP4 antiserum did not recognize native antigens. To facilitate localization studies, we constructed an N-terminal FLAG-tagged VAMP4 expression plasmid and generated a stable AtT-20 cell line expressing this construct (referred to as the FV4 cell line). Lysate from this cell line showed an additional band immunoreactive toward both anti-VAMP4 and anti-FLAG monoclonal antibody M2 (Fig. 2B,lanes 2, 3); this exogenous FLAG–VAMP4 was expressed at an approximately fivefold higher level than the endogenous protein. Indirect immunofluorescence analysis of FV4 cells with anti-FLAG M2 antibody showed that FLAG–VAMP4 was found in the juxtanuclear region of the cell body (Fig. 2Ca–Cc, arrowheads), but it was absent from process tips where mature secretory granules concentrated in AtT-20 cells (arrows). Confocal analysis demonstrated that the staining in the cell body exhibited cisternal patterns and was colocalized precisely with POMC/ACTH, with very little punctate or cell surface staining (Fig. 2Cd–Cf, arrowheads). In brefeldin A-treated (BFA) cells, FLAG–VAMP4 staining no longer exhibited the characteristic cisternal pattern and instead collapsed to a condensed structure near the microtubuloorganizing center (MTOC) region (data not shown), a behavior characteristic of TGN proteins. In transfected HeLa cells and CHO cells FLAG–VAMP4 also was found to be colocalized precisely with the TGN marker TGN38 (data not shown). The same staining pattern was observed regardless of the expression levels, suggesting that the TGN localization is not an artifact of overexpression. These data suggest that VAMP4 is associated with regulated secretory marker in the early part of the pathway (the TGN), but it is not found on the mature secretory organelles.

Characterization of VAMP4 in AtT-20 cells.A, RT-PCR analysis performed on AtT-20 cDNAs with primers specific to VAMP4 (lane 2). Lane 1, Molecular weight standards; lane 3, negative control performed with no added DNA template; lanes 4, 5, controls performed with plasmids harboring either a VAMP2 cDNA or a VAMP4 cDNA as the template. A single band of 426 bp was observed only in reactions performed with either AtT-20 cDNAs or the VAMP4 plasmid. Digestion of this product with HindIII gave the predicted restriction pattern as deduced by the rat VAMP4 sequence. B, Immunoblot analysis of endogenous and transfected VAMP4. PNS was prepared from wild-type AtT-20 (wt, lane 1) and cells stably transfected with a FLAG-tagged VAMP4 plasmid (FV4, lanes 2, 3). Lysate (10 μg) was subject to SDS-PAGE and immunoblot analysis with either polyclonal antisera against a soluble form of recombinant rat VAMP4 (lanes 1, 2) or M2 monoclonal antibody against the FLAG epitope (lane 3). The anti-VAMP4 antisera recognized a single band of ∼18 kDa in wild-type cells and an additional band that also was recognized by the anti-FLAG antibody in FLAG–VAMP4 transfected cells. C, Colocalization of VAMP4 with ACTH immunoreactivities in the Golgi region, but not in the process tips. AtT-20 cells stably transfected with FLAG–VAMP4 were processed for indirect immunofluorescence. Fixed and permeabilized cells were doubly stained with anti-FLAG M2 monoclonal antibody (Cb, Ce) and anti-ACTH (Ca, Cd) polyclonal antibodies.Ca–Cc, A representative field of cells visualized by an Axiophot microscope, showing colocalization of staining patterns in the cell body (arrowheads), but not in the process tips (arrows) where MSGs accumulated. More than 50 cells were examined, and all show similar patterns. Cd–Cf, Higher resolution analysis of the staining patterns in the Golgi region by confocal microscopy. Scale bar, 10 μm.
Subcellular fractionation of FV4 cells on velocity gradients indicated that VAMP4 was distributed not only in TGN but also in vesicular fractions (data not shown), suggesting that it may be present on post-Golgi organelles such as ISGs. To determine more precisely the VAMP4 trafficking within the regulated pathway, we developed a pulse–chase/immunoabsorption procedure. Briefly, FV4 cells were pulse-labeled for 5 min with [35S]sulfate and then chased for various time periods to allow for labeling of granules at different stages of maturity. Then the cells were homogenized, and FLAG–VAMP4-bearing organelles were isolated by using anti-FLAG-coupled (M2) Sepharose beads. Captured organelles were solubilized, and their contents of labeled peptides were analyzed by SDS-PAGE and PhosphorImager quantitation. We first demonstrated that FLAG–VAMP4-bearing organelles could be isolated specifically on M2 beads (Fig.3A). Immunoblot analysis of bead-bound organelles that used anti-FLAG polyclonal antibodies showed that 58% of the immunoreactivity present in the input PNS was immunoisolated on M2 beads (Fig. 3A, lanes 1, 2). The efficiency was increased to 80% when Triton X-100 was included during the isolation procedure (Fig. 3A, lane 4), indicating that the binding of intact organelles to beads was less efficient than the solubilized protein. Inclusion of a peptide containing the FLAG epitope effectively competed away the binding of FLAG–VAMP4 to beads (Fig. 3A, lane 3), demonstrating the specificity of the immunoabsorption procedure.
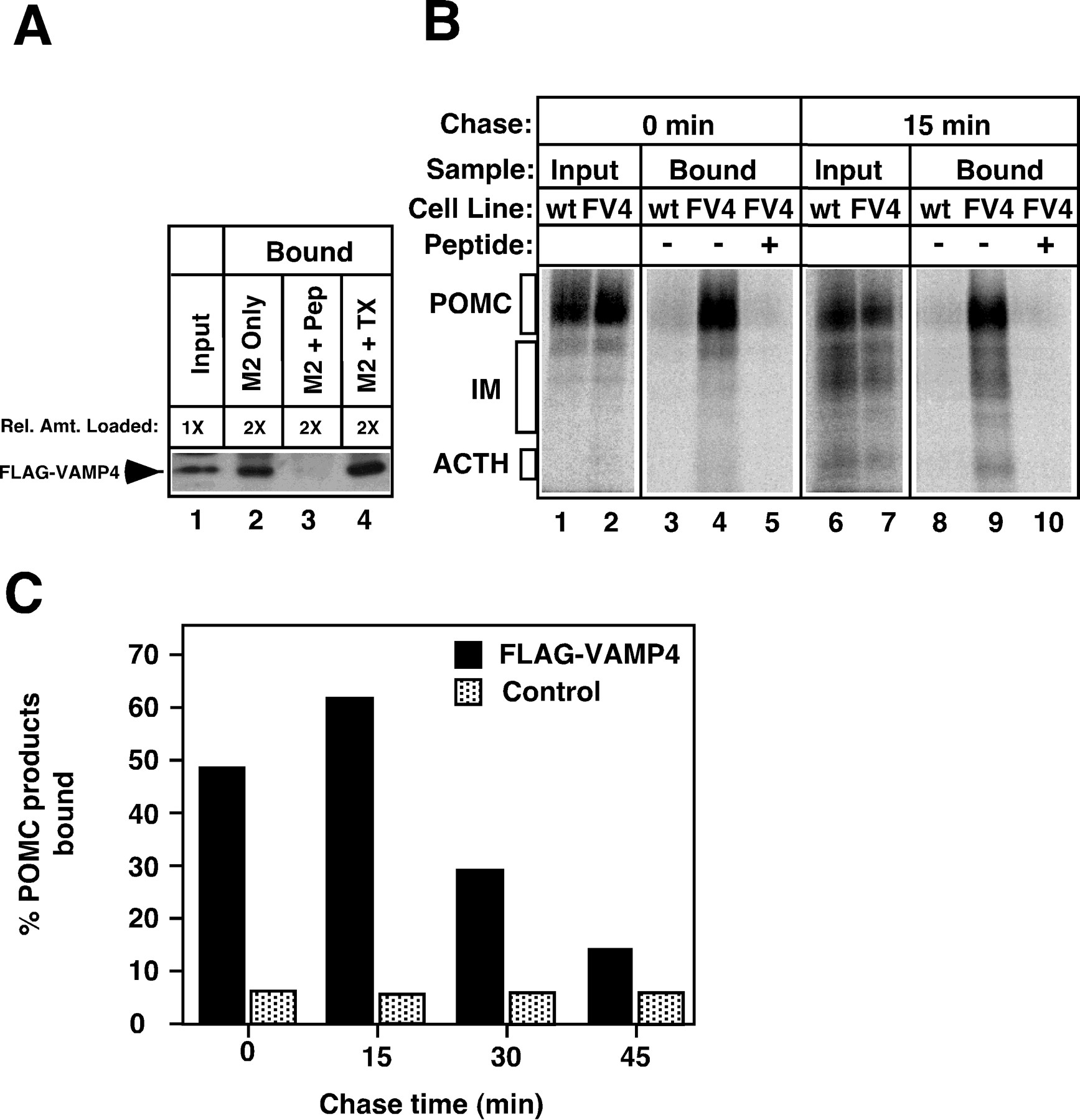
Sorting of VAMP4 from the regulated secretory pathway during granule maturation. A, VAMP4-containing organelles could be immunoisolated on anti-FLAG beads (M2 beads). Cells stably transfected with FLAG–VAMP4 were homogenized, and the PNS was incubated with M2 beads. After washing, the organelles bound to the beads were solubilized, and the amount of FLAG–VAMP4 was determined by immunoblotting with polyclonal anti-FLAG antibodies.Lane 1, An aliquot of PNS equivalent to one-half of the amount used for immunoisolation was analyzed as the input. Lane 2, Material bound to M2 beads. Lane 3, Material bound to M2 beads in the presence of a competing FLAG-containing peptide (Pep). Lane 4, Material bound to M2 beads when 1% Triton X-100 (TX) was included during the immunoisolation procedure. The addition of the FLAG peptide abolished binding, indicating that the capture of FLAG–VAMP4 by M2 beads was specific. Inclusion of Triton X-100 increased the recovery of FLAG–VAMP4 on the beads. B, VAMP4 was present on the TGN and on newly formed ISGs. FLAG–VAMP4 stably transfected cells (FV4) or wild-type AtT-20 cells (wt, as negative controls) were pulse-labeled with [35S]sulfate for 5 min to label POMC in the TGN. The cells either were harvested immediately (lanes 1–5) or were chased for 15 min before homogenization to allow labeled POMC to migrate into ISGs (lanes 6–10). FLAG–VAMP4-containing organelles were immunoisolated with M2 beads as described in A, and bead-bound material was eluted with Triton X-100 and concentrated by acetone precipitation. Labeled POMC products recovered from the beads were analyzed by SDS-PAGE and PhosphorImager. Lanes 1, 2, 6, 7, An aliquot of PNS (
 ) was analyzed as the input. Lanes 4, 9, Material bound to the beads from FV4-transfected cells. Lanes 3, 8, Material bound to the beads from untransfected wild-type cells. IM, Intermediate forms of POMC-processing products. Specific capture of labeled POMC products by M2 beads was observed with FV4-transfected cells at both time points, but not with wild-type cells. The inclusion of FLAG peptides also abolished the signals seen in FV4 cells (lanes 5, 10).C, Association of VAMP4 with markers of the regulated secretory pathway was dependent on chase times. A time course experiment similar to B was performed to determine the amount of labeled POMC/ACTH captured by M2 beads with increasing chase times. Control values were obtained with wild-type cells or by competition with FLAG peptides. Values are expressed as the percentage of total input that was recovered on M2 beads after correction for loading, lysis, immunoadsorption efficiency, and cell line expression (see Materials and Methods). Each time point was performed at least twice with similar results.
) was analyzed as the input. Lanes 4, 9, Material bound to the beads from FV4-transfected cells. Lanes 3, 8, Material bound to the beads from untransfected wild-type cells. IM, Intermediate forms of POMC-processing products. Specific capture of labeled POMC products by M2 beads was observed with FV4-transfected cells at both time points, but not with wild-type cells. The inclusion of FLAG peptides also abolished the signals seen in FV4 cells (lanes 5, 10).C, Association of VAMP4 with markers of the regulated secretory pathway was dependent on chase times. A time course experiment similar to B was performed to determine the amount of labeled POMC/ACTH captured by M2 beads with increasing chase times. Control values were obtained with wild-type cells or by competition with FLAG peptides. Values are expressed as the percentage of total input that was recovered on M2 beads after correction for loading, lysis, immunoadsorption efficiency, and cell line expression (see Materials and Methods). Each time point was performed at least twice with similar results.
Using this procedure, we have found that FLAG–VAMP4 is associated with the TGN and newly formed ISGs (Fig. 3B). Pulse-labeled cells were harvested immediately (labeled POMC in the TGN) or chased for 15 min (labeled POMC products in newly formed ISGs) before homogenization. At both time points the labeled POMC products were captured by the M2 beads (Fig. 3B, lanes 4, 9), indicating the association of VAMP4 with these two organelles. The observed binding was specific, because inclusion of a competing FLAG peptide in the absorption mix (Fig. 3B, lanes 5, 10) or mock immunoabsorption with wild-type cells that had not been transfected with FV4 (Fig. 3B, lanes 3, 8) showed little binding. Importantly, the association of radiolabeled peptides with VAMP4 diminished as the chase times extended beyond 15 min (quantitated in Fig. 3C). At 0–15 min of chase, 47–60% of total labeled POMC products was captured by M2 beads. By 45 min of chase, the level dropped to 12.5%. Together, these data suggest that VAMP4 enters the regulated pathway but is removed during the early stages of granule maturation.
We next asked whether VAMP4 is sorted away from the ISGs by vesicle budding. Because BFA has been shown to inhibit a variety of ARF-mediated vesicle budding events (Peyroche et al., 1999), we tested whether BFA interfered with VAMP4 removal from the regulated pathway. The immunoabsorption experiments shown in Figure 3 were repeated, except that after 7 min of chase (to allow entry of POMC into ISGs) BFA was added to the cells and granule maturation was allowed to proceed in the continued presence of BFA. Cells were harvested at 45 min of chase and processed for immunoabsorption (Fig.4). In BFA-treated cells, ∼60% of labeled ACTH was still captured by M2 beads after 45 min of chase (Fig.4A, lanes 4–6, and quantitation in 4B), similar to the level before the addition of BFA (see Fig. 3C). By contrast, the level of immunoabsorption in control untreated cells dropped to ∼10% after 45 min (Fig.4A, lanes 1–3, 4B). These data demonstrated a persistent association of VAMP4 with secretory granule markers when granules were allowed to age in the presence of BFA. Thus, an ARF-mediated vesicle budding event underlies the VAMP4 sorting pathway, and BFA provides a useful reagent to block this pathway.

Inhibition of VAMP4 sorting by BFA.A, An immunoisolation experiment similar to Figure 3 was performed by using FV4-transfected cells except that, after the pulse-labeling, the cells were chased first for 7 min to allow labeled POMC to reach ISGs before they were chilled on ice. Then BFA was added to one plate; the cells were warmed to 37°C and chased in the continued presence of BFA for a total of 45 min (lanes 4–6). A parallel dish was not treated with BFA as the control (lanes 1–3). VAMP4-containing organelles were immunoisolated, and POMC products bound to M2 beads were analyzed.Lanes 1, 4, An aliquot of input PNS (
 ).Lanes 2, 5, Material bound to the beads. Lanes 3, 6, Material bound to the beads in the presence of competing FLAG peptides. The increased amount of unprocessed POMC seen inlane 4 as compared with lane 1(corresponding to 24% of the total label) is attributable to material remaining in the Golgi at the time of BFA addition (Fernandez et al., 1997). B, Quantitation of data showing increased association of VAMP4 with granule markers in BFA-treated cells. Only mature ACTH was quantitated to avoid contributions from POMC trapped in the TGN. This experiment was performed three times with similar results.
).Lanes 2, 5, Material bound to the beads. Lanes 3, 6, Material bound to the beads in the presence of competing FLAG peptides. The increased amount of unprocessed POMC seen inlane 4 as compared with lane 1(corresponding to 24% of the total label) is attributable to material remaining in the Golgi at the time of BFA addition (Fernandez et al., 1997). B, Quantitation of data showing increased association of VAMP4 with granule markers in BFA-treated cells. Only mature ACTH was quantitated to avoid contributions from POMC trapped in the TGN. This experiment was performed three times with similar results.
Interference of the VAMP4 pathway with BFA prevents ISGs from acquiring the proper secretory behavior
VAMP4 is removed from ISGs (see Fig. 3C) during the same time window as when granules acquire their stimulus-responsiveness (see Fig. 1). We therefore asked whether blocking the VAMP4 pathway by BFA would lead to alterations in the secretory response of aged granules (Fig. 5). Pulse-labeled cells were chased first for 15 min to allow labeled POMC to enter ISGs. Then BFA was added and the cells were allowed to chase for a maturation period of 3 hr in the continuous presence of BFA (Chase 2). At the end of the maturation period the cells were stimulated with 8-Br-cAMP for 3 hr (Chase 3) in the presence of BFA to induce regulated secretion. 8-Br-cAMP required longer treatment but induced an overall stronger stimulatory response than SNF and thus was used in this experiment. Granules that matured in the absence of BFA exhibited a strong secretory response to the secretagogue (Fig. 5A, comparelanes 3, 4 and 5, 6) and a concomitant decrease of the intracellular ACTH pool (Fig. 5A, comparelanes 7, 8 and 9, 10). In contrast, granules aged in the presence of BFA failed to respond to 8-Br-cAMP stimulation. The amount of labeled ACTH released into the medium was not significantly different between stimulated and unstimulated cells (Fig.5B, compare lanes 3, 4 and 5, 6), and very little depletion of intracellular stored ACTH was detected (Fig. 5B, compare lanes 7, 8 and9, 10). These results were quantitated in Figure5C. BFA affected granule maturation in two major respects. First, granules aged in BFA were unable to undergo regulated exocytosis. 8-Br-cAMP elicited a 5.72-fold stimulation of ACTH release from control cells, whereas it elicited only a 0.94-fold stimulation from BFA-treated cells. Second, granules aged in BFA exhibited a high basal release rate. In control cells the rate of unstimulated release dropped to ∼3% during the late chase period (3–6 hr). In BFA-treated cells the unstimulated secretion remained high during the same 3–6 hr of chase; ∼9% mature ACTH continued to be secreted in the absence of stimulation. Therefore, granules aged in BFA appear to be arrested in a state with high unstimulated release and poor responsiveness to secretagogues. Note that, although BFA causes a high rate of basal release, the amount of total label that was recovered at the end of the 6 hr chase was not lower than in the controls. This is attributable to the fact that BFA also inhibits constitutive-like secretion, thus increasing the cellular contents of labeled hormone (Fernandez et al., 1997).

Effects of BFA on the maturation of the exocytotic behavior of granules. Duplicate cultures of wild-type cells were pulse-labeled and chased in BFA-free DMEM for 15 min to allow for entry of labeled POMC into ISGs (Chase 1, C1). Then BFA was added during a subsequent 3 hr chase to test its effect on granule maturation (Chase 2, C2). Afterward, the cells were treated with 8-Br-cAMP for 3 hr to induce regulated secretion (Chase 3,C3). BFA was present throughout Chase 2 and Chase 3. Control cells were chased in the absence of BFA (A) and compared with cells that were treated with BFA (B). Lanes 1, 2, Labeled POMC products secreted during Chase 2; lanes 3, 4, labeled POMC products secreted during Chase 3 from unstimulated cells;lanes 5, 6, labeled POMC products secreted during Chase 3 from stimulated cells. Lanes 7, 8, Extracts from unstimulated cells; lanes 9, 10, extracts from stimulated cells after Chase 3. As noted in Figure4A, in BFA-treated cells some labeled POMC was trapped in the TGN and was recovered as unprocessed POMC. In this experiment this amount corresponded to <10% of the total label.C, The results in B were quantitated, and the amount of labeled ACTH that was secreted in response to 8-Br-cAMP during Chase 3 was normalized to the amount of total label that was recovered in all POMC and POMC-derived peptides. BFA-treated cells were not responsive to 8-Br-cAMP and exhibited a higher rate of unstimulated release of ACTH during Chase 3. The data represent the average of two experiments.
Very similar results also were obtained when SNF was used as a secretagogue instead of 8-Br-cAMP (data not shown). In control cells SNF induced a 3.7-fold stimulation of ACTH secretion above the unstimulated level. By contrast, SNF elicited only a 1.38-fold of stimulation from cells chased in the presence of BFA. Together, these results support the notion that conversion of ISGs from an unregulated to a regulated exocytotic state requires a BFA-sensitive trafficking event.
The effects of BFA are reversible and occur only during a critical time window
If BFA exerts its effects on secretory activity by blocking the VAMP4 pathway, one would expect that the time course of BFA inhibition on granule maturation would mimic the kinetics of VAMP4 removal. We therefore have analyzed the time window during which BFA exerts its effects on the secretory behavior of the granule. Cells were pulse-labeled and chased for varying periods of time (30 min–3 hr) before the addition of BFA (Fig. 6). Then the cells were chased in the presence of BFA for an additional 2 hr, and their responses to 8-Br-cAMP stimulation were tested. When cells were chased for 30 min before BFA treatment, subsequent incubations resulted in granules that were only weakly responsive to 8-Br-cAMP (Fig. 6A, lanes 1–4). Extending the chase time to 1–2 hr before BFA addition led to a much higher degree of regulated secretion and depletion of intracellular ACTH (Fig. 6A, lanes 5–12). A summary of these experiments is shown in Figure 6B. The addition of BFA 15–30 min after the pulse label arrested the newly formed ISGs in a state of high unregulated secretion and low responsiveness to secretagogues. The BFA-sensitive step occurred between 15 and 60 min after budding of the ISGs from the TGN, a time window that coincided with the removal of VAMP4 from the regulated pathway (see Fig.3C). The close correlation in kinetics of VAMP4 removal and passage of granules through the BFA block suggests that the two events are interrelated. The finding that BFA added at late times did not affect the exocytotic response of the granule (Fig. 6) also indicated that BFA did not exert its effects by blocking regulated exocytosis directly.

Time course of the BFA block to the acquisition of stimulus-responsiveness by maturing granules. Duplicate cultures of wild-type cells were pulse-labeled (P) and chased in DMEM (Chase 1, C1) for increasing periods of time before BFA addition. Then the cells were chased for 2 hr to allow the granules to age in the presence of BFA. Their secretory responses were tested by exposure to 8-Br-cAMP in DMEM for 3 hr in the continued presence of BFA (Chase 2, C2). A, Cells were chased for 30 min (lanes 1–4), 1 hr (lanes 5–8), and 2 hr (lanes 9–12) before BFA treatment. Lanes 1, 2, 5, 6, 9, 10, Labeled POMC products secreted during Chase 2 in the absence or presence of 8-Br-cAMP. Lanes 3, 4, 7, 8, 11, 12, Corresponding extracts from unstimulated or stimulated cells. B, The response to secretagogues is plotted as a function of the chase time before BFA addition. The amount of labeled ACTH secreted from stimulated and unstimulated cells during Chase 2 was quantitated and expressed as the percentage of labeled ACTH remaining within cells at the start of Chase 2. Maturing granules passed through the BFA block within 1 hr of chase. The data represent the average of two experiments.
It is possible that BFA induced irreversible changes to the ISG membrane. We therefore investigated the reversibility of the BFA effects on ISG membrane maturation (Fig.7). Cells were pulse-labeled, chased for 15 min to allow ISG formation, and treated with BFA for 2 hr. Then BFA was washed out of the cells during three subsequent 1 hr chase periods, after which the secretory responses to 8-Br-cAMP were tested. The rate of unstimulated release of mature ACTH during BFA treatment was calculated as the percentage of total label that was secreted per hour and compared with those during the washout periods. As shown in Figure7A, secretion of ACTH increased transiently on removal of BFA, peaking within 1 hr of BFA washout, and then declined to a low level. Control mock-treated cells showed a very low level of secretion during the same time period (Fig. 7A). This phasic secretion after BFA washout is reminiscent of the transient secretion that normally occurred during 15–60 min of chase (see Fig.1B). Importantly, stimulus-dependent secretion of labeled ACTH was observed in BFA washout cells (Fig. 7B). The addition of 8-Br-cAMP induced a 6.6-fold of stimulated release of ACTH, compared with 7.3-fold in mock-treated cells. This experiment indicates that the effects of BFA on membrane remodeling are mainly reversible.

Reversibility of the effects of BFA on granule maturation. Duplicate cultures of wild-type cells were pulse-labeled (P), chased in BFA-free DMEM for 15 min (C1), and then treated for 2 hr with BFA (C2). To test for reversibility, we washed BFA out of the cells during the subsequent three 1 hr chases (C3,C4, C5). Then regulated secretion was induced with 8-Br-cAMP for 2.5 hr (C6). The amount of labeled ACTH that was secreted during each period was normalized to the total label recovered in all POMC and POMC-derived peptides and was calculated as the percentage secreted per hour.A, Unstimulated secretion during BFA treatment (0–2 hr) and during the washout period (2–3 hr, 3–4 hr, 4–5 hr) is shown for treated cells (right panel) and compared with control untreated cells (left panel). On the removal of BFA a surge of labeled ACTH was secreted. Then this secretory rate rapidly declined to a low level similar to control cells. B, Stimulated secretion of labeled ACTH after BFA washout. The amount of labeled ACTH that was secreted during Chase 6 was normalized to labeled ACTH remaining within the cells at the start of Chase 6. Granules regained their ability to undergo regulated exocytosis after washout. The data represent the average of two experiments.
Synaptotagmin IV follows the trafficking patterns of VAMP4 and modulates Ca2+-triggered exocytosis during granule maturation
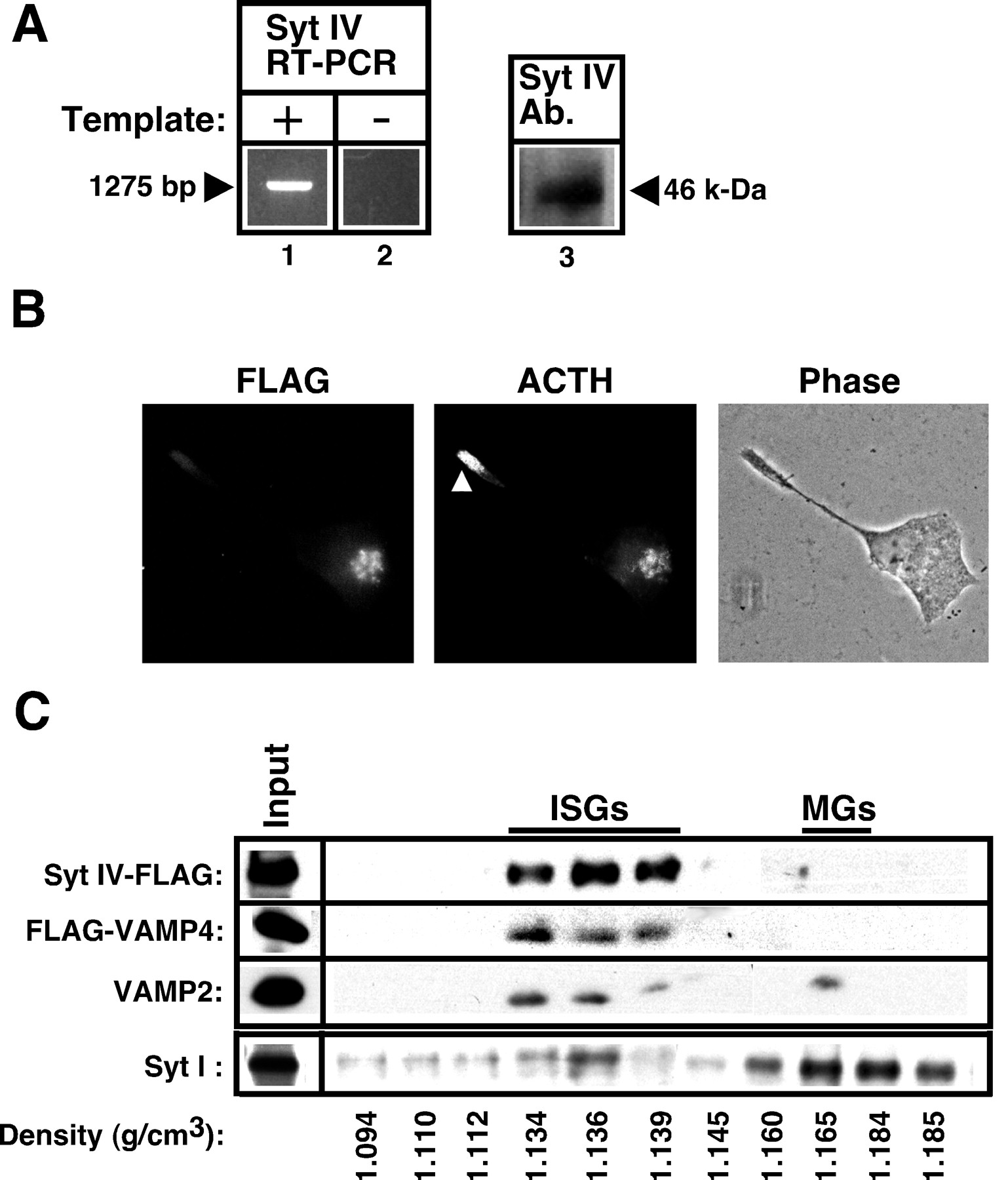
The above data suggest that granule membrane maturation may require the removal of a key inhibitor of Ca2+-triggered exocytosis from the ISG membrane. Overexpression of VAMP4 itself had no effect on granule membrane maturation, nor does it have detectable effects on constitutive-like secretion or regulated secretion (data not shown). This suggests that some other component(s) removed along the VAMP4 pathway may modulate Ca2+-triggered exocytosis. Recently, Syt IV, a Ca2+-independent member of the synaptotagmin protein family, was shown to inhibit Ca2+-triggered exocytosis of synaptic vesicles by interacting with the calcium-sensing synaptotagmin I (Littleton et al., 1999; Thomas et al., 1999). We therefore investigated whether Syt IV played a role in the maturation of secretory granules in AtT-20 cells. RT-PCR analysis with isoform-specific primers demonstrated that a single product of the correct size for Syt IV was amplified specifically (Fig.8A, lanes 1, 2). Syt IV immunoreactivity also was detected on purified Golgi membranes with a polyclonal antiserum specific for rat Syt IV (Fig.8A, lane 3).

Synaptotagmin IV and VAMP4 share a similar distribution and trafficking pattern in AtT-20 cells. A, Syt IV is on Golgi membranes in AtT-20 cells. RT-PCR analysis that used the AtT-20 cDNA library with Syt IV-specific primers showed the presence of a single reaction product of the correct size (lane 1). Reactions performed in the absence of the template yielded no product (lane 2). Immunoblot analysis of Golgi-enriched fractions from a velocity gradient performed on wild-type AtT-20 cells showed a band with the correct size that was immunoreactive toward a Syt IV-specific antiserum (lane 3). B, Colocalization of Syt IV and ACTH immunoreactivity in the Golgi region, but not at process tips (arrowhead). Cells stably transfected with Syt IV–FLAG were processed for double immunofluorescence with the anti-FLAG M2 antibody (left panel) and polyclonal ACTH antibodies (middle panel). The phase contrast image shows position of neurite and nucleus (right panel). C, Localization of both Syt IV–FLAG and FLAG–VAMP4 to ISGs. PNS from stably transfected Syt IV–FLAG or FLAG–VAMP4 cells were pooled and subjected to velocity sucrose gradient fractionation. Fractions enriched in ISGs (1, 2) were pooled and subject to a second equilibrium sucrose gradient fractionation. The fractions were analyzed by immunoblot analysis with anti-FLAG (to detect Syt IV and VAMP4) or a monoclonal antibody against rat VAMP2. The Input lane is equivalent to
 of total PNS (10 μg of proteins). Fractions containing peak Syt IV and VAMP4 immunoreactivities coincided with a peak of VAMP2 at 1.13–1.14 gm/cm3—the characteristic density of immature secretory granules (labeled ISGs). Only VAMP2 was recovered in mature secretory granule fractions (labeledMGs) with higher densities (>1.16). For comparison, fractions from a velocity gradient enriched in MSGs (3, 4) also were subjected to the same equilibrium gradient centrifugation, and the resulting fractions were immunoblotted with an anti-Syt I monoclonal antibody (bottom row). Syt I immunoreactivities were found in both ISG and MSG fractions. This experiment was repeated twice with similar results.
of total PNS (10 μg of proteins). Fractions containing peak Syt IV and VAMP4 immunoreactivities coincided with a peak of VAMP2 at 1.13–1.14 gm/cm3—the characteristic density of immature secretory granules (labeled ISGs). Only VAMP2 was recovered in mature secretory granule fractions (labeledMGs) with higher densities (>1.16). For comparison, fractions from a velocity gradient enriched in MSGs (3, 4) also were subjected to the same equilibrium gradient centrifugation, and the resulting fractions were immunoblotted with an anti-Syt I monoclonal antibody (bottom row). Syt I immunoreactivities were found in both ISG and MSG fractions. This experiment was repeated twice with similar results.
Endogenous Syt IV was expressed at a low level and was not easily detected by immunofluorescence with anti-rat Syt IV antiserum. To facilitate localization studies, we attached a FLAG epitope to the C terminus of this type I membrane protein and isolated an AtT-20 cell line stably expressing this construct. Immunofluorescence microscopy with the anti-FLAG M2 antibody shows that Syt IV–FLAG has a similar staining pattern as VAMP4. Specifically, immunoreactivity was detected in the Golgi region, which colocalized with POMC, but no immunoreactivity was observed on mature secretory granules that accumulated at the tip of neurites (Fig. 8B). In transfected COS7 cells Syt IV was colocalized with the TGN marker sialyltransferase (data not shown).
In preliminary studies we found that Syt IV–FLAG on intact membranes failed to bind to M2 beads and thus could not be used in immunoisolation experiments. We therefore used the gradient isolation procedure developed by Tooze et al. (1991) to determine whether Syt IV also was localized to ISGs. Briefly, Syt IV–FLAG-expressing cells were mixed with FLAG–VAMP4-expressing cells and fractionated on a velocity gradient. Pools of fractions from the velocity gradient then were purified further on an equilibrium gradient that separates MSGs from ISGs. An example of the equilibrium gradient separation of a slow-migrating membrane pool (enriched in ISGs) from the velocity gradient is shown in Figure 8C (top three rows). Immunoblot analysis demonstrated that both Syt IV–FLAG and FLAG–VAMP4 peaked at the same fractions containing ISGs (equilibrium densities between 1.13 and 1.14 gm/ml), but they were absent from MSGs fractionating at higher densities (>1.16). By contrast, when these same fractions were blotted with an anti-VAMP2 antibody, the endogenous VAMP2 (a SNARE protein involved in regulated exocytosis of MSGs) was present on both ISGs and MSGs (Fig. 8C, third row). For comparison, the distribution of endogenous synaptotagmin I on the equilibrium gradient was examined by using a faster migrating membrane pool (enriched in MSGs) from the velocity gradient (Fig.8C, bottom row). The synaptotagmin I immunoreactivities were found on both ISGs and MSGs, as expected. These data suggest that, like VAMP4, Syt IV is present on nascent granules but becomes removed during the maturation process.
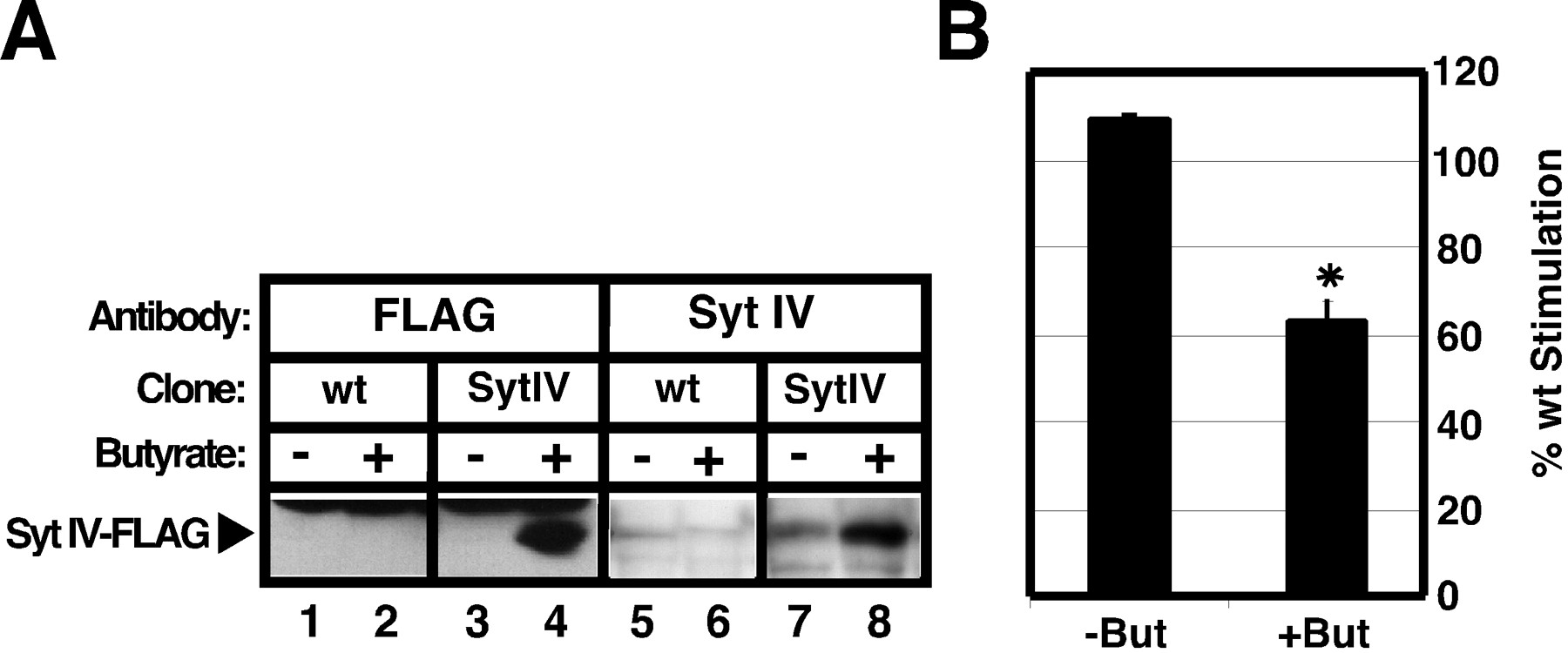
We reasoned that, if Syt IV indeed inhibits Ca2+-triggered exocytosis of ISGs, then overexpression of this protein should perturb the proper acquisition of Ca2+-triggered exocytosis during maturation. To control Syt IV expression, we isolated an inducible AtT-20 cell line that expressed Syt IV–FLAG on butyrate treatment (Grote and Kelly, 1996). Incubation of this cell line with 6 mm butyrate for 12 hr resulted in the specific expression of a protein reactive to both the FLAG antibody and the Syt IV antibody (Fig. 9A). Untransfected wild-type cells expressed much lower levels of Syt IV and showed little reactivity toward either antibody under these conditions. Wild-type cells and Syt IV–FLAG cells were induced in the presence of butyrate for 12 hr. Then the cells were pulse-labeled with [35S]sulfate for 5 min and chased for an additional 60 min before being stimulated with SNF. Control cells that were not subjected to butyrate treatment were processed in parallel for both cell types. In the absence of butyrate induction both transfected and untransfected cells showed similar stimulation indexes (Fig.9B, left bar); the stimulation index of Syt IV–FLAG cells was 110% of the stimulation index of wild-type cells. By contrast, when both cell types were treated with butyrate, a significant decrease was seen in regulated secretion from the Syt IV–FLAG cell line as compared with wild-type cells (Fig. 9B, right bar); the stimulation index of Syt IV–FLAG cells was 62% of the stimulation index of wild-type cells. Induction of Syt IV expression had little effect on constitutive/constitutive-like secretion during the 15–30 min chase, the rate of POMC processing, or POMC staining in the Golgi (data not shown). These data support a model in which Synaptotagmin IV modulates Ca2+-triggered exocytosis of maturing secretory granules in AtT-20 cells.

Synaptotagmin IV inhibits Ca2+-triggered exocytosis in AtT-20 cells.A, Twelve-well dishes containing either wild-type cells (wt; lanes 1, 2, 5, 6) or inducible Syt IV–FLAG cells (Syt IV; lanes 3, 4, 7, 8) were treated with 6 mm butyrate (But) for 12 hr (lanes 2, 4, 6, 8) or left untreated (lanes 1, 3, 5, 7) before harvesting with 1× Sample Buffer. Then the samples were subjected to immunoblot analysis by using either polyclonal antiserum against the FLAG epitope (lanes 1–4) or polyclonal antiserum against Syt IV (lanes 5–8). Specific expression of Syt IV–FLAG is seen only in Syt IV cells that were treated with butyrate (lanes 4, 8). B, Wild-type cells and Syt IV cells were induced with butyrate before a 5 min pulse labeling with [35S]sulfate. Next the cells were chased for 60 min in the continued presence of butyrate, and ACTH secretion was stimulated by the addition of SNF for 15 min. Control cells were processed in parallel except that they were not treated with butyrate. Then the samples were subjected to SDS-PAGE and quantified by using PhosphorImager analysis. The fold stimulation was determined as described before, and the percentage of wild-type stimulation was calculated as the ratio of the fold stimulation of Syt IV cells to that of wild-type cells. The asterisk indicates statistically significant differences between wild-type and Syt IV cells (p = 0.01). Values for−But represent the average of two experiments (n = 2) and the values for +Butrepresent the average of four experiments (n = 4).
DISCUSSION
The secretory state of ISGs changes as they mature
In this study we show that nascent secretory granules in AtT-20 cells are essentially stimulus-nonresponsive on budding from the TGN. Stimulus-responsiveness develops only after a crucial maturation period. At first sight, this appears to be in direct contradiction to the numerous reports showing that ISGs are stimulus-responsive (Tooze et al., 1991). However, close examination of the literature shows that there is a quantitative difference in stimulus-dependent secretion on granule maturation in all systems. In PC-12 cells the secretion of [35S]sulfate-labeled secretogranin II in response to a depolarizing stimulus was three- to fourfold at 30 min of chase but increased to >30-fold at 4 hr of chase (Tooze et al., 1991). In the rat exocrine pancreas the secretion of [35S]methionine-labeled proteins in response to carbachol stimulation was approximately threefold at 30–60 min but increased to >10-fold after 2 hr (Arvan and Castle, 1987). Likewise, the stimulated release of [35S]methionine-labeled peptidylglycine α-amidating monooxygenase, PAM-3, from AtT-20 cells increased from approximately twofold at 1 hr to approximately eightfold at 4 hr (Milgram et al., 1994). Although these studies cannot be compared quantitatively because of the use of different radiolabels (and hence cellular sites of incorporation) and secretagogues, a similar qualitative trend was found in all systems. However, the basis for the time-dependent increase in stimulation index was not investigated in these previous studies.
In our study we have established first that the increase in stimulation index corresponds to a period of granule maturation. This conclusion is based on the fact that transport of POMC from the TGN to ISGs takes place quickly (t1/2 ∼3 min;Fernandez et al., 1997), whereas changes in stimulation index occur significantly later, between 15 min to 1 hr of chase (see Fig. 1). One possible explanation is that the presence of constitutive-like vesicles (exhibiting characteristics of constitutive secretion) might have lowered the apparent “stimulation index.” With increasing chase time periods this pool diminishes, but the granule pool remains; this could result in an increase in the apparent stimulation index. The experiments with BFA, however, do not support this explanation. BFA reduces secretion from constitutive-like vesicles to 30–50% of the control levels (De Lisle et al., 1996; Fernandez et al., 1997) (see Fig. 5A,B, lanes 1, 2), but it does not affect regulated exocytosis of MSGs (see Fig. 6A,lanes 9–12). If the above explanation were correct, one would expect the overall stimulation index to increase on BFA treatment. On the contrary, we found that BFA dramatically reduced the stimulation index from 5.72 to 0.94 (see Fig. 5C). An alternative model we prefer is that granule maturation involves an actual change in the fusion characteristics of granules. Newly formed ISGs contain a mixture of vesicle trafficking proteins that confer on them an intermediate behavior between those of constitutive and regulated vesicles: secretion from these granules exhibits a lower stimulation index than bona fide regulated granules and a high basal secretory rate reminiscent of constitutive vesicles. A key aspect of granule maturation, then, is further sorting of these vesicle trafficking proteins from the ISG membrane. Such modifications are crucial in the production of granules that exhibit tight regulation by secretagogues.
The VAMP4 pathway and its relationship to granule membrane remodeling
One line of evidence supporting our model is that BFA inhibits granule membrane remodeling and locks the granules in a stimulus-insensitive secretory state (see Fig. 5). On washout of BFA, normal remodeling resumes and the granules become stimulus-responsive (see Fig. 7). Because BFA inhibits ARF exchange activities (Peyroche et al., 1999), these data suggest that granule membrane remodeling involves an ARF-mediated sorting pathway. In further support of this model, we have found that the vesicle trafficking protein VAMP4 is sorted from the ISG membrane in a BFA-sensitive manner (see Fig. 4). Moreover, the kinetics of BFA-sensitive maturation of the exocytotic behavior of granules (see Fig. 6) closely paralleled the kinetics of BFA-sensitive removal of VAMP4 from the ISG (see Fig.3C)—both showing t1/2∼30 min. The simplest explanation for our data is that VAMP4 provides a marker for the sorting pathway that is critical for remodeling the secretory response of the granule. Our observation of a VAMP4 sorting pathway from the ISG is unlikely an overexpression artifact. Because of limitations of reagents that are usable for immunoisolation experiments, we could analyze only the trafficking of exogenous FLAG–VAMP4 protein that was expressed at an approximately fivefold higher level than the endogenous protein (see Fig.2B). However, recent immunoelectron microscopic studies have shown that endogenous VAMP4 immunoreactivity in PC-12 cells was found on ISGs in addition to the TGN and endosomes (Steegmaier et al., 1999), consistent with our immunoisolation studies of exogenous FLAG–VAMP4 in AtT-20 cells. At present it is unknown whether constitutive-like secretion and granule membrane remodeling result from the same or a different trafficking event and what role VAMP4 plays in these processes.
It should be noted that, although nascent secretory granules exhibit properties reminiscent of constitutive vesicles, they are not bona fide constitutive carriers. The half-time for constitutive secretion of GAG chains from the TGN to the cell surface is ∼30 min or ∼2% secreted per minute (Fernandez et al., 1997). If granules aged in BFA behave as true constitutive carriers, their contents would be secreted rapidly and virtually nothing would remain in the cells after 6 hr of chase. This is clearly not the case (see Fig. 5B, lanes 7–10). Instead, the unstimulated rate of ACTH secretion from BFA-treated cells was significantly slower than constitutive secretion, estimated to be ∼0.1% per min.
Synaptotagmin IV inhibits Ca2+-triggered exocytosis
Our studies have uncovered a key step during secretory granule biogenesis that involves membrane sorting along the VAMP4 pathway. Because remodeling results in an increase in the exocytotic response of granules to Ca2+, an attractive hypothesis is that a key inhibitor of Ca2+-triggered exocytosis is removed from the granule membrane during this process. One candidate for this inhibitor is Syt IV, which recently has been shown to inhibit Ca2+-triggered exocytosis of synaptic vesicles in neurons as well as in PC-12 cells (Littleton et al., 1999; Thomas et al., 1999). In support of this model we find that Syt IV is expressed in AtT-20 cells and has a trafficking pattern identical to VAMP4. Specifically, Synaptotagmin IV is found on the Golgi and ISGs, but not on the mature granule, suggesting removal of this protein during maturation (see Fig. 8). Furthermore, overexpression of Syt IV during maturation reduces the ability of granules to respond to SNF by ∼40–50% (see Fig. 9) similar to what is seen with BFA (∼60% inhibition of SNF response; see Interference of the VAMP4 Pathway in Results). Taken together, these data suggest that removal of Syt IV via the VAMP4 pathway is a key step in the biogenesis of Ca2+-responsive secretory granules in AtT-20 cells. A working model for our findings is shown in Figure 10.

Shown is a working model for granule membrane remodeling during maturation (see Discussion).
Recently, Ibata et al. (2000) examined the subcellular distribution of Syt IV in PC12 cells and cultured hippocampal neurons and found that Syt IV was localized to the Golgi and the tips of neurites. However, in contrast to the studies of Thomas et al. (1999), they found that Syt IV signals were not colocalized well with Syt I. Our finding that Syt IV is present on ISGs, but not Syt I-containing MSGs in AtT-20 cells (see Fig. 8C), is consistent with the results of Ibata et al. (2000). Because the Syt IV mRNA level is upregulated during perinatal development and by depolarization, our data raised an interesting possibility that Syt IV-containing ISGs may play a role in synaptic growth during neuronal development and/or induction of synaptic plasticity.
Comparison with other systems
As discussed above, granule maturation generally involves an increase in the stimulation index. There are, however, significant differences among cell types. In AtT-20 cells we observed a period between 15 and 30 min of chase during which very little regulated exocytosis could be induced from ISGs (see Fig. 1A). In PC-12 and pancreatic islet cells, regulated exocytosis could be detected as early as 10 min after vesicle budding from the TGN (Tooze et al., 1991; Kuliawat and Arvan, 1992). These differences may reflect differences in membrane trafficking patterns because of differing physiological demands. For instance, most tumor cells exhibit a relatively high level of constitutive and constitutive-like secretion: ∼50% of pulse-labeled POMC was secreted from AtT-20 cells via the constitutive and the constitutive-like pathway (Fernandez et al., 1997), compared with <10% for secretogranin II in PC-12 cells or proinsulin in islet cells (Tooze et al., 1991; Kuliawat and Arvan, 1992). Upregulation of the constitutive-like pathway may provide a useful mechanism for tumors to enhance their secretion and stimulate cell growth in an autocrine manner. In this regard it is interesting to note that VAMP4 contains an acidic dileucine motif and an acidic cluster motif at its N terminus, both of which are potential phosphorylation sites. The presence of these motifs also suggests that VAMP4 may be removed from the ISGs via an AP3 or PACS-mediated (Molloy et al., 1998) mechanism. A better understanding of these trafficking steps during granule maturation will help to elucidate how they are regulated dynamically in different physiological states.
In summary, we have identified a pathway that functions to remove an inhibitor of Ca2+-triggered exocytosis during the maturation of secretory granules. Our identification of VAMP4 as a marker of this sorting pathway should facilitate the future study of granule membrane remodeling and its dynamic regulation in different physiological states.
Footnotes
This work was supported by grants from the Public Health Service (GM 35239), National Science Foundation (MCB-9983342), University of California Cancer Research Coordinating Committee, and University of California Faculty Research Grant (to H.-P.M.); a postdoctoral fellowship from the American Heart Association (to M.H.); Molecular Cell Biology Predoctoral Training Grant (to B.A.E. and D.L.); and Cancer Research Lab Training Grant (to B.A.E.). We thank Dr. Harvey Herschman for the generous gift of the anti-synaptotagmin IV antibodies, and we thank members of the Moore lab for helpful discussion.
Correspondence should be addressed to Dr. Hsiao-Ping Moore, Department of Molecular and Cell Biology, University of California at Berkeley, 142 Life Sciences Addition #3200, Berkeley, CA 94720-3200. E-mail:hpmoore{at}uclink4.berkeley.edu.
Dr. Eaton's present address: Department of Biochemistry Biophysics, University of California, San Francisco, CA 94143.
Dr. Haugwitz's present address: Clontech, 1020 East Meadow Circle, Palo Alto, CA 94303.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}