

Abstract
The orphan nuclear receptor Nurr1 is required for the development of ventral mesencephalic dopaminergic neurons in mice. One of the possible mechanisms that might contribute to the regulation activity of Nurr1 is through interaction with other proteins. To identify potential partners of Nurr1, we screened a yeast two-hybrid library from developing mouse embryonic mesencephalon with the Nurr1 ligand-binding domain (NLBD). We identified a novel interacting protein, termed the Nurr1-interacting protein (NuIP). We demonstrate that it specifically interacts with NLBD using the mammalian two-hybrid assay and coimmunoprecipitation studies in MN9D cells. In addition, we show that NuIP interacts with Nurr1 in lysates from substantia nigra. Coexpression of NuIP with Nurr1 results in potentiation of the transcriptional activity of Nurr1 on an nerve growth factor inducible-B response element reporter, as well as reporters driven by the endogenous tyrosine hydroxylase promoter. The mechanism underlying the regulatory action of NuIP on Nurr1 is demonstrated to be through assembly of distinct helical domains of the NLBD. Using a NuIP specific antibody, we show that expression of NuIP protein is mainly colocalized with Nurr1 in adult midbrain dopaminergic neurons. Finally, we demonstrate that suppression of NuIP expression in MN9D cells by NuIP-specific small interfering RNA leads to decreased cell division and decreased expression of a Nurr1 target gene, the dopamine transporter. These results suggest NuIP interacts with and positively regulates the activity of Nurr1 protein and modulates the phenotype of dopaminergic cells.
Introduction
Development of mesencephalic dopaminergic neurons in mice requires the expression of the transcription factor Nurr1 (also known as NR4A2). Loss of Nurr1 function through gene targeting results in the failure of midbrain progenitors to complete specification of the dopaminergic lineage (Zetterström et al., 1997). Nurr1, an orphan member of the nerve growth factor inducible-B subfamily of nuclear receptors (NRs), has no known activating ligand. Recent crystallographic analysis of the Nurr1 ligand-binding domain (NLBD) demonstrates a ligand pocket that is apparently too confining to accommodate lipophilic ligands, such as steroids (Wang et al., 2003). This suggests that Nurr1 functions in a ligand-independent manner but does not address how its transcriptional activity is regulated. Among the mechanisms that may contribute to modulating its transcriptional activity include posttranslational modification and/or interaction with other proteins that induce the adoption of an “activated” NLBD structure. Indeed, studies on the assembly of an active NLBD in HEK293 cells has implicated c-ret signaling as a negative regulator (Wang et al., 2003). With the exception of formation of transactivating heterodimeric complexes with other NRs, such as retinoid-X receptor α (RXRα) (Perlmann and Jansson, 1995), all other interactors identified thus far, such as p57kip2 and PIASγ (Joseph et al., 2003; Galleguillos et al., 2004), are negative regulators of Nurr1 transcriptional activity.
Given the postulated ligand independent regulation of Nurr1, we sought to identify additional proteins that would interact with, and modulate the action of, Nurr1 in the developing midbrain. Screening a yeast two-hybrid library prepared from developing mouse embryonic mesencephalon for NLBD interactors has resulted in the identification of a new family of gene products that interact with and regulate the activity of Nurr1. This family of gene products arises from alternative splicing from a single gene, among which we have termed the longest product the Nurr1-interacting protein (NuIP). Using a mammalian two-hybrid assay in a dopaminergic cell line, MN9D cells, we now report that NuIP indeed interacts with both the full-length NLBD and the AF2-deleted NLBD, suggesting it interacts with Nurr1 by a mechanism dissimilar from other known modulators such as RXRα. Interaction of Nurr1 and NuIP is further demonstrated by coimmunoprecipitation in MN9D cells and endogenous substantia nigra (SN) lysates. When coexpressed with Nurr1, NuIP protein potentiates the transcriptional activity of Nurr1 using either an nerve growth factor inducible-B response element (NBRE)-containing reporter or endogenous tyrosine hydroxylase (TH) promoter reporter constructs. To test the mechanism by which NuIP regulates Nurr1 activity, we used an NLBD assembly assay and demonstrated that NuIP can indeed promote the assembly of NLBD. Using a newly developed polyclonal antibody generated against an NuIP peptide, we show that the protein is extensively colocalized with Nurr1 in adult midbrain dopaminergic neurons. Finally, to evaluate the endogenous function of NuIP in dopaminergic cells, we suppressed the expression of NuIP gene in MN9D cells by small interfering RNA (siRNA) and found that loss of NuIP function led to decreased cell number in culture and decreased expression of a Nurr1 target gene, the dopamine transporter (DAT). Together, these results suggest that a novel gene encodes a protein termed NuIP, which interacts and positively regulates the activity of Nurr1 protein possibly by promoting the assembly of the NLBD.
Materials and Methods
Two-hybrid screening.
pPC97-NLBD589A was transformed into the yeast strain YRG-2 (Mata ura 3-52his 3-200ade 2-101 lys 2-801trp 1-901leu 2-3 112gal 4-542gal 80-538LYS2::UASGAL1-TATAGAL1-HIS3 URA3::UASGAL4 17mer(x3)-TATACYC1-lacZ) (Stratagene) together with a 13.5 d embryonic mouse ventral mesencephalic cDNA library fused to a Gal4 activation domain pPC86 vector (Chevray and Nathans, 1992). Library screening was performed by using HIS3 and LacZ reporters as described (Stratagene). Candidate interacting clones were purified and retransformed into yeast to confirm the interaction with bait constructs. Confirmed positive clones were identified by sequencing analysis.
Plasmids.
The different bait constructs were generated by cloning variant Nurr1 cDNA fragments in-frame with the Gal4 DNA-binding domain coding sequence in pPC97 vector. Oligonucleotides listed below were used to amplify the full-length NLBD or the truncated NLBD (NLBD583) from the vector pBSNurr1 (kind gift from Dr. B. Hoffer, National Institutes of Health–National Institute on Drug Abuse, Bethesda, MD) by PCR using pfu polymerase: Nurr1, 5′-(5′-TAGAGTCGACGGCAGCCATGCCTTGTGTTCAGGCG), 3′-(5′-CTAGGCGGCCGCGGGAGAAGGTCTTAGAAAGGTAA); NLBD, 5′-(5′-TAGAGTCGACCCAGGATCCCTCTCCCCCCTCACCT), 3′-(5′-CTAGCGGCCGCGGGAGAAGGTCTTAGAAAGGTAA); NLBD583, 5′-(5′-TAGAGTCGACCCAGGATCCCTCTCCCCCCTCACCT), 3′-(5′-CTAGCGGCCGCTTATGGTACCAAGTCTTCCAATTT).
For each of these oligonucleotides, the 5′ primer introduced a unique SalI site to the 5′ of the coding sequence and the 3′ primer introduced a unique NotI site downstream of the stop codon. The Nurr1 fragment and the pPC97 vector were double digested with SalI and NotI, respectively, and the fragments purified and ligated by T4 DNA ligase. The pPC97Nurr1589A and pPC97NLBD589A plasmids were generated by site-directed mutagenesis.
The pPC86RXR construct was generated by cloning full-length human RXRα cDNA in-frame with the Gal4 DNA activation domain coding sequence in the pPC86 vector. Oligonucleotides listed below were used to amplify the full-length hRXRα from the vector pCMXhRXRα by PCR using pfu polymerase: (5′-CTG GGA ATT CAC ATG GAC ACC AAA CAT TTC) and (5′-CTA AGC GGC CGC CTA AGT CAT TTG GTG CGG). Amplified PCR fragments were inserted into the pPC86 vector. Oligonucleotides used in all studies were synthesized by Integrated DNA Technologies. All sequences were verified by sequencing (Integrated DNA Technologies).
The pHSV-green fluorescent protein (GFP)/Nurr1 construct contains the coding sequences for both humanized Renilla reniformis GFP and Nurr1 under the control of separate promoters in the plasmid HSVPrPucλ3CMV and was generated as described previously (Luo and Federoff, 2003). A reporter construct containing three copies of NBRE upstream of a minimal promoter driving luciferase was generated as described previously (Luo and Federoff, 2003). Reporter constructs containing −3kb, −6kb, or −9kb for the rat TH promoter were constructed by Dr. Kathleen Maguire-Zeiss (Georgetown University, Washington, DC). Briefly, the fragment of endogenous rat TH promoter of −9kb, −6kb, or −3kb was cloned into the HSVlacZ vector upstream of a LacZ reporter gene. Flag-tagged full-length Nurr1 expression vector was generated by cloning a Nurr1 PCR fragment in-frame fused to the 3′ terminal of a Flag sequence into a pcDNA3 vector (Clontech).
The full-length NuIP gene was cloned by reverse transcriptase (RT)-PCR using cDNA library from embryonic 13.5 (E13.5) mouse ventral midbrain as template and the following oligos as primers: (5′-TAGAGTCGACGGAACCGGGCACCGACCAGCTTGAGCCA) and (5′-CTAGTCTAGACTTGTTCTCAATTAGAATCTG). The amplified PCR fragment was inserted into the SalI and XbaI sites of the HSVPrPuc expression vector, and the sequence was confirmed by sequencing analysis (Integrated DNA Technologies). VP16NuIP was generated by cloning a PCR fragment of NuIP into the SalI and XbaI sites of pVP16 vector (Clontech) in-frame with the VP16 activation domain using a different 5′ primer: (5′-TAG AGT CGA CGG CAC CGA CCA GCT TCA GCC A-3′). NuIPV5 was generated by cloning a NuIP PCR fragment in-frame fused to the 5′ terminal of a V5 sequence into a modified pVP16 vector in which the VP16 activation domain sequence is deleted.
The Gal4H1 construct was cloned by inserting a PCR fragment containing the helix 1 (H1) sequence of NLBD into the pPM vector (Clontech) in-frame with the Gal4 DB sequence. The VP16H3–12 construct was cloned by inserting a PCR fragment containing the helix 3–12 sequence of NLBD into the pVP16 vector (Clontech) in-frame with the VP16 activation domain sequence. The following primers were used to generate the PCR fragments: H1 5′-(5′-TAGAGTCGACCGAAGAGCCCACAGGATCCCTCT), 3′-(5′-CTAGTCTAGAATCTCCACTCATCTGATAGTCAGG); H3–12 5′-(5′-TAGAGTCGACATGATACCCAACATATCCAGCAG), 3′-(5′-CTAGTCTAGAGGGAGAAGGTCTTAGAAAGGTAA).
Oligonucleotides used in all studies were synthesized by Integrated DNA Technologies. All constructions were verified by sequencing (Integrated DNA Technologies).
Transfections and reporter assay.
MN9D cells were plated at 1.5 × 105 cells/well in 24-well plates coated with polyethyleneimine 24 h before transfection. Cells were washed with Optimem (Invitrogen) and incubated with DNA and 1.5 μl of Lipofectamine 2000 (Invitrogen) in Optimem for 8 h. Each well was transfected with 100 ng of individual reporter construct, 500 ng of pHSV-GFP/Nurr1, and 250 ng of HSVNuIP or pBS as carrier DNA. Fifty ng of pRL-Null reference plasmid containing the Renilla luciferase gene was used as an internal control and for normalization of transfection efficiency. After an 8 h incubation, the transfection reagents were removed and replaced with DMEM with 10% FBS. Luciferase and Renilla luciferase activities were assayed 24 h later using the Dual-Luciferase reporter assay system according to the manufacture's instructions (Promega). Luciferase activities were normalized to the Renilla luciferase activity. Each assay was performed a minimum of three times for each condition and average values ±SD are shown for each sample.
Mammalian two-hybrid assay.
The interaction between Nurr1 and NuIP was examined using the mammalian two-hybrid assay. MN9D cells were cotransfected with pGal4NLBD or pGal4NLBD583 and pVP16NuIP constructs along with a reporter gene driven by five copies of the Gal4-binding sites. The cells were subsequently harvested and analyzed as described above for reporter activity.
Nurr1 LBD assembly assay.
The assembly of H1 and H3–12 domain of NLBD was assessed by using the NLBD assembly assay (Wang et al., 2003). HEK293 cells were cotransfected with the reporter construct, Gal4H1, and VP16H3–12 together with HSVX NuIP or pBS as carrier DNA. The cells were then harvested and analyzed for luciferase activity and normalized to reference Renilla luciferase activity. Each condition was measured with triplicate samples, and each experiment was repeated at least three times with similar results.
Development and characterization of NuIP specific antibody.
An NuIP specific peptide (CVMDGWPGEADKPSRA) was used to immunize rabbits (Affinity Bioreagents). After three immunization boosts, immune sera containing IgG were purified and tested for specificity for the NuIP protein. Briefly, a clonal MN9D cell line was transfected with either a control plasmid (HSVlacZ) or a NuIP-expressing vector (HSVNuIP). Twenty four hours after transfection, cell lysates were prepared using modified radioimmunoprecipitation assay (RIPA) buffer. Total protein (20 μg) from each condition was separated on an 8% SDS-PAGE gel and transferred to a polyvinylidene fluoride (PVDF) membrane for Western blot analysis. The membrane was incubated with 1 μg/ml NuIP antibody for 1 h at room temperature, followed by a 45 min incubation of secondary HRP-conjugated anti-rabbit antibody (Jackson ImmunoResearch Laboratories). For peptide preabsorption, the NuIP antibody solution was preincubated with an excess of the NuIP specific peptide (10 μg/ml) for 30 min at room temperature before the incubation with membrane.
Coimmunoprecipitation.
MN9D cells were cotransfected with a NuIP-V5 expression vector and a Flag-Nurr1 expression vector. Twenty four hours after transfection, nuclear extracts were prepared by NE-PER nuclear extraction reagent (Pierce Chemical). Nuclear lysate was precleared with Protein A beads and was incubated with rabbit polyclonal anti-V5 antibody (Novus Biologicals) in a modified RIPA buffer or with RIPA buffer only (no antibody) as a control. Protein A beads were incubated with the antibody/buffer:lysates for 1 h, washed five times with RIPA buffer, and bound proteins eluted by boiling in Laemmli sample buffer. Proteins were subjected to denaturing PAGE (6% SDS-PAGE), transferred to PVDF membrane, and probed with a monoclonal V5 antibody (Invitrogen) or a monoclonal Flag antibody (Sigma-Aldrich). Coimmunoprecipitation of endogenous Nurr1-NuIP complexes was performed by incubating 120 μg of mouse SN lysates with rabbit polyclonal anti-Nurr1 antibody (Novus Biologicals) in PBS or with PBS only (no antibody) as a control. Use of protein A beads, their incubation, and washing procedures were performed as described above. Proteins were separated by SDS-PAGE and blotted with rabbit polyclonal anti-NuIP antibody (described above).
RT-PCR.
Total RNA from cells was extracted by Trizol reagent (Invitrogen). Total RNA (2 μg) was treated with RQ-1 RNase-free DNase I and reverse transcribed into cDNA using random hexamers by avian myleoblastis virus reverse transcriptase as recommended by manufacturer's protocol (Roche Diagnostics). Synthesized cDNAs were then subject to PCR amplification using the following primers to detect different transcripts: NuIP: forward primer (5′-TTGAAGTGGACACCCAATCAGC), reverse primer (5′-CAGGTCTGGAGACACGATCATGTG); NuIPa: forward primer (5′-CTCTGGCTTCCCACAGTCTCCC), reverse primer (5′-CAGGTCTGGAGACACGATCATGTG); NuIPb: forward primer (5′-CTCTGGCTTCCCACAGTCTCCC), reverse primer (5′-GCTTCTTGCAGACAACAGCAGG); NuIPc: forward primer (5′-TTGAAGTGGACACCCAATCAGC), reverse primer (5′-GCTTCTTGCAGACAACAGCAGG); Nurr1: forward primer (5′-ATTCCAATCCGG CAATGACC), reverse primer (5′-TTGCAACCTGTGCAAGACCAC).
Supression of NuIP expression by siRNA.
MN9D cells were maintained at 37°C and 5% CO2 in DMEM with l-glutamine and 4500 mg/L glucose (Sigma-Aldrich) and 3.7 g of sodium bicarbonate per litter added, pH 7.4, and 10% fetal bovine serum. Inducible cell lines harboring NuIP short hairpin RNA (shRNA) constructs were prepared in MN9D cells by stable transfection with pcDNA6/TR vector, selection and expansion in the presence of 5 μg/ml blasticidin for 2 weeks, and screening for induction of expression of a vector-based lacZ reporter gene (pcDNA4/TO/lacZ) by β-galactosidase assay (Invitrogen). The clone expressing the highest level of functional tetracycline repressor was then used to establish inducible NUIP RNA interference (RNAi) cell lines. Short hairpin DNA sequences against the coding sequence of NuIP mRNA were designed using two different approaches (Reynolds et al., 2004; Huesken et al., 2005) and cloned into pSUPERIOR.neo+GFP vector (Oligoengen) following manufacturer's instructions. Multiple hairpin constructs were screened for effective knockdown of NuIP. Of these different constructs two were used in this study and are described as follows: RNAi#1, 5′-GTACCAGATCCTCTCCAGA-3′; and RNAi#2, 5′-CCCGGGACCTCGTGCATAA-3′.
To check NuIP mRNA knockdown, total RNA was extracted using the RNeasy kit (QIAGEN) from appropriate cell samples 24 h after induction of shRNA expression, followed by cDNA synthesis using the high capacity cDNA Archive kit (Applied Biosystems) and quantitative RT-PCR (qRT-PCR). The probes for qRT-PCR of NuIP (Taqman Gene Expression Assays Mm00554850 m1; Applied Biosystems) amplify 140 nt around exon boundary 21–22 with the assay location at 2845 of the cDNA (NM_172718.1). NuIP mRNA levels were normalized to 18 s rRNA using probes purchased from Applied Biosystems. qRT-PCR was performed in triplicate.
Cell number counting.
Three days after induction of shRNA expression in tet repressor-expressing MN9D cells in 12-well plates, the cells were trypsinized and counted using a hemocytometer. The percentage of the number of the cells with shRNA expressed (+DOX) versus the cells without shRNA expressed (−DOX) was calculated. The data shown in Figure 10 represented an average of seven independent assays each performed in duplicate.
Western blotting and immunocytochemistry.
To measure suppression of NuIP protein by siRNA, tet repressor-expressing MN9D cells were plated 1 d before transfection in polyethyleneimine-coated dishes. The cells were then transfected with either null pSUPERIOR.neo+GFP vector (mock) or constructs containing RNAi#1 and RNAi#2 sequences, using Transfectamine 2000 diluted in Opti-MEM (Invitrogen), according to the manufacturer's protocol. Doxycycline (DOX) was added to a final concentration of 2 μg/ml 24 h after transfection to induce shRNA expression. The cells were collected 72 h after induction for Western blot analysis of various proteins. Total protein (20 μg) from each condition was separated on an 8% SDS-PAGE gel and transferred to a PDVF membrane for Western blot analysis. The blots were then probed by various antibodies: anti-NuIP Ab (described above), anti-DAT (Novus Biologicals), anti-TH (Millipore) and anti-α Tubulin (Santa Cruz Biochemicals).
For NuIP and TH double-labeling immunocytochemistry, adult male C57BL/6J mice (2–3 months) were anesthetized and perfused with 4% paraformaldehyde (PFA), pH 7.2. Brains were removed and postfixed in 4% PFA for 24 h, followed by immersion in 20% sucrose in 0.1 m phosphate buffer, pH 7.2, for 3 d and immersion in 30% sucrose for 1 week. The brains were sectioned at 30 μm on a microtome and mounted on gelatin-coated slides. Immunohistochemistry was performed to identify TH-immunoreactive (IR) and NuIP-IR cells in the SN. Briefly, sections were blocked in PBS containing 10% normal goat serum (Jackson ImmunoResearch Laboratories), 2% BSA, and 0.3% Triton X-100 for 3 h, followed by incubation in primary antibody (mouse anti-TH, 1:500; rabbit anti-NuIP, 1:1000; INCSTAR) overnight at 4°C. After washes in PBS, sections were incubated with Alexa Fluor 594 conjugated goat anti-rabbit secondary antibody (1:1000; Invitrogen) and Alexa Fluor green 488 conjugated goat anti-mouse secondary antibody (1:1000; Invitrogen) for 1 h at 25°C. After additional washes (3 ×) with PBS, the slides were examined under a confocal microscope (Olympus BX50WI; Olympus FluoView).
Results
Identification of a Nurr1 interacting protein
We set out to identify potential Nurr1 interacting proteins using a yeast two-hybrid systems. A mouse E13.5 ventral midbrain library was constructed in the yeast two-hybrid vector PC86. A set of Gal4-Nurr1 fusion constructs were prepared (Fig. 1), which included the full-length Nurr1 (GalNurr1), the entire Nurr1 LBD (GalNLBD), Nurr1 LBD deleted of AF2 (Gal4NLBDρ583), and a full-length Nurr1 LBD carrying a transcription inactivating mutation in AF2 (Gal4NLBD589A). Attributable to the intrinsic activity of the AF2 domain within the Nurr1 LBD domain (Table 1), the library was screened with a construct containing the inactivating mutation Gal4NLBD589A. The selection produced several interactors, only one of which was subsequently confirmed on replication. This clone, provisionally termed NuIP, contained a partial opening reading frame corresponding to a gene of unknown function. To confirm the specificity of the interaction and determine whether the interaction interface with Nurr1 LBD was similar to that of RXRα, additional studies in yeast were undertaken. As shown in Table 2, pPC86-NuIP, unlike RXRα, interacted with the LBD of Nurr1 devoid of its AF2 domain. This suggests that the protein interface in the NLBD that interacts with NuIP is different from that of RXRα.

Nurr1 bait constructs used in the yeast two-hybrid assay. Different forms of Nurr1 cDNA were cloned in-frame with the Gal4 DNA binding domain and were subsequently tested in yeast two-hybrid assay as bait constructs.
Activity of reporter genes with various Nurr1 bait constructs
Identification of a positive interactor for Nurr1 protein
In silico analysis (Vector NTI; Invitrogen) of the partial NuIP ORF indicated that it belongs to a family of transcripts that is expressed in both human and mouse. Reconstruction of the transcription unit from disparate DNA sequence data suggests a single gene with four alternatively processed transcripts (Fig. 2A). To confirm the existence of each transcript, specific PCR primers were designed, and RNA from different adult tissues from the mouse was examined by RT-PCR. As shown in Figure 2B, all transcripts were expressed, but the patterns of expression indicate preferential abundance of some isoforms in some tissues and not others. There was general concordance of expression of full-length NuIP, NuIPa, and NuIPc in midbrain cortex, striatum, cerebellum, pons/medulla, and embryonic ventral mesencephalon. This overall pattern mirrors the expression of Nurr1. The expression of NuIPb was observed only in cerebellum and embryonic ventral mesencephalon. No expression of any NuIP isoform was detected in the spleen or the heart, but weak expression patterns of full-length NuIP and NuIPc, as well as Nurr1, were observed in kidney. Based on our analysis of mRNA products, we cloned a full-length cDNA encoding the longest isoform of NuIP from mouse prenatal midbrain (E13.5). This gene has a predicted ORF spanning 3.3 kb and encodes a protein product of 1093 aa (Fig. 3). Importantly, the high degree of correlation between Nurr1 and NuIP mRNA expression (Fig. 2B) in multiple brain regions provides a biological context for their potential to interact at the protein level in these tissues.

Alternative splicing of NuIP gene and the tissue distribution of different transcript isoforms. A, Schematic representation of the putative alternatively spliced isoforms of NuIP gene. B, RT-PCR, using primers specific for the alternatively spliced NuIP transcripts, Nurr1, and GAPDH, were performed on mRNAs extracted from different mouse tissues. Nurr1 and full-length NuIP transcripts were consistently coexpressed in the various tissues examined.

NuIP ORF. The conceptual amino acid sequence of the longest ORF encoded by a full-length NuIP transcript.
Nurr1 and NuIP functionally interact in mammalian cells
To examine the functional relationship of Nurr1 and NuIP, a mammalian two-hybrid interaction approach was used. The strong transcriptional activator HSV VP-16 was fused to full-length NuIP and cotransfected with the Gal4DB, Gal4NLBD, or GAL4NLBDΔ583, together with a Gal4 responsive reporter into the MN9D dopaminergic cell line (Hermanson et al., 2003). As shown in Figure 4, significant transactivation was observed with both NLBD constructs, suggesting a functional interaction in this mammalian dopaminergic cell line. In contrast to the reported RXR interaction with Nurr1, NuIP interacts with an NLBD construct lacking the AF2 domain (NLBD583) (Fig. 4), an observation in agreement with the yeast interaction data.

NuIP interacts with full-length NLBD and NLBD583. The interaction between NuIP and Nurr1 was confirmed by a mammalian two-hybrid assay. MN9D cells were transfected with a luciferase reporter gene driven by five UAS Gal4-binding sites and with bait constructs (Gal4DB, GAL4NLBD, or GAL4NLBD583). Putative interacting constructs (VP16, control; VP16-NuIP) were also cotransfected to determine whether they would stimulate transcription by bringing together the TAD of VP16 with the GAL4 DNA binding domain of the bait constructs. Data are expressed as average relative units ±SD. Significant differences (*p < 0.01) were observed in the presence of VP16-NuIP using Student's t test. RLU, Relative light units.
Evidence for interaction of NuIP and NLBD in mammalian cells was also demonstrated by coimmunoprecipitation studies. In these experiments, MN9D cells were transfected with full-length and epitope-tagged constructs of Nurr1 and NuIP and then subjected to epitope specific immunoprecipitation followed by SDS-PAGE and Western blotting. As shown in Figure 5, immunoprecipitation of NuIPV5 resulted in the coprecipitation of protein Nurr1FLAG. To test whether endogenous Nurr1 and NuIP protein interact with each other in SN tissue, rich in dopaminergic neurons, we also performed coimmunoprecipitation in tissue lysates. The result shows that endogenous NuIP protein is coimmunoprecipitated by a rabbit polyclonal anti-Nurr1 antibody (Fig. 5C).

Coimmunoprecipitation of NuIP and Nurr1. MN9D cells were cotransfected with a NuIP-V5 expression vector and a Flag-Nurr1 expression vector. Twenty four hours after transfection, nuclear extracts were prepared by NE-PER nuclear extraction reagent (Pierce Chemical). Nuclear lysates were subjected to coimmunoprecipitation as described in Materials and Methods. A, B, Immunoprecipitated NuIP-V5 protein was detected with a monoclonal V5 antibody (A), and Flag-Nurr1 protein that coimmunoprecipitated with NuIP protein was detected with a monoclonal anti-Flag antibody (B). Arrows indicate the migration positions of NuIP-V5 and Flag-Nurr1 proteins. To test the interaction of endogenous Nurr1 and NuIP protein, mouse SN tissue lysates were incubated with rabbit anti-Nurr1 antibody or no antibody control and pulled down by Protein A/G beads. C, Immunoprecipitated proteins were separated by SDS-PAGE gel and blotted by a rabbit anti-NuIP protein as described in Materials and Methods. Arrows indicate the migration position of NuIP protein.
NuIP augments the transcriptional activity of Nurr1
The transactivation function of Nurr1 has been shown using a hybrid promoter containing NBRE cis-elements (Wilson et al., 1991) and, more recently, using the promoter driving expression of TH (Iwawaki et al., 2000). To examine whether NuIP can modify the functional properties of Nurr1 full-length constructs of each were prepared in mammalian expression vectors and then used in standard promoter reporter transfection studies in MN9D cells. Transfection of the NBRE-reporter construct alone produced a small but detectable amount of gene expression (Fig. 6). Cotransfection with full-length Nurr1 resulted in a marked transcriptional enhancement. In contrast, cotransfection of full-length protein NuIP along with the reporter construct alone produced no change in reporter gene expression. Interestingly, cotransfection of Nurr1 plus NuIP resulted in significantly increased gene expression. This was not attributable to an effect on the abundance of Nurr1 in these transfected cells as assessed by Western blotting (data not shown). A similar set of experiments was undertaken in MN9D cells, testing whether the same set of effector constructs, Nurr1 and NuIP, would modify the transcriptional activity of the TH promoter. As shown in Figure 7, NuIP consistently increased the transcriptional activity of Nurr1 on all three TH constructs. Together, these results indicate that NuIP augments the transcriptional activity of Nurr1.

NuIP potentiates the activity of Nurr1 on NBRE. MN9D cells were cotransfected with an NBRE-containing luciferase reporter construct and Nurr1 or Nurr1 plus NuIP. Cell extracts were subsequently assayed for luciferase activity in triplicate samples. The data are presented as mean values ±SD, and the experiments were repeated three times with similar results. *p < 0.05 compared with Nurr1 alone using Student's t test. RLU, Relative light units.

NuIP augments Nurr1 transcriptional activity on the TH promoter in MN9D cells. MN9D cells were transfected with different length TH promoter constructs (TH3, TH6, and TH9 kb promoters driving β-galactosidase) together with either expression vectors encoding Nurr1, NuIP, or Nurr1 plus NuIP to determine which constructs would stimulate TH promoter-reporter gene expression. Nurr1 stimulated transcription of the 3kb, 6kb, and 9kb promoter-reporter constructs. Addition of the NuIP construct augments Nurr1-dependent transcription. NuIP transfection alone produced no stimulation. The data are presented as mean values ±SD for triplicate samples, and the experiments were repeated three times with similar results. *p < 0.05 compared with Nurr1 alone using Student's t test. RLU, Relative light units.
NuIP protein promotes the assembly of helical domains 1 and 3–12 of the Nurr1 LBD
To explore the possible mechanism whereby NuIP potentiates the transcriptional activity of Nurr1, we tested whether NuIP can promote the assembly of H1 domain and H3–12 domains of the NLBD, which have been shown to correlate with the transcriptional activity of NLBD (Wang et al., 2003). The assay is designed to examine whether NuIP can serve as a scaffold for the binding of the separate helical domains and promote their assembly into a transcriptionally competent complex (Fig. 8A). As shown in Figure 8B, and consistent with previously reports, the H1 domain and H3–12 domains interact with each other in transfected HEK293 cells, as demonstrated by increased activity of the reporter gene. Importantly, when cotransfected with NuIP, this interaction is significantly augmented (p < 0.001).

NuIP potentiates the assembly of H1 and H3–12 domains of NLBD. The ability of NuIP to augment the assembly of H1 and H3–12 of the NLBD was examined using the assembly assay in HEK293 cells. Cells were transfected with Gal4H1 with either VP16 alone or VP16H3–12 in the presence or absence of NuIP. Addition of NuIP construct further augments the assembly of the NLBD (*p < 0.001, Student's t test; mean ±SD). Each condition consisted of triplicate samples, and the experiments were repeated three times with similar results. RLU, Relative light units.
NuIP protein is expressed in Nurr1 containing SN dopaminergic neurons
To further test whether NuIP is expressed in Nurr1 containing cells, specifically SN dopaminergic neurons, we raised polyclonal antisera to a unique NuIP peptide. The specificity of the NuIP antibody was confirmed by Western blot and peptide preabsorption. This antibody reveals a band that is the approximate predicted molecular weight of the NuIP protein (150 kDa) only in the cell lysates that are transfected with a NuIP expressing construct (HSVNuIP) (Fig. 9A) and not in lysates from cells transfected with a control plasmid (HSVlacZ). When we preincubated the antibody with an NuIP specific peptide, the NuIP signal was lost, indicating that the antibody specifically recognizes NuIP. Blotting with a β-actin antibody confirmed equivalent protein loading (Fig. 9B). immunohistochemical analysis of adult brain tissue with the anti-NuIP antibody discloses expression in the ventral midbrain dopaminergic group and extensive colocalization with TH immunoreactivity (Fig. 9C), which is readily apparent when the labeling for NuIP and TH are overlaid (Fig. 9Ca,f). This result strongly suggests that Nurr1 and NuIP are coexpressed within the dopaminergic cells in the adult mouse SN.
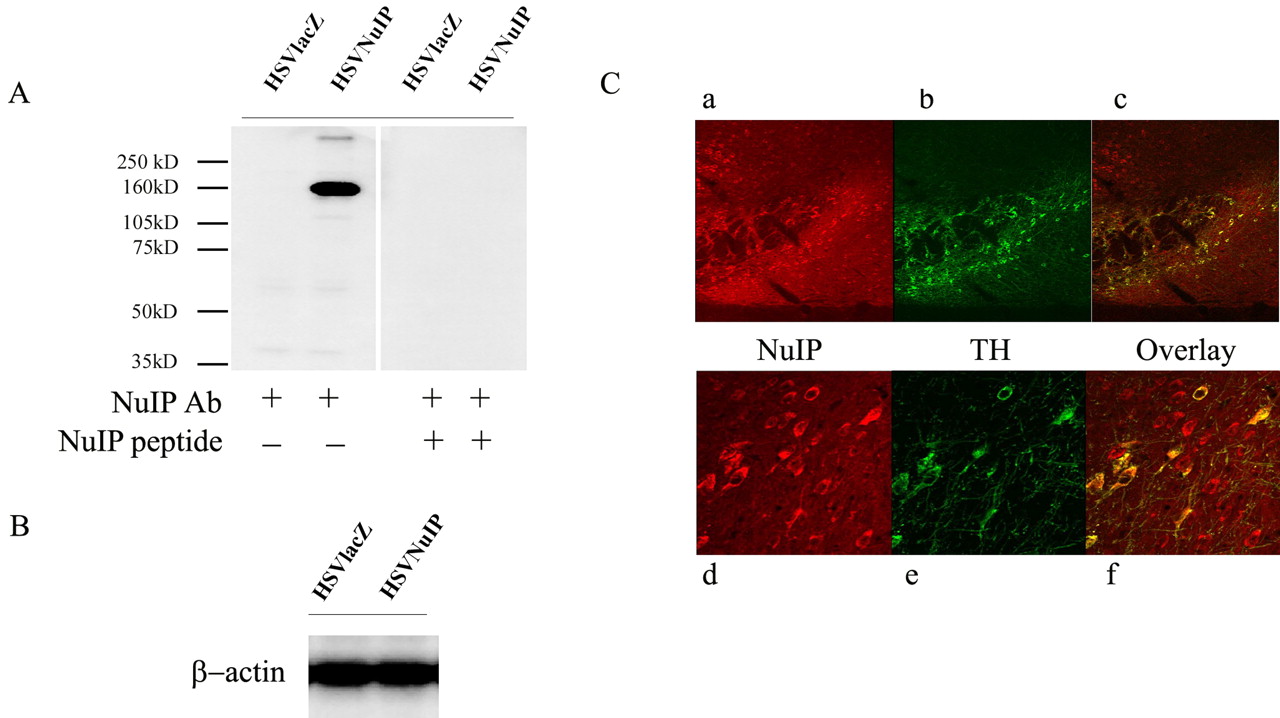
Coimmunolocalization of NuIP with TH in adult substantia nigra. A NuIP specific antibody was developed, tested, and used to detect NuIP protein expression. A, Cell lysates from a clonal MN9D cell line that were transfected with either HSVlacZ (lane 1) or HSVNuIP (lane 2) were subject to Western blotting using a NuIP antibody as described in Materials and Methods. Lanes 3 and 4 are incubated with NuIP antibody solution that has been preincubated by a NuIP peptide as described in Materials and Methods. B, The same cell lysates described as above were subject to Western blotting using a β-actin antibody to confirm equivalent protein loading. C, Adult mice (3 months old; n = 3) midbrains were sectioned and immunostained with anti-NuIP (Ca,d) and anti-TH (Cb,e) and each developed with a different fluorophore coupled secondary antibody. Sections were visualized by confocal microscopy at the level of the substantia nigra. Colocalization of expression in dopaminergic neurons is shown in the overlay channel (Cc,f at higher magnification).
Suppression of endogenous NuIP function results in decreased cell proliferation and expression of Nurr1 target gene
To investigate the outcome of NuIP loss of function, we used inducible siRNA to suppress the expression of NuIP protein. As shown in Figure 10A, two siRNA sequences target regions between the RUN and TBC domain that are specific for NuIP gene. Induction of siRNA expression by DOX led to a decrease in NuIP expression in MN9D cells both on the protein (Fig. 10B) and mRNA (Fig. 10C) levels, whereas the mock construct did not alter NuIP expression (Fig. 10B,C). Because Nurr1 has been demonstrated to be important in promoting a dopaminergic phenotype in cells and in regulating several downstream targets that are involved in dopamine synthesis and recycle, we tested whether suppression of NuIP function would affect the expression level of these Nurr1 target genes. The data show that suppression of NuIP function led to a reduction of DAT protein, a known Nurr1 target (Fig. 10B,E). To determine whether decreased NuIP function would alter dopaminergic MN9D cell phenotype, we examined cell number in siRNA induced and mock transfected cells. The data, shown in Figure 10D, reveal that reduction in NuIP function results in diminished cell numbers 72 h after siRNA induction. Whether the decrease in cell number is attributable to decreased cell proliferation or increased cell death needs further investigation.

Effects of NuIP knockdown in an engineered MN9D cell line. A, Domain structure of NuIP and sites targeted by inducibly expressed siRNA in corresponding mRNA. RUN domain, aa 44–189; TBC domain, 881–1053. siRNA generated from RNAi#1 vector targets the junction of exons 12 and 13, and siRNA from RNAi#2 targets exon 8. B, Inducible knockdown of NuIP protein. Stable MN9D cells expressing tetracycline repressor were transfected with pSUPERIOR constructs with or without siDNA inserts. Twenty-four hours after transfection, 2 μg/ml of DOX was added to induce siRNA expression. The cells were harvested 72 h after induction. Cell lysates were analyzed for NuIP, DAT, TH, and α-tubulin by Western blot. C, Inducible knockdown of NuIP mRNA. Stable MN9D cells expressing tetracycline repressor were transfected with pSUPERIOR constructs with or without siDNA inserts. Twenty four hours after transfection, 2 μg/ml of DOX was added to induce siRNA expression, and RNA was isolated 72 h after induction. NuIP transcript levels were quantified with qRT-PCR and normalized to 18S ribosomal RNA; averages of triplicate quantification are shown. Error bars indicate SD. D, MN9D cell numbers after inducible RNAi knockdown of NuIP mRNA. Stable MN9D cells expressing tetracycline repressor were treated the same way as described above. The results are means from seven independent experiments; error bars indicate SD. E, Downregulation of the DAT in stably transfected MN9D cells in which NuIP was knocked down. Cells grown as above were harvested 72 h after RNAi induction, protein lysates prepared, samples analyzed via Western blots, lanes scanned, and the intensity of DAT and TH expression determined. A significant difference was observed between mock and RNAi for DAT (p = 0.006). The results shown represent the means of 2–4 independent experiments. Error bars indicate SD. *p < 0.05; **p < 0.006; ***p < 0.01.
Discussion
Using the NLBD as bait in the yeast two-hybrid screen, we identified a novel NuIP. The NUIP gene generates a variety of transcription products via alternative splicing with the full-length NuIP producing a protein of ∼150 kDa, which is expressed primarily in the CNS and in a pattern that is similar to Nurr1 expression. The interaction of NuIP with Nurr1 in mammalian cells was confirmed both by a mammalian two-hybrid assay and coimmunoprecipitation experiments in the MN9D dopaminergic cell line and in endogenous mouse SN tissue lysates. Furthermore, NuIP appears to be a Nurr1 transcriptional coregulator. When coexpressed in mammalian cells, it promotes the transcriptional activity of Nurr1 both using NBRE driven reporters and endogenous TH promoters, which is a known target gene of Nurr1 (Iwawaki et al., 2000). Suppression of NuIP protein expression in dopaminergic MN9D cell resulted in decreased cell numbers and in decreased protein levels of another Nurr1 downstream target, the DAT, suggesting that endogenous NuIP protein function is required in maintaining the dopaminergic phenotype of these cells. Finally, immunostaining with a NuIP specific antisera demonstrated that NuIP is expressed in the adult midbrain dopaminergic neurons, in which Nurr1 functions in the development and maintenance of this cellular population. Together, these results indicate the identification of a novel protein that could function as a positive regulator of Nurr1 in vivo.
NuIP protein interacts with Nurr1 in a different manner compared with RXRα, because RXRα cannot form a heterodimer with the AF2-deleted NLBD (Perlmann and Jansson, 1995; Castro et al., 1999), whereas NuIP interacts with both the full-length and truncated forms of NLBD. This suggests that NuIP interacts with Nurr1 at a unique site from RXRα. Unlike other protein interactors that negatively regulate Nurr1 activity, such as p57kip2 (Joseph et al., 2003) and PIASγ (Galleguillos et al., 2004), NuIP potentiates the transcriptional activity of Nurr1 on both NBRE-containing and endogenous TH promoter reporter constructs. The mechanism underlying the ability of NuIP to positively regulate the transcriptional activity of Nurr1 is not clear. Nurr1 is a ligand-independent nuclear receptor whose active conformation is maintained by intramolecular interactions between the amino acid side chain of the NLBD (Wang et al., 2003). In the same study, Wang et al. (2003) evaluated the assembly of two LBD fragments and demonstrated that the assembly of the H1 and H3–12 domains of the NLBD correlates well with the transcriptional activity of Nurr1. Using this assembly assay, we tested the hypothesis that NuIP protein promotes the transcriptional activity of Nurr1 by promoting the assembly of NLBD domains and by doing so stabilizes the NLBD. Indeed, when NuIP was coexpressed with the two ligand-binding helical domains, its transcriptional activity was enhanced nearly twofold, suggesting that NuIP protein facilitates the assembly of NLBD, perhaps by acting as a scaffold.
To elucidate the precise function of NuIP protein, we showed in this study that overexpression of NuIP promoted Nurr1 function on TH promoter activity, whereas suppression of NuIP protein in dopaminergic cells decreased DAT gene expression. Cell numbers are also affected after 72 h of NuIP siRNA induction. However, whether this decrease in cell number is attributable to decreased cell proliferation or increased cell death is not clear. Our preliminary data show that, in addition to Nurr1, NuIP also interacts with Centrin 2 and Sec31 in a different yeast two-hybrid screening (F. Xing and H. J. Federoff, unpublished observations). Centrin 2 has been determined to localize in the centrosome of chromosomes and is involved in chromatin segregation, and Sec31 is a partner of Complex 1 of the ER to Golgi vesicle and is involved in protein trafficking. These data suggest that NuIP may play an important role in cell proliferation and in the biochemistry of neurotransmitter phenotype.
The NuIP gene was described previously as small G-protein signaling modulator 1 (Yang et al., 2007) and is found in a variety of species including Caenorhabditis elegans, mouse, rat, and human, suggesting that it is highly conserved across species (GenBank sequence analysis). Domain analyses identified an N-terminal RUN domain (Callebaut et al., 2001) and a C-terminal TBC domain (Nakamura et al., 1992) in NuIP amino acid sequences. Both the RUN and TBC domains are found in proteins that are actively involved in the GTPase signaling pathway (Callebaut et al., 2001). RUN domain-containing proteins are often found in Ras-related pathways (Takai et al., 2001) and could function as specific effectors for some proteins in the Ras superfamily (Callebaut et al., 2001). However, structural analysis also suggests the possibility that the RUN domain can harbor a catalytic activity (Rittinger et al., 1997; Scheffzek et al., 1997). TBC domains have been identified in many GTPase-activating proteins and have been demonstrated to function as GTPase-activators (Neuwald, 1997; Albert et al., 1999). The presence of both RUN and TBC domains in NuIP protein argues for a role in a GTPase signaling pathway. Identification of NuIP as an interactor and regulator of Nurr1 protein suggests the potential linkage of Nurr1 to Ras-like GTPase signaling pathways. We also observed that neither the RUN domain nor the TBC domain is sufficient for the interaction with Nurr1. Instead, certain NuIP specific sequences, together with the RUN domain, are required for its interaction with the Nurr1 protein (data not shown). Given the fact the NuIP protein is present in both cytosolic and nuclear compartments of the cell, it is very likely that the NuIP protein is a unique messenger for cross-talk between the Nurr1 protein and the Ras-like GTPase signaling pathway. It is known that NRs can be regulated by ligand-independent mechanisms, including several signaling pathways (Escriva et al., 1997). The orphan nuclear receptor, steroidogenic factor 1 (SF-1), is an example. It has been shown that the LBD activity and conformation of SF-1 can be regulated as a result of cross-talk with the mitogen-activated protein kinase signaling pathway (Desclozeaux et al., 2002). It is interesting to note that the signaling receptor tyrosine kinase Ret (Mason, 2000) can regulate the activity of Nurr1. A constitutively active oncogenic derivative of Ret suppresses the transcriptional activity of Nurr1 (Wang et al., 2003). Because NuIP contains TBC domains that may perhaps turn off a signaling cascade through its GTPase-activating function (Takai et al., 2001), it is tempting to speculate that it may modulate the action of Ret. Additional experiments are required to test this possibility.
NuIP is expressed in the midbrain dopaminergic neurons placing it in an appropriate cellular context to function as a Nurr1 coregulator. The RT-PCR results demonstrate that NuIP transcripts and protein are present in Nurr1 positive midbrain dopaminergic tissue. The immunocytochemistry data demonstrate that NuIP expression in cells overlaps with those dopaminergic TH positive cells in the midbrain. Sustained expression of NuIP in midbrain dopaminergic neurons suggests it might have a role in the cell biology of these cells. As NuIP may be a member of a G-protein-related signaling pathway, it is postulated that it is involved and linked to multiple downstream pathways. Therefore, it is not surprising that NuIP protein is not exclusively expressed in TH-expressing cells. Searching in the GeneBank sequence revealed the presence of NuIP homologous genes in a variety of species. The roles of these related molecules await further investigation.
In summary, we have identified a novel interactor and coregulator of Nurr1, termed NuIP, which positively regulates the transcriptional activity of Nurr1 and which may mediate cross- talk between Nurr1 and GTPase mediated signaling pathways.
Footnotes
-
This work was supported by Department of Defense Grant DAMD17-02-1-0695 (H.J.F.). We thank members of the Federoff laboratory for useful discussions, particularly the Parkinson's Disease Group, and Dr. Maguire-Zeiss. In particular, special thanks to Dr. Linda Callahan for invaluable assistance with this manuscript and figure preparation. We thank Dr. Anthony A. Lanahan for the yeast 2-hybrid plasmids. We also thank Drs. Tim Mhyre and Barry Hoffer for critical reading of this manuscript.
- Correspondence should be addressed to Dr. Howard J. Federoff at his present address: Office of the Executive Vice President and Executive Dean, Georgetown University Medical Center, 4000 Reservoir Road, NW, 120 Building D, Washington, DC 20007. hjf8{at}georgetown.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}